
Incorporation of the New anti-Octadecaborane Laser Dyes into Thin Polymer Films: A Temperature-Dependent Photoluminescence and Infrared Spectroscopy Study

 , ,
, ,  , , and
, , and 
Abstract
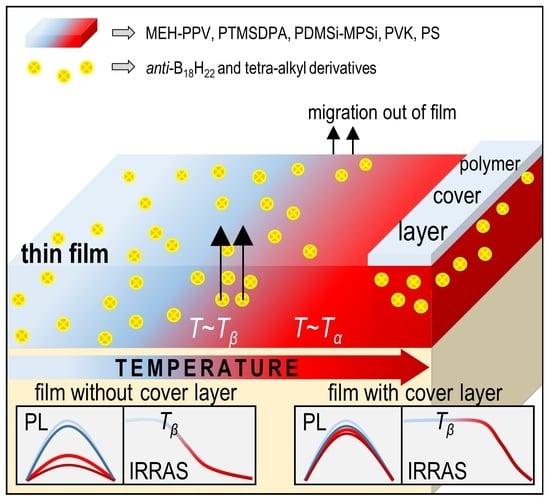
:
1. Introduction
2. Results and Discussion
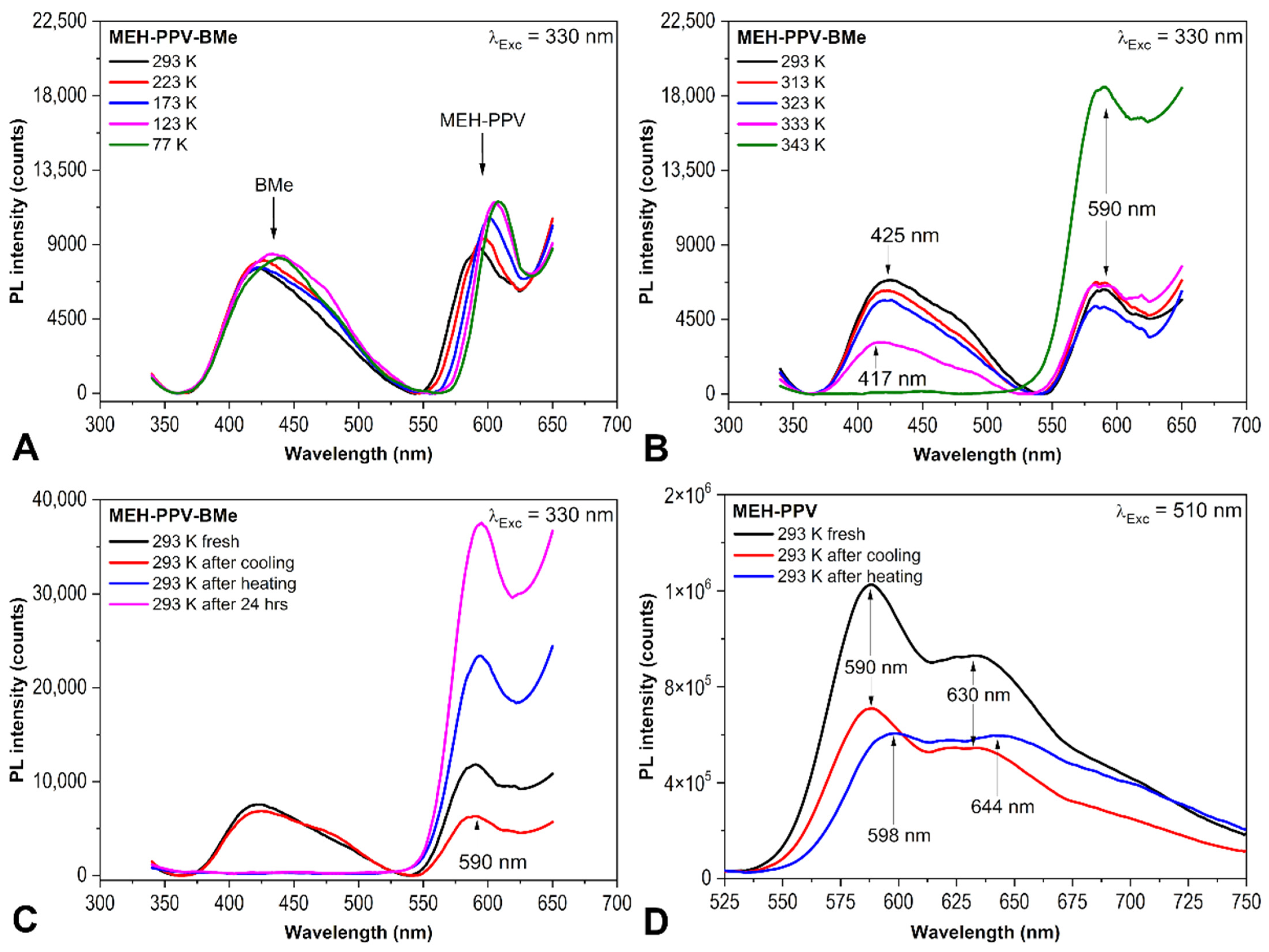
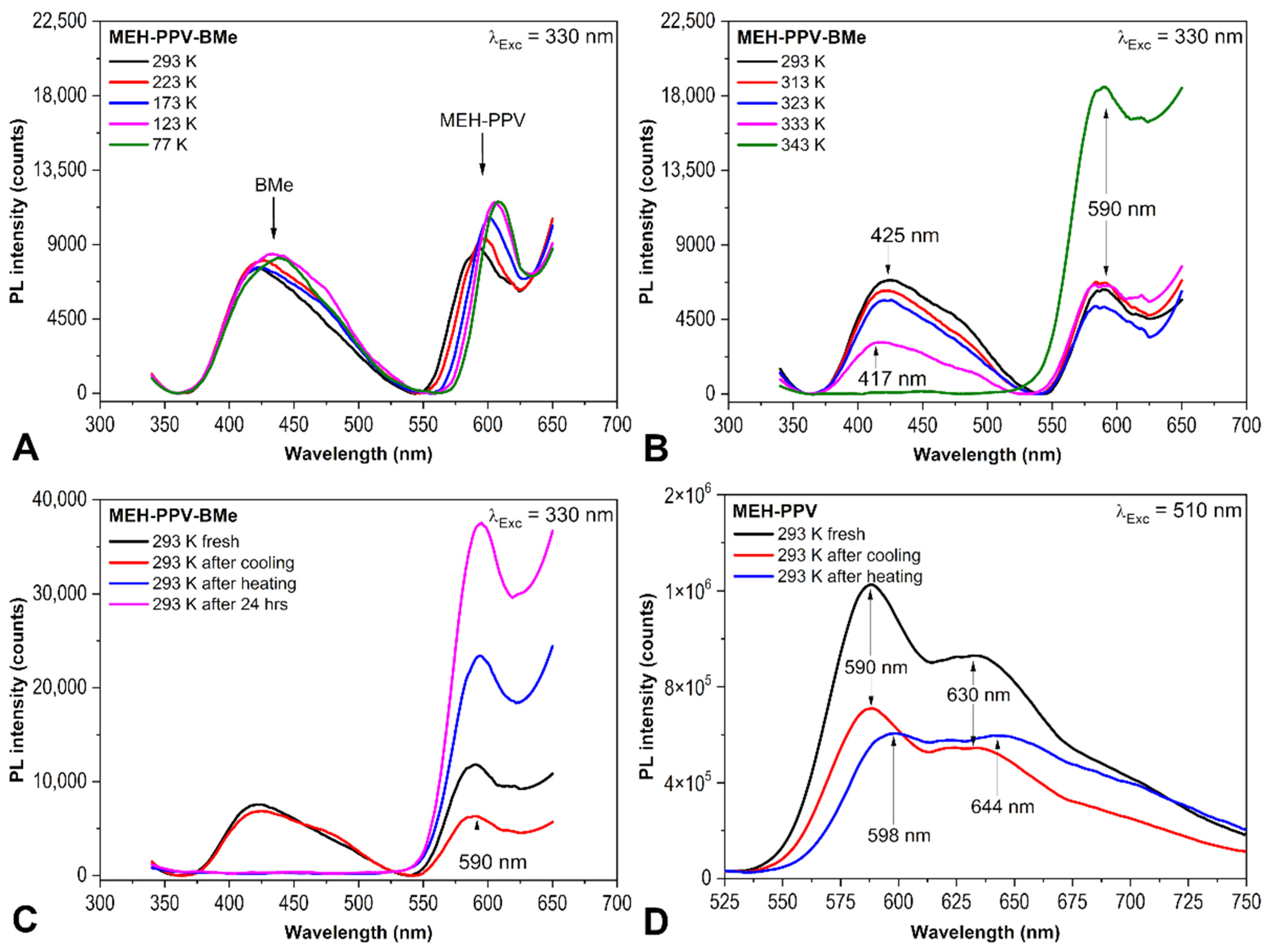
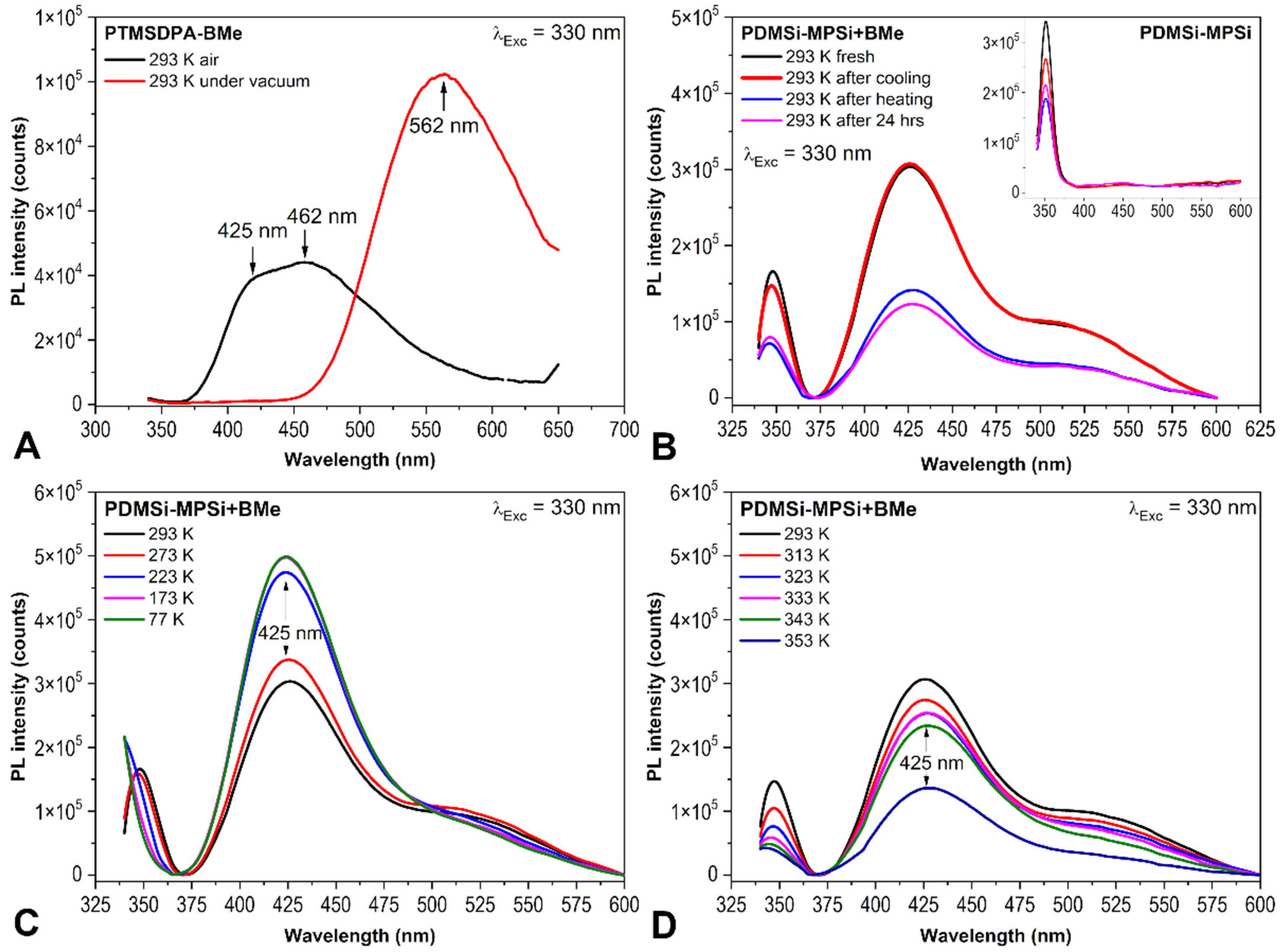
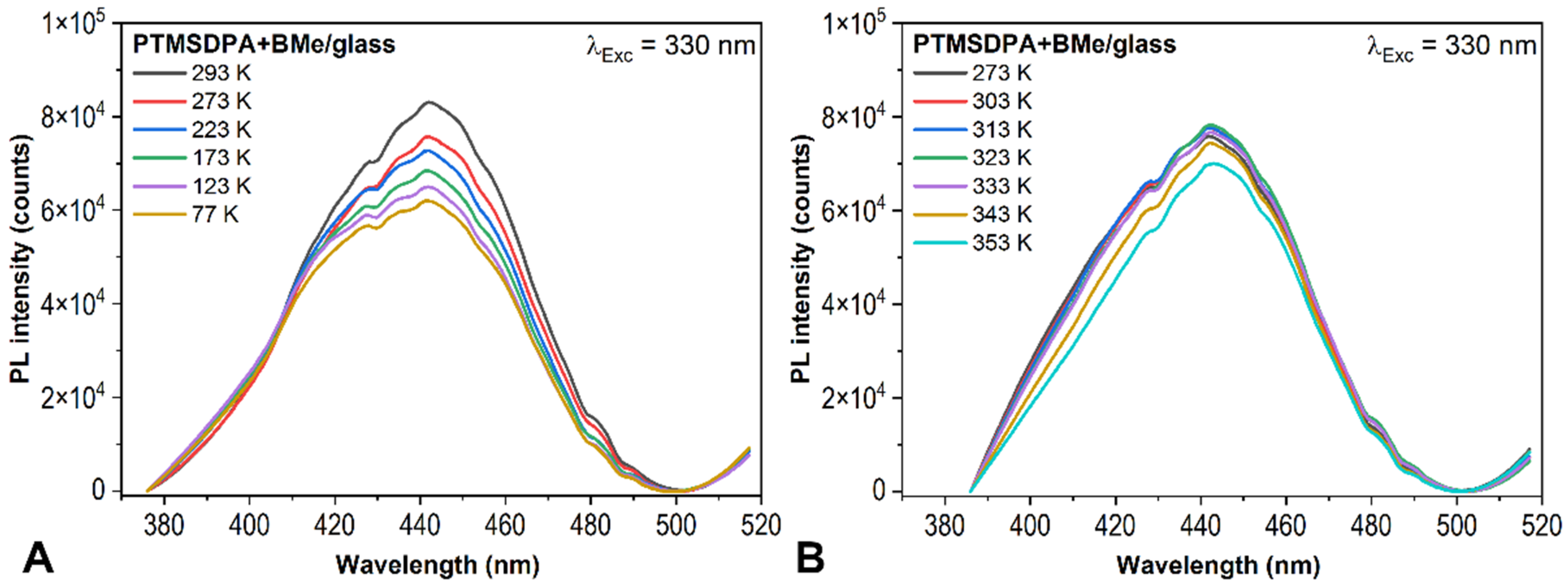
2.1. PL Spectroscopy Experiments
2.2. PM-IRRAS Experiments
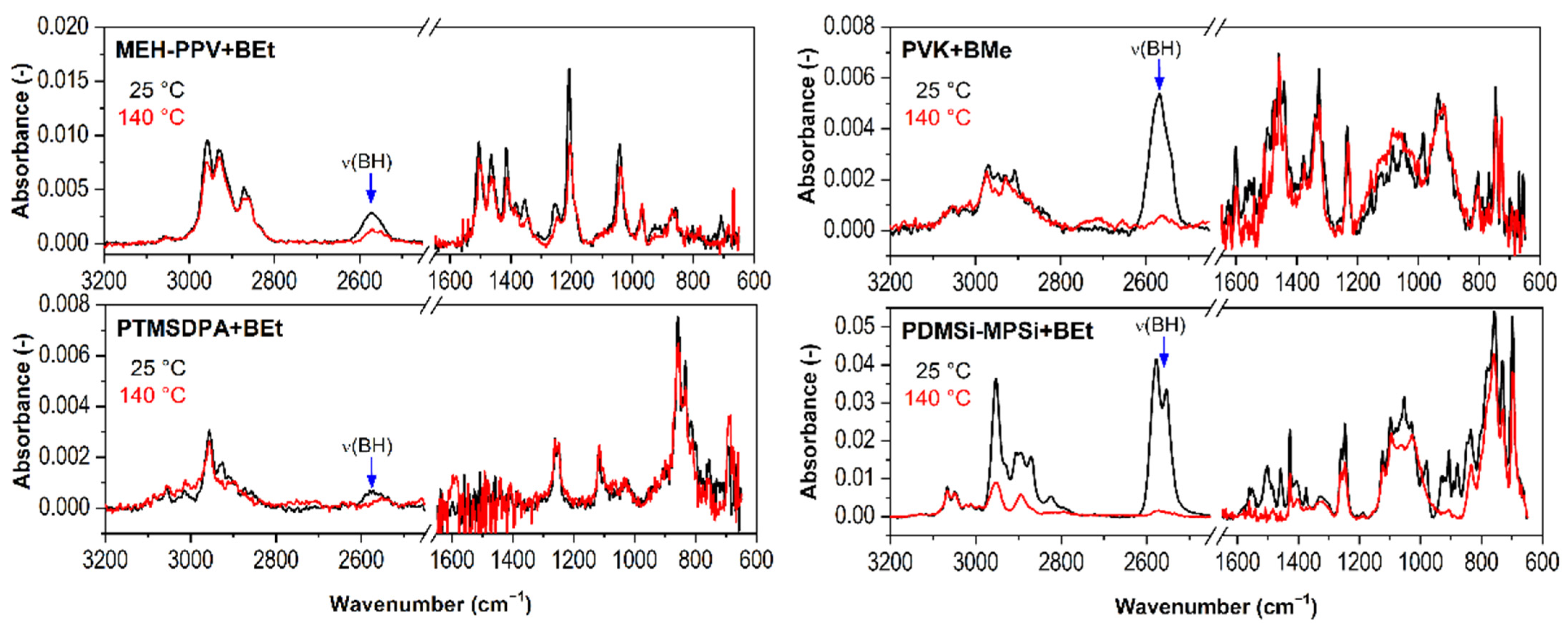
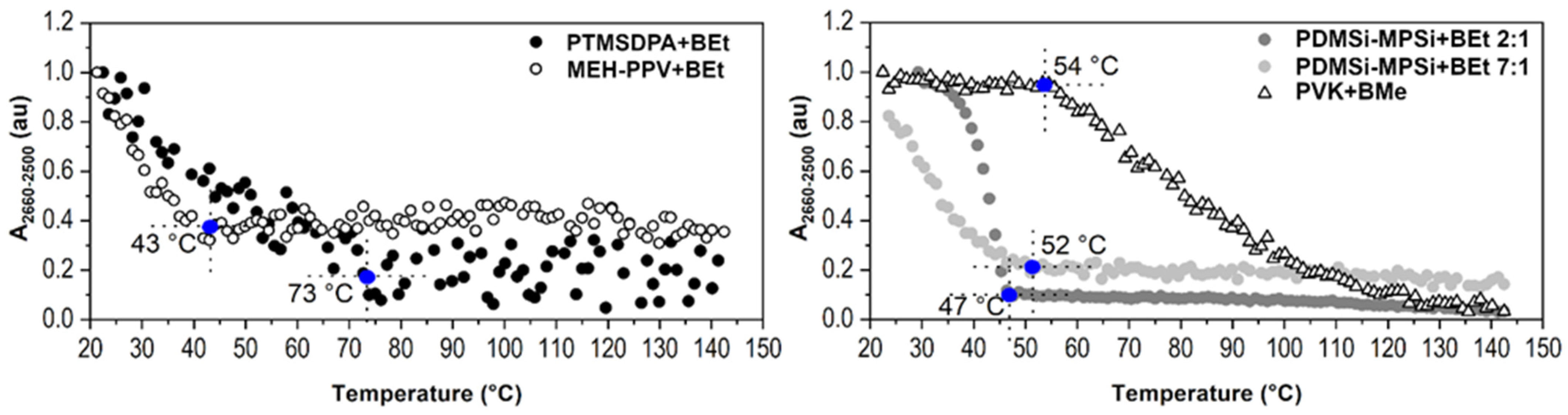
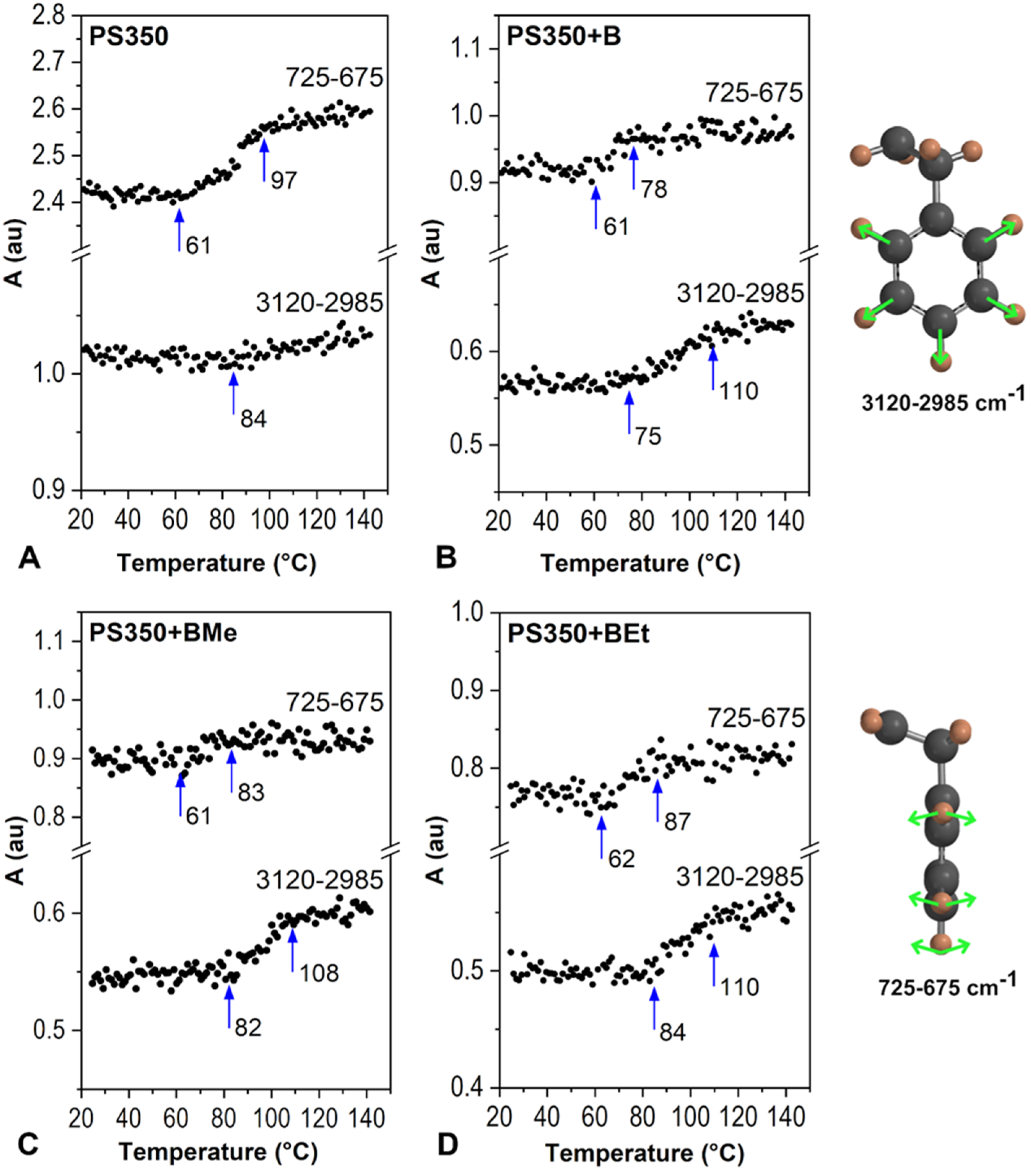
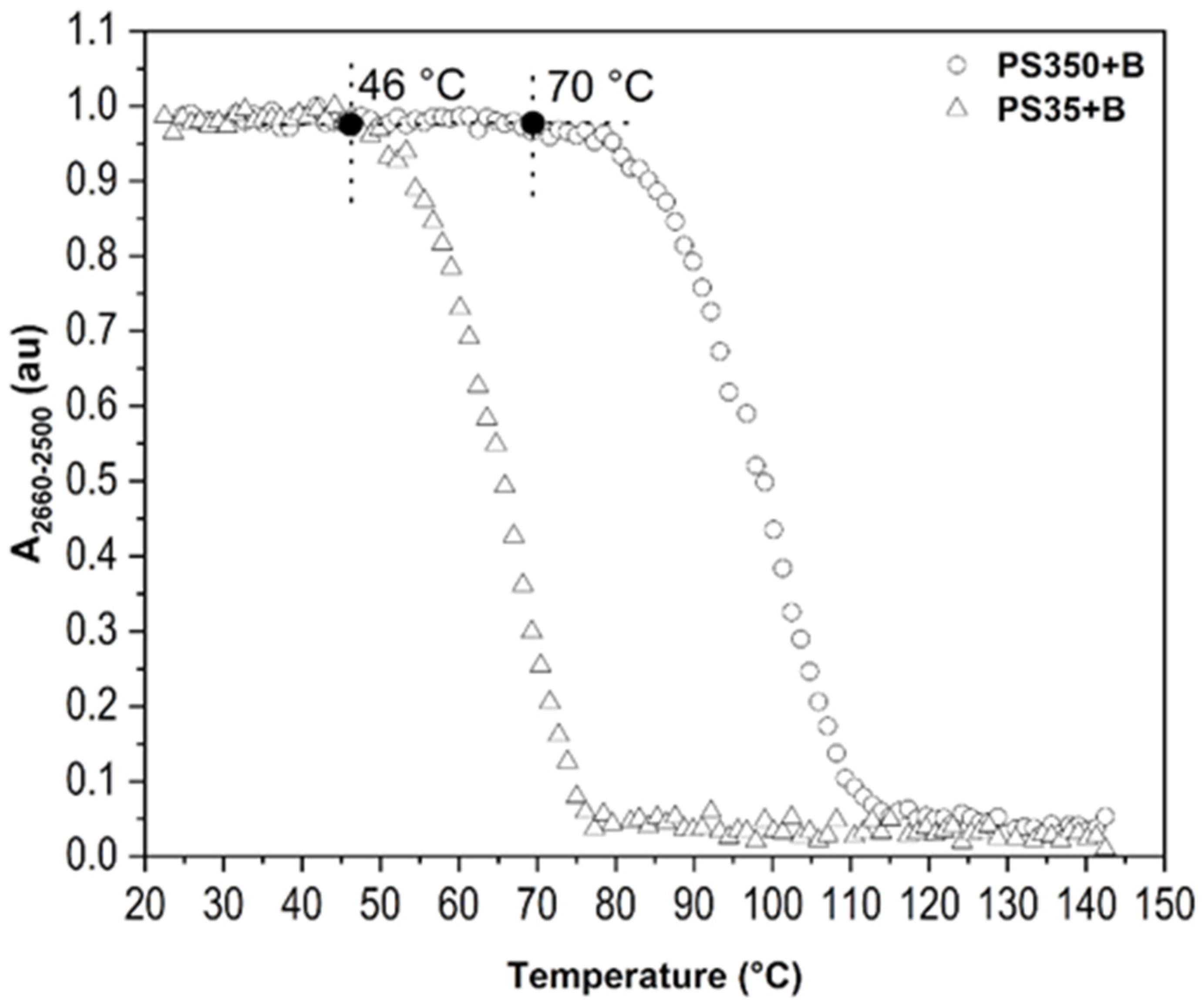
2.2.1. Type of Borane Cluster
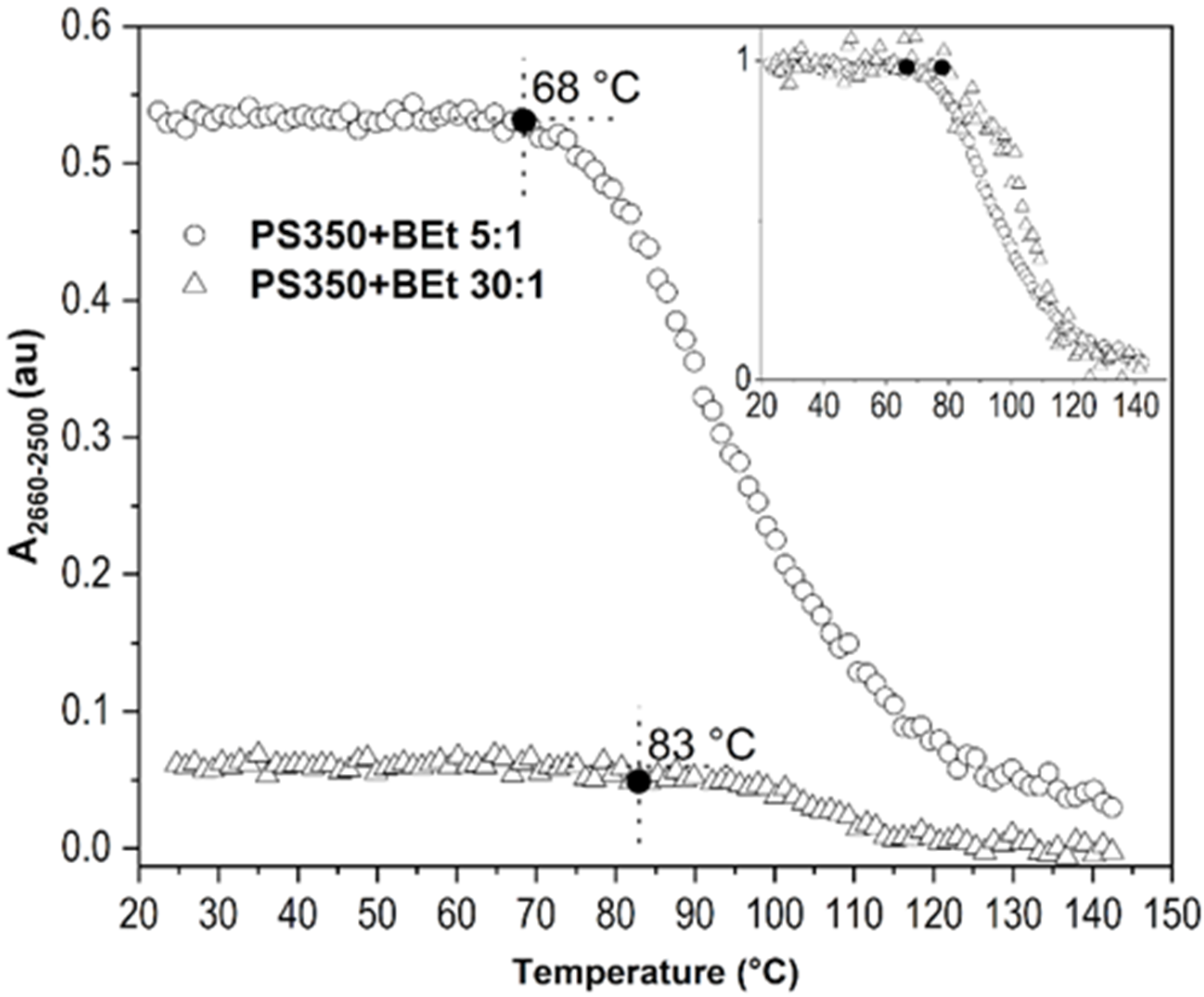
2.2.2. Concentration of Borane Cluster
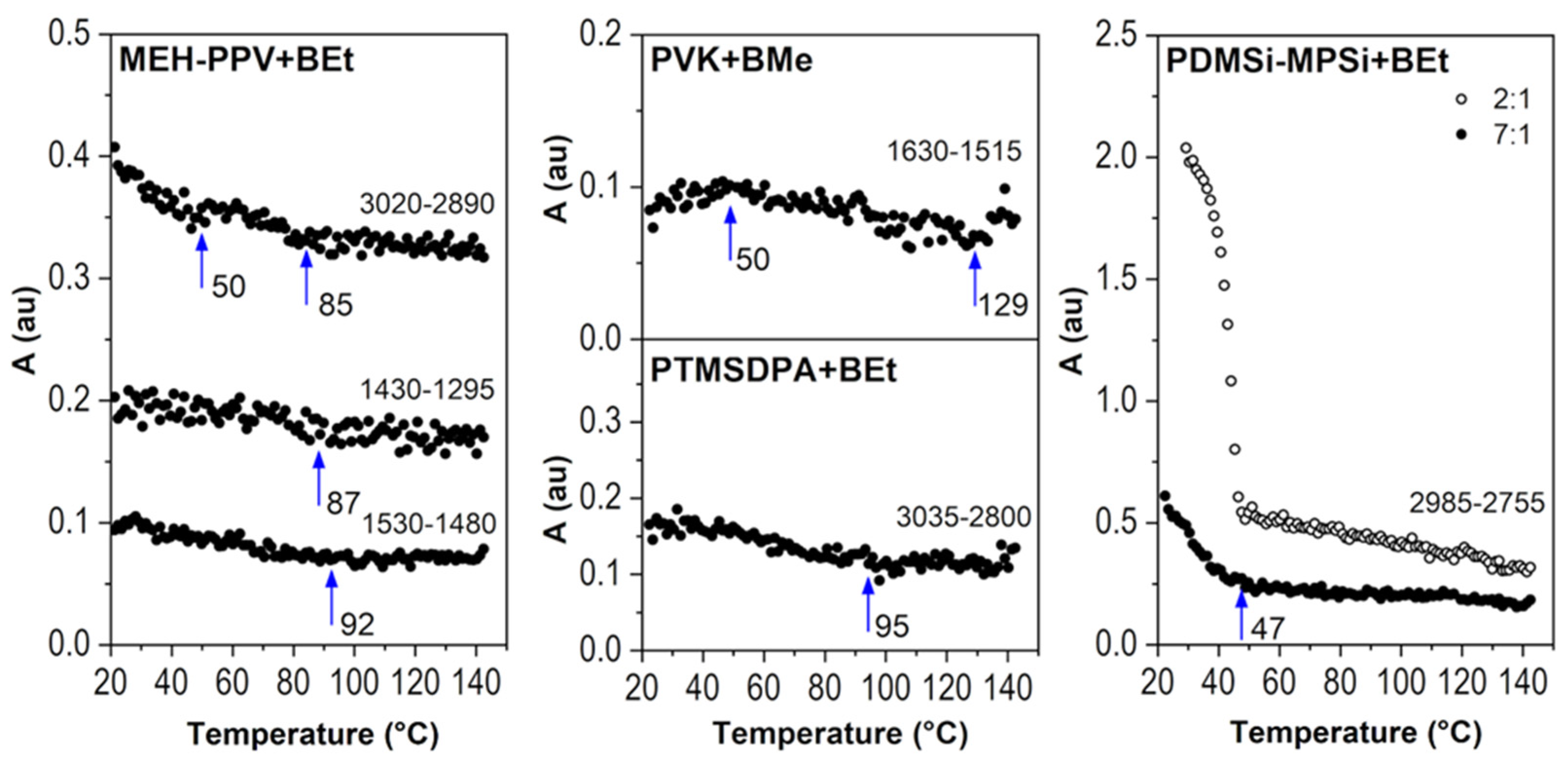
2.2.3. Molecular Weight of Polymer
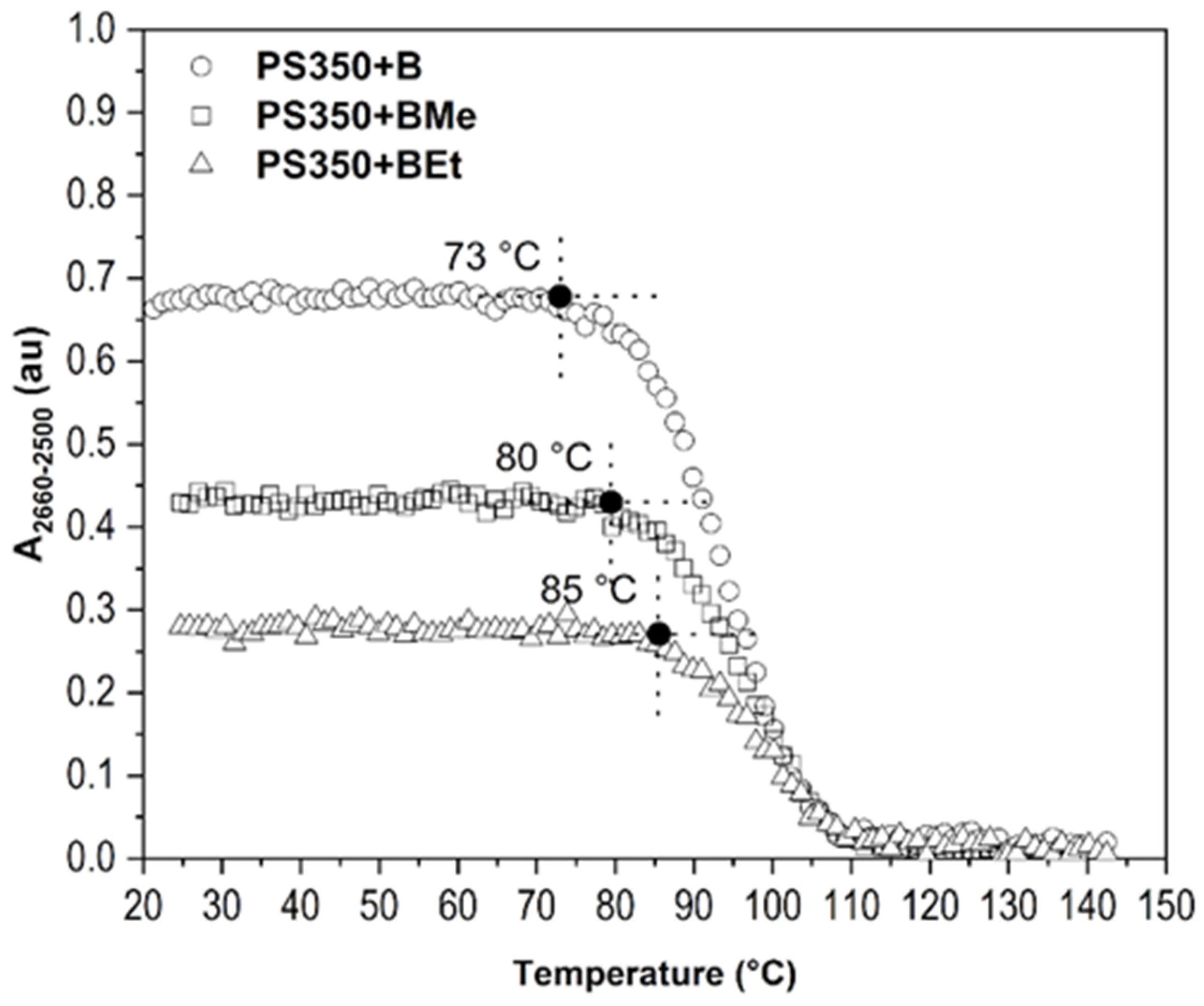
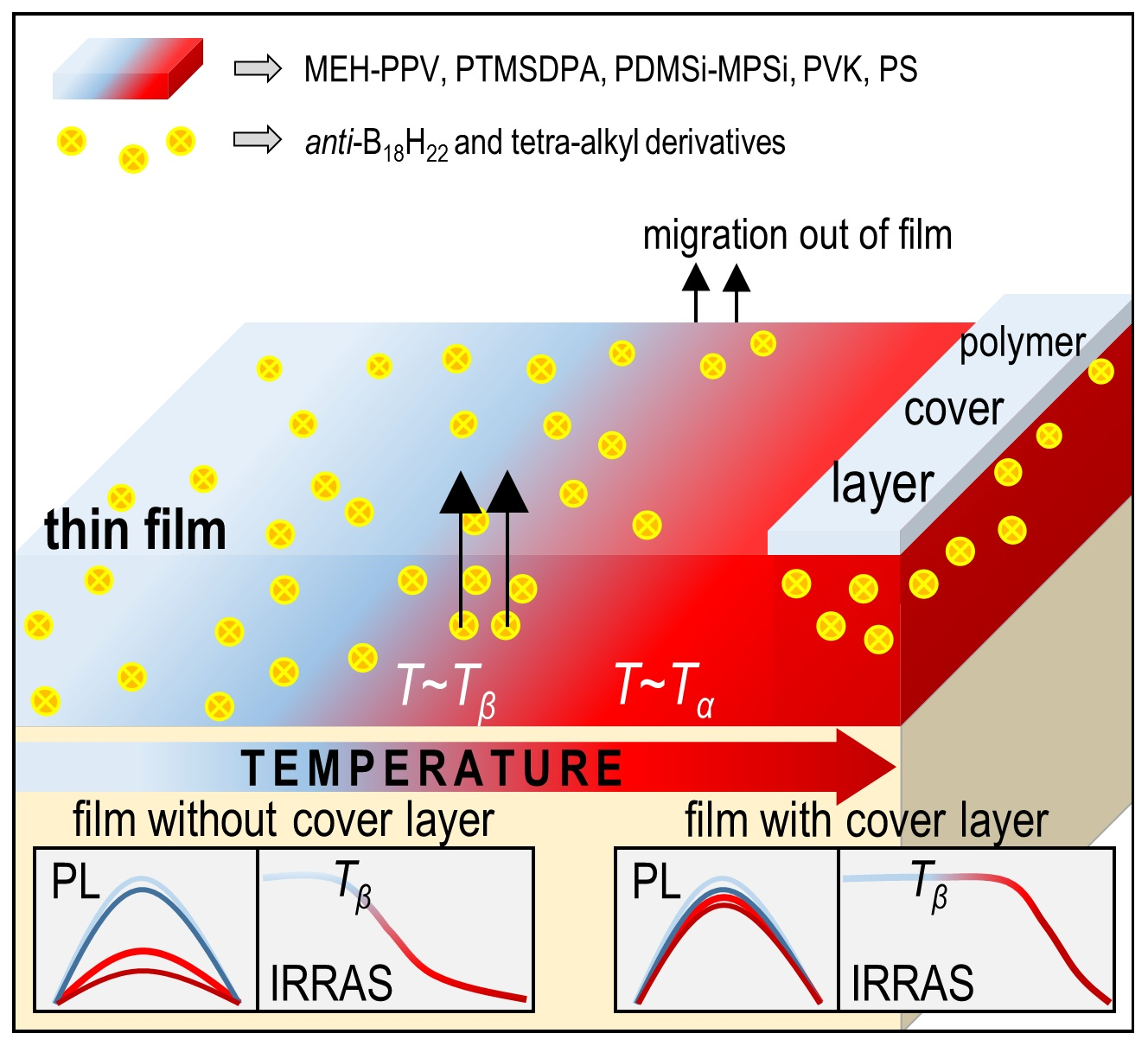
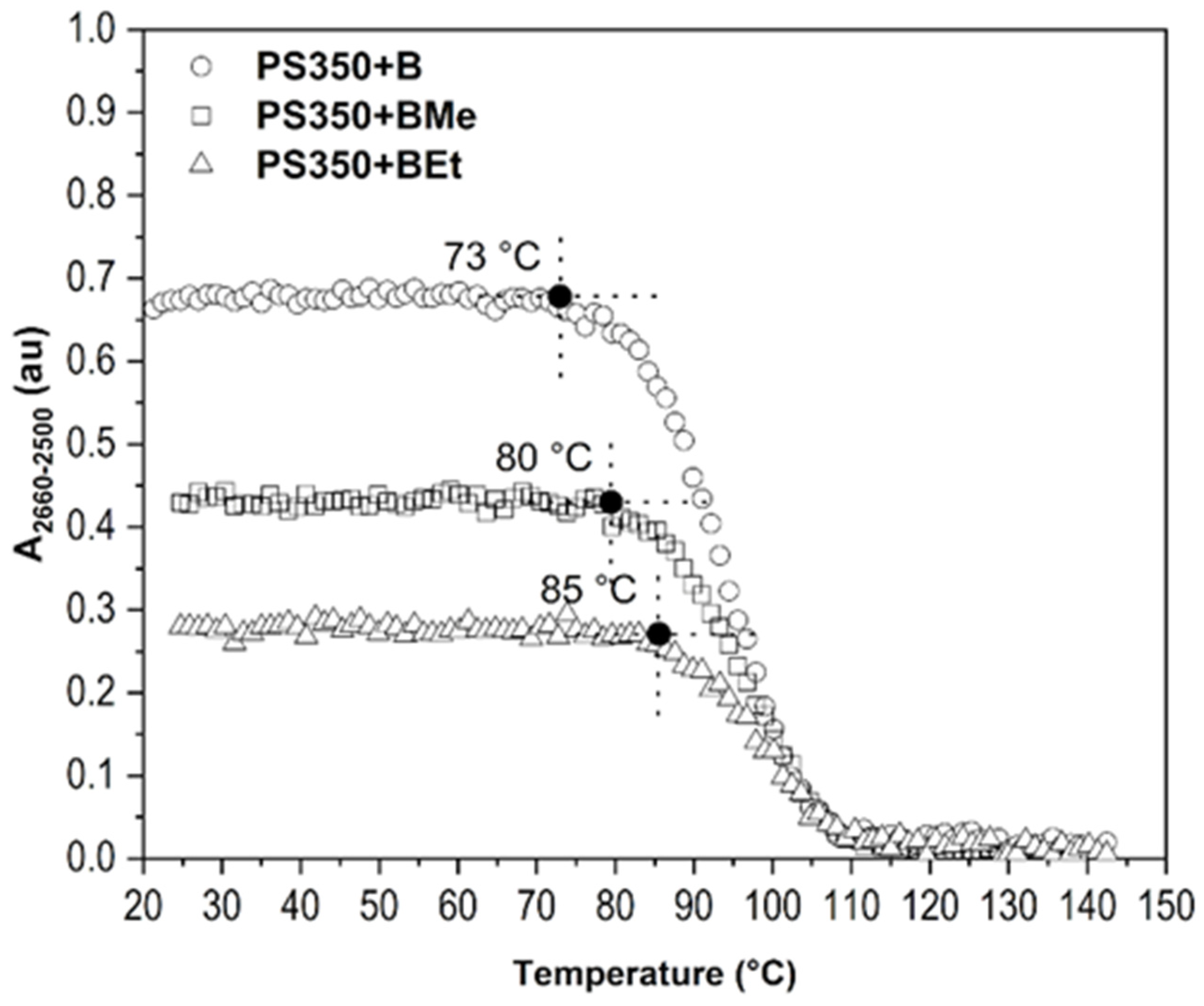
2.3. Improvement of Thin Film Temperature Stability
3. Materials and Methods
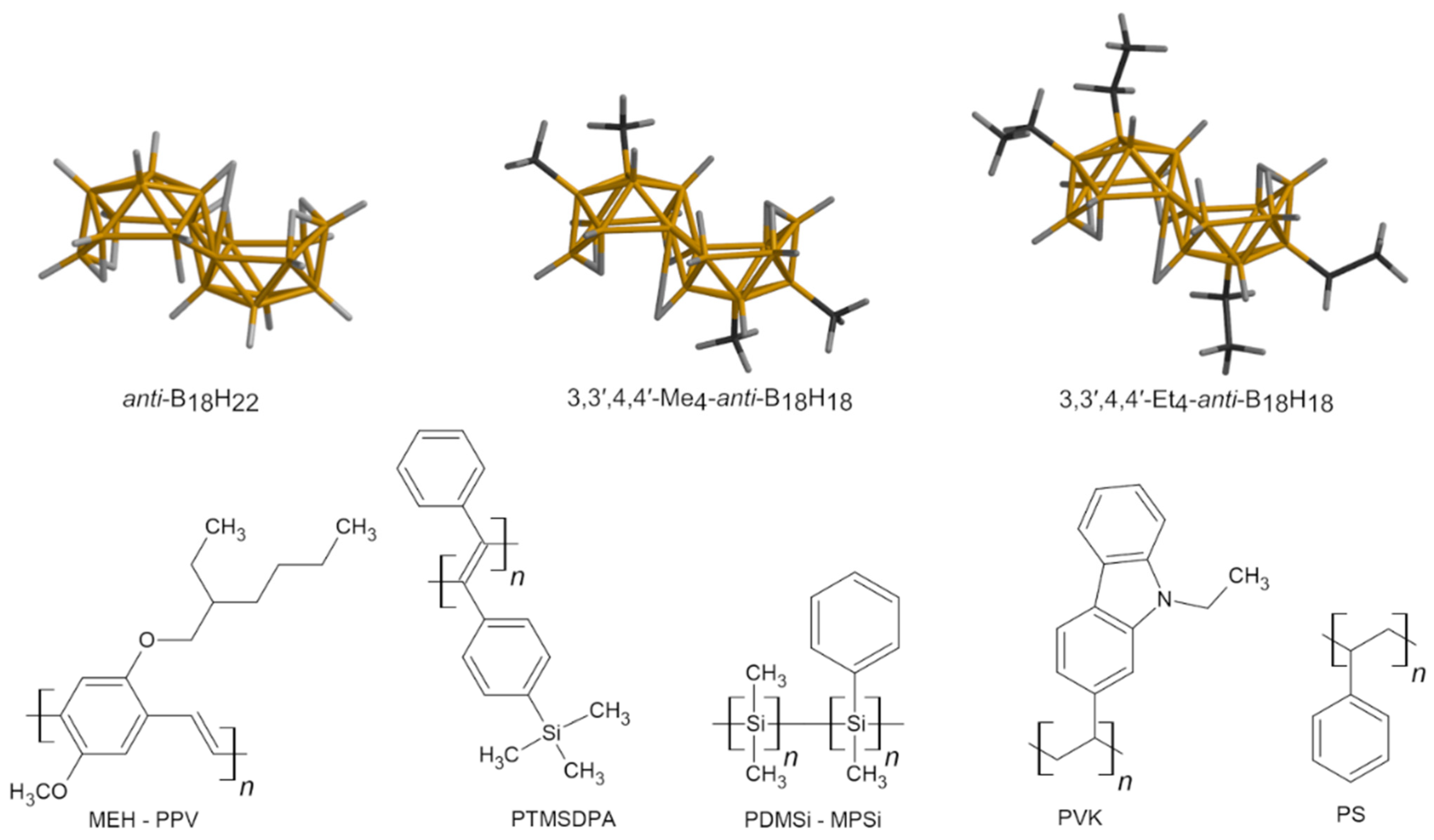
3.1. Materials
3.2. Thin Film Preparation
3.3. Film Thickness and Surface Structure
3.4. Photoluminescence Spectroscopy
3.5. Polarization-Modulation Infrared Reflection–Absorption Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sevcik, J.; Urbanek, P.; Skoda, D.; Jamatia, T.; Nadazdy, V.; Urbanek, M.; Antos, J.; Munster, L.; Kuritka, I. Energy resolved-electrochemical impedance spectroscopy investigation of the role of Al-doped ZnO nanoparticles in electronic structure modification of polymer nanocomposite LEDs. Mater. Des. 2021, 205, 109738. [Google Scholar] [CrossRef]
- Lee, J.H.; Chen, C.H.; Lee, P.H.; Lin, H.Y.; Leung, M.K.; Chiu, T.L.; Lin, C.F. Blue organic light-emitting diodes: Current status, challenges, and future outlook. J. Mater. Chem. C 2019, 7, 5874–5888. [Google Scholar] [CrossRef]
- Zhu, M.R.; Yang, C.L. Blue fluorescent emitters: Design tactics and applications in organic light-emitting diodes. Chem. Soc. Rev. 2013, 42, 4963–4976. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.R.; Zou, Y.; Chen, W.Q.; Huang, Y.J.; Yao, C.J.; Zhang, Q.C. The Role of Weak Molecular Dopants in Enhancing the Performance of Solution-Processed Organic Field-Effect Transistors. Adv. Electron. Mater. 2019, 5, 1800547. [Google Scholar] [CrossRef]
- Cerdan, L.; Braborec, J.; Garcia-Moreno, I.; Costela, A.; Londesborough, M. A borane laser. Nat. Commun. 2015, 6, 5958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevcik, J.; Urbanek, P.; Hanulikova, B.; Capkova, T.; Urbanek, M.; Antos, J.; Londesborough, M.; Bould, J.; Ghasemi, B.; Petrkovsky, L.; et al. The Photostability of Novel Boron Hydride Blue Emitters in Solution and Polystyrene Matrix. Materials 2021, 14, 589. [Google Scholar] [CrossRef]
- Ma, M.; Guo, Y. Physical Properties of Polymers Under Soft and Hard Nanoconfinement: A Review. Chin. J. Polym. Sci. 2020, 38, 565–578. [Google Scholar] [CrossRef]
- Jin, K.L.; Torkelson, J.M. T-g-confinement effects in strongly miscible blends of poly(2,6-dimethyl-1,4-phenylene oxide) and polystyrene: Roles of bulk fragility and chain segregation. Polymer 2017, 118, 85–96. [Google Scholar] [CrossRef]
- Forrest, J.A.; DalnokiVeress, K.; Stevens, J.R.; Dutcher, J.R. Effect of free surfaces on the glass transition temperature of thin polymer films. Phys. Rev. Lett. 1996, 77, 4108. [Google Scholar] [CrossRef]
- Kim, S.; Roth, C.B.; Torkelson, J.M. Effect of Nanoscale Confinement on the Glass Transition Temperature of Free-Standing Polymer Films: Novel, Self-Referencing Fluorescence Method. J. Polym. Sci. Part B Polym. Phys. 2008, 46, 2754–2764. [Google Scholar] [CrossRef]
- Tsui, O.; Russell, T.P.; Hawker, C.J. Effect of interfacial interactions on the glass transition of polymer thin films. Macromolecules 2001, 34, 5535–5539. [Google Scholar] [CrossRef]
- Glor, E.C.; Angrand, G.V.; Fakhraai, Z. Exploring the broadening and the existence of two glass transitions due to competing interfacial effects in thin, supported polymer films. J. Chem. Phys. 2017, 146, 203330. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, J.; Forrest, J.A.; Borjesson, L. Quantifying glass transition behavior in ultrathin free-standing polymer films. Phys. Rev. E 2000, 62, 5187–5200. [Google Scholar] [CrossRef] [PubMed]
- Priestley, R.D.; Broadbelt, L.J.; Torkelson, J.M.; Fukao, K. Glass transition and alpha-relaxation dynamics of thin films of labeled polystyrene. Phys. Rev. E 2007, 75, 061806. [Google Scholar] [CrossRef] [Green Version]
- Priestley, R.D.; Ellison, C.J.; Broadbelt, L.J.; Torkelson, J.M. Structural relaxation of polymer glasses at surfaces, interfaces and in between. Science 2005, 309, 456–459. [Google Scholar] [CrossRef]
- Li, Q.F.; Hua, R.; Cheah, I.J.; Chou, K.C. Surface structure relaxation of poly(methyl methacrylate). J. Phys. Chem. B 2008, 112, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Serghei, A.; Mikhailova, Y.; Huth, H.; Schick, C.; Eichhorn, K.J.; Voit, B.; Kremer, F. Molecular dynamics of hyperbranched polyesters in the confinement of thin films. Eur. Phys. J. E 2005, 17, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Badia, J.D.; Teruel-Juanes, R.; Acebo, C.; Gil-Castell, O.; Serra, A.A. Ribes-Greus, Dielectric spectroscopy of novel thiol-ene/epoxy thermosets obtained from allyl-modified hyperbranched poly(ethyleneimine) and diglycidylether of bisphenol A. Eur. Polym. J. 2019, 113, 98–106. [Google Scholar] [CrossRef]
- Burroughs, M.J.; Christie, D.; Gray, L.A.G.; Chowdhury, M.; Priestley, R.D. 21st Century Advances in Fluorescence Techniques to Characterize Glass-Forming Polymers at the Nanoscale. Macromol. Chem. Phys. 2018, 219, 1700368. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.M.; Lu, Y.L.; Duan, Y.X.; Yan, S.K.; Shen, D.Y. Glass transition temperature determination of poly(ethylene terephthalate) thin films using reflection-absorption FTIR. Macromolecules 2004, 37, 2532–2537. [Google Scholar] [CrossRef]
- Hajduk, B.; Bednarski, H.; Trzebicka, B. Temperature-dependent spectroscopic ellipsometry of thin polymer films. J. Phys. Chem. B 2013, 54, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Singh, L.; Ludovice, P.J.; Henderson, C.L. Influence of molecular weight and film thickness on the glass transition temperature and coefficient of thermal expansion of supported ultrathin polymer films. Thin Solid Films 2004, 449, 231–241. [Google Scholar] [CrossRef]
- Haramina, T.; Kirchheim, R.; Tibrewala, A.; Peiner, E. Mechanical spectroscopy of thin polystyrene films. Polymer 2008, 49, 2115–2118. [Google Scholar] [CrossRef]
- Londesborough, M.G.S.; Hnyk, D.; Bould, J.; Serrano-Andres, L.; Sauri, V.; Oliva, J.M.; Kubat, P.; Polivka, T.; Lang, K. Distinct Photophysics of the Isomers of B18H22 Explained. Inorg. Chem. 2012, 51, 1471–1479. [Google Scholar] [CrossRef] [Green Version]
- Bould, J.; Lang, K.M.; Kirakci, K.; Cerdan, L.; Roca-Sanjuan, D.; Frances-Monerris, A.; Clegg, W.; Waddell, P.G.; Fuciman, M.; Polivka, T.; et al. A Series of Ultra-Efficient Blue Borane Fluorophores. Inorg. Chem. 2020, 59, 17058–17070. [Google Scholar] [CrossRef]
- Hanulikova, B.; Capkova, T.; Antos, J.; Urbanek, M.; Urbanek, P.; Sevcik, J.; Kuritka, I. Temperature dependence of vibrational motions of thin polystyrene films by infrared reflection-absorption spectroscopy: A single measurement tool for monitoring of glass transition and temperature history. Polym. Test. 2021, 101, 107305. [Google Scholar] [CrossRef]
- Maliakal, A.J. Characterization of Dopant Diffusion within Semiconducting Polymer and Small-Molecule Films Using Infrared-Active Vibrational Modes and Attenuated Total Reflectance Infrared Spectroscopy. ACS Appl. Mater. Interfaces 2013, 5, 8300–8307. [Google Scholar] [CrossRef] [PubMed]
- Reiser, P.; Muller, L.; Sivanesan, V.; Lovrincic, R.; Barlow, S.; Marder, S.R.; Pucci, A.; Jaegermann, W.; Mankel, E.; Beck, S. Dopant Diffusion in Sequentially Doped Poly(3-hexylthiophene) Studied by Infrared and Photoelectron Spectroscopy. J. Phys. Chem. C 2018, 122, 14518–14527. [Google Scholar] [CrossRef]
- Sauri, V.; Oliva, J.M.; Hnyk, D.; Bould, J.; Braborec, J.; Merchan, M.; Kubat, P.; Cisarova, I.; Lang, K.; Londesborough, M.G.S. Tuning the Photophysical Properties of anti-B18H22: Efficient Intersystem Crossing between Excited Singlet and Triplet States in New 4,4′-(HS)(2)-anti-B18H20. Inorg. Chem. 2013, 52, 9266–9274. [Google Scholar] [CrossRef]
- Londesborough, M.G.S.; Dolansky, J.; Bould, J.; Braborec, J.; Kirakci, K.; Lang, K.; Cisarova, I.; Kubat, P.; Roca-Sanjuan, D.; Frances-Monerris, A.; et al. Effect of Iodination on the Photophysics of the Laser Borane anti-B18H22: Generation of Efficient Photosensitizers of Oxygen. Inorg. Chem. 2019, 58, 10248–10259. [Google Scholar] [CrossRef]
- Cossiello, R.F.; Kowalski, E.; Rodrigues, P.C.; Akcelrud, L.; Bloise, A.C.; deAzevedo, E.R.; Bonagamba, T.J.; Atvars, T. Photoluminescence and relaxation processes in MEH-PPV. Macromolecules 2005, 38, 925–932. [Google Scholar] [CrossRef]
- Botiz, I.; Freyberg, P.; Leordean, C.; Gabudean, A.M.; Astilean, S.; Yang, A.; Stingelin, N. Enhancing the Photoluminescence Emission of Conjugated MEH-PPV by Light Processing. ACS Appl. Mater. Interfaces 2014, 6, 4974–4979. [Google Scholar] [CrossRef]
- Moncada, J.; Terlier, T.; Ivanov, I.N.; Dadmun, M.D. Correlation of the Structure with Performance in MEH-PPV/dPS Thin Films Illuminated during Processing. ACS Appl. Polym. Mater. 2021, 3, 3821–3830. [Google Scholar] [CrossRef]
- Varshni, Y.P. Temperature dependence of energy gap in semiconductors. Physica 1967, 34, 149–154. [Google Scholar] [CrossRef]
- Kuritka, I.; Sedlarik, V.; Harea, D.; Harea, E.; Urbanek, P.; Sloufova, I.; Coufal, R.; Zednik, J. Polymer Labelling with a Conjugated Polymer-Based Luminescence Probe for Recycling in the Circular Economy. Polymers 2020, 12, 1226. [Google Scholar] [CrossRef] [PubMed]
- Toy, L.G.; Nagai, K.; Freeman, B.D.; Pinnau, I.; He, Z.; Masuda, T.; Teraguchi, M.; Yampolskii, Y.P. Pure-gas and vapor permeation and sorption properties of poly[1-phenyl-2-[p-(trimethylsilyl)phenyl]acetylene] (PTMSDPA). Macromolecules 2000, 33, 2516–2524. [Google Scholar] [CrossRef]
- Kwak, G.; Fukao, S.; Fujiki, M.; Sakaguchi, T.; Masuda, T. Temperature-dependent, static, and dynamic fluorescence properties of disubstituted acetylene polymer films. Chem. Mater. 2006, 18, 2081–2085. [Google Scholar] [CrossRef]
- Pipertzis, A.; Hossain, M.D.; Monteiro, M.J.; Floudas, G. Segmental Dynamics in Multicyclic Polystyrenes. Macromolecules 2018, 51, 1488–1497. [Google Scholar] [CrossRef]
- van Krevelen, D.W. (Ed.) Chapter 4—Volumetric Properties. In Properties of Polymers, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 1997; pp. 71–107. [Google Scholar]
- Suzuki, H. Temperature dependence of the electroluminescent characteristics of light-emitting diodes made from poly(methylphenylsilane). Adv. Mater. 1996, 8, 657–659. [Google Scholar] [CrossRef]
- Meenu, K.M.; Bag, D.S.; Lagarkha, R. Synthesis of Functional Photoactive Polysilane Copolymers with Disperse Yellow 7 Methacrylate and Study of their Optical, Photophysical and Thermal Properties. J. Polym. Mater. 2019, 36, 275–292. [Google Scholar] [CrossRef]
- White, R.P.; Lipson, J.E.G. Polymer Free Volume and Its Connection to the Glass Transition. Macromolecules 2016, 49, 3987–4007. [Google Scholar] [CrossRef]
- Kazukauskas, V.; Cyras, V.; Pranaitis, M.; Apostoluk, A.; Rocha, L.; Sicot, L.; Raimond, P.; Sentein, C. Influence of polar molecular chain orientation on optical and carrier transport properties of polymer blends. Org. Electron. 2007, 8, 21–28. [Google Scholar] [CrossRef]
- Giro, G.; di Marco, P.G.; Pizzoli, M.; Ceccorulli, G. Detection of polymer thermal transitions by excimer fluorescence, poly-n-vinylcarbazole. Chem. Phys. Lett. 1988, 150, 159–164. [Google Scholar] [CrossRef]
- Deppe, D.D.; Dhinojwala, A.; Torkelson, J.M. Small molecule probe diffusion in thin polymer films near the glass transition: A novel approach using fluorescence nonradiative energy transfer. Macromolecules 1996, 29, 3898–3908. [Google Scholar] [CrossRef]
- Dai, A.; Wan, A.; Magee, C.; Zhang, Y.; Barlow, S.; Marder, S.R.; Kahn, A. Investigation of p-dopant diffusion in polymer films and bulk heterojunctions: Stable spatially-confined doping for all-solution processed solar cells. Org. Electron. 2015, 23, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Kawana, S.; Jones, R. Character of the glass transition in thin supported polymer films. Phys. Rev. E 2001, 63, 021501. [Google Scholar] [CrossRef]
- Yang, Q.; Chen, X.; He, Z.; Lan, F.; Liu, H. The glass transition temperature measurements of polyethylene: Determined by using molecular dynamic method. RSC Adv. 2016, 6, 12053–12060. [Google Scholar] [CrossRef]
- Tan, C.H.; Zhang, B.K.; Chen, J.; Zhang, L.N.; Huang, X.G.; Meng, H.Y. Study of Hydrolysis Kinetic of New Laser Material [anti-B18H22]. Russ. J. Inorg. Chem. 2019, 64, 1359–1364. [Google Scholar] [CrossRef]
- Kim, J.H.; Jang, J.; Zin, W.C. Thickness dependence of the glass transition temperature in thin polymer films. Langmuir 2001, 17, 2703–2710. [Google Scholar] [CrossRef]
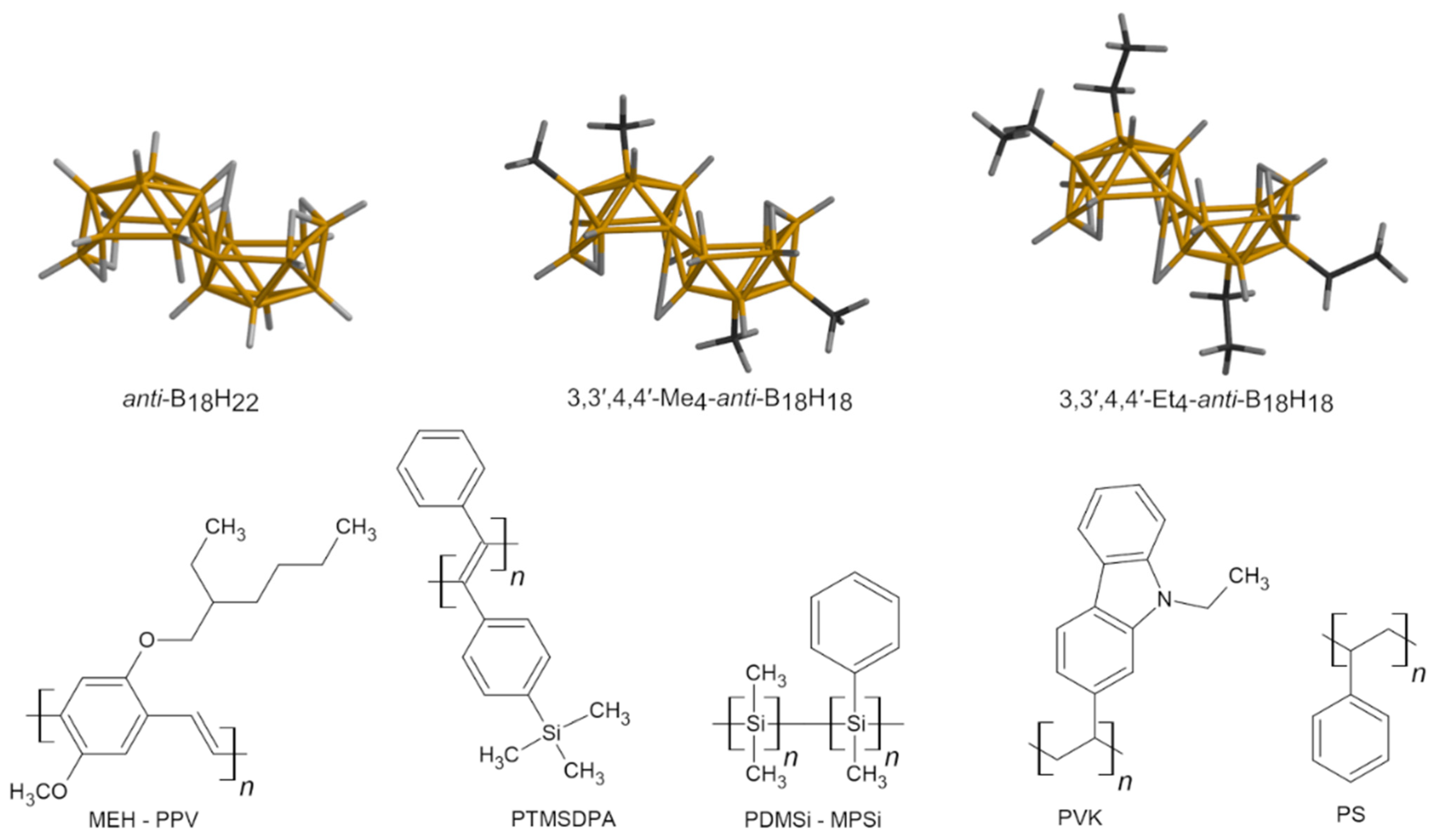




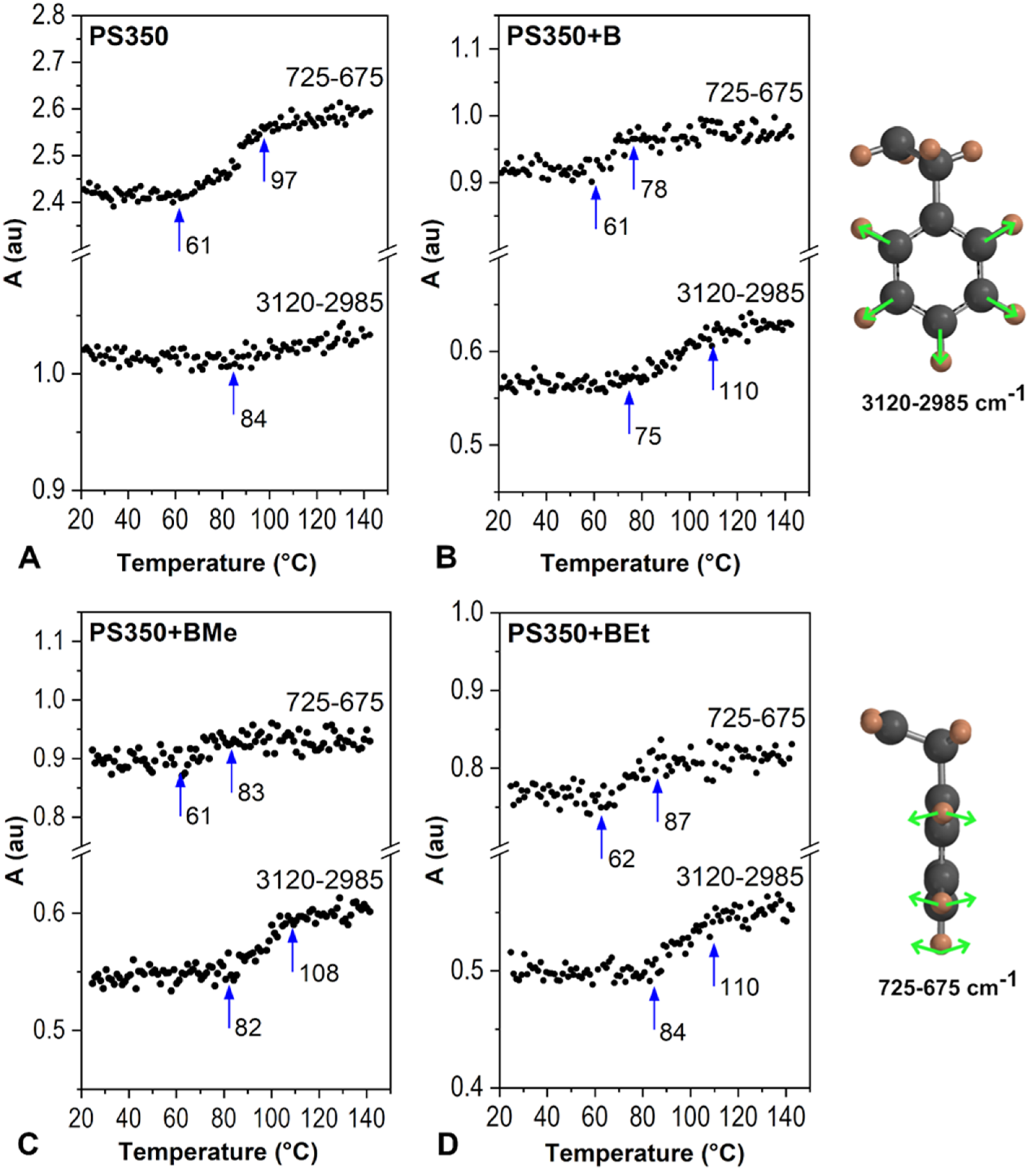
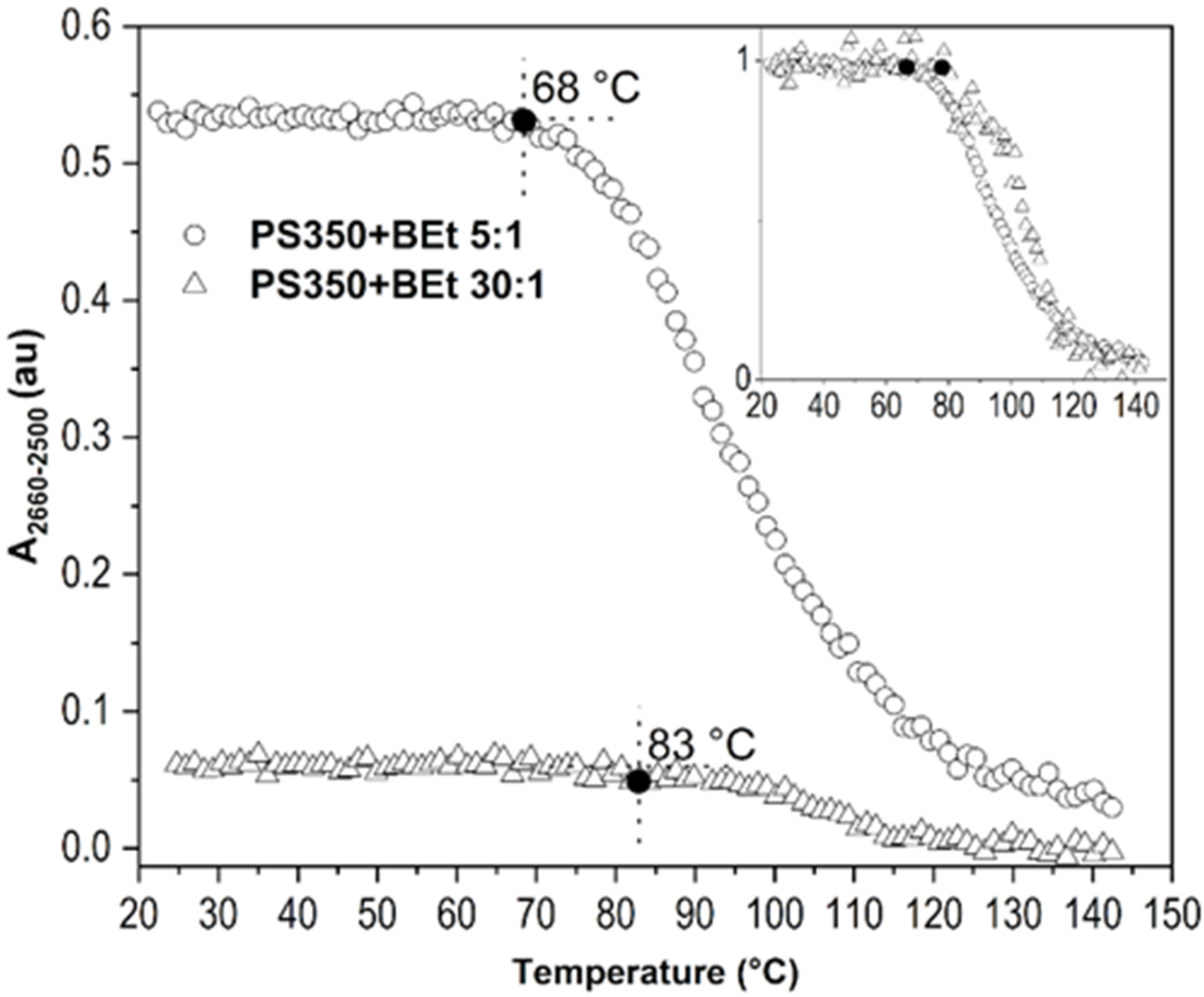
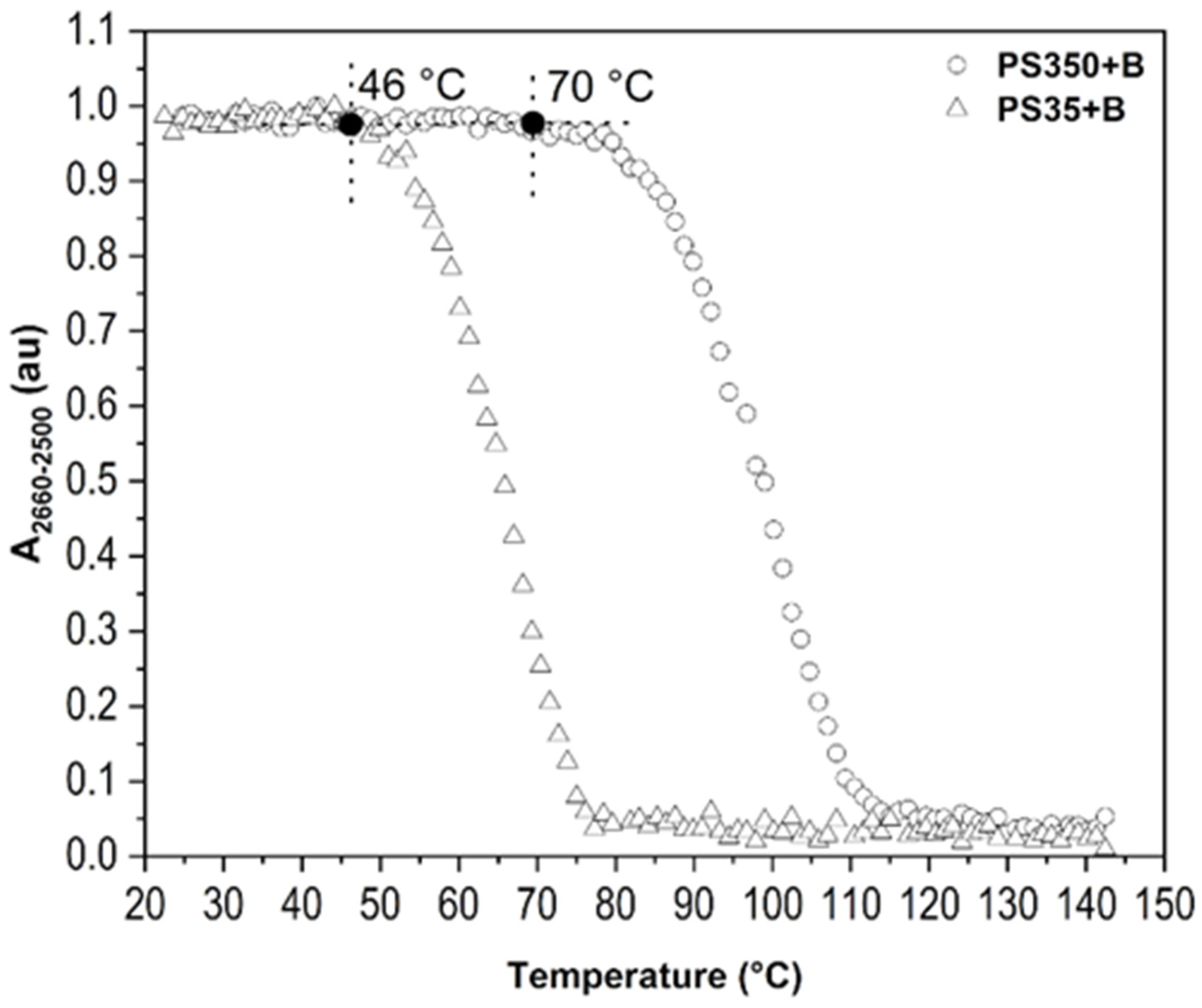
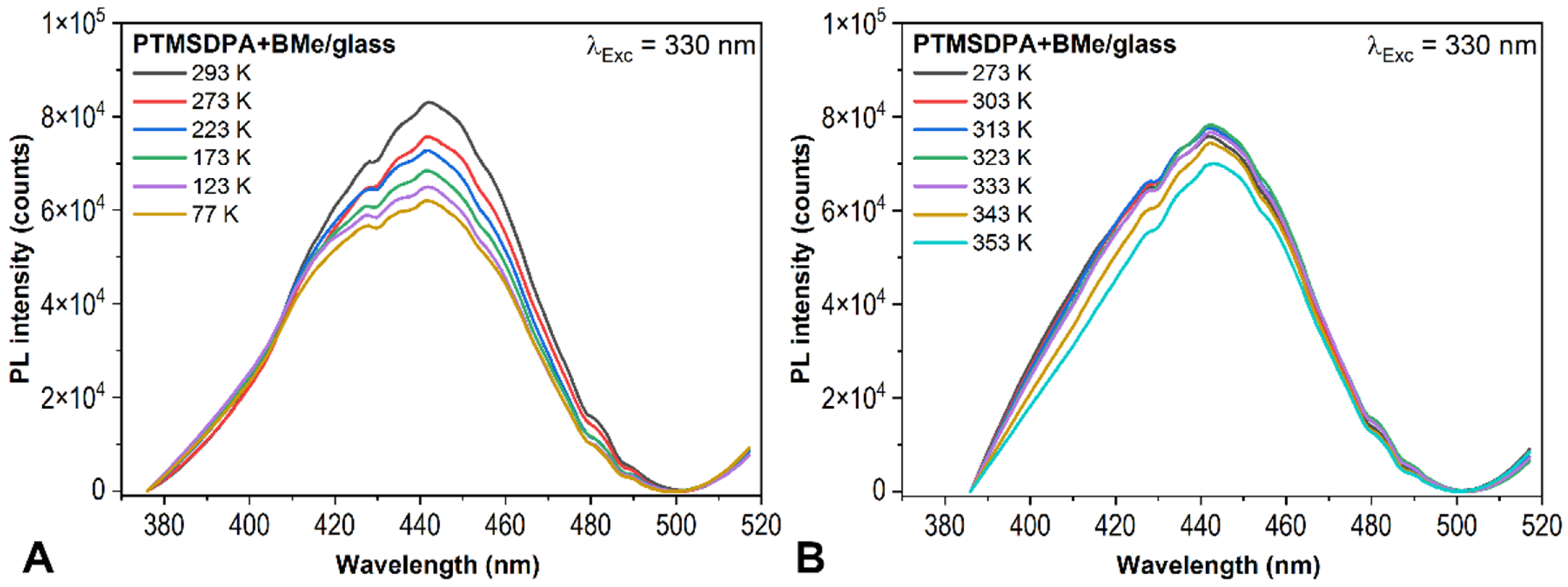


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
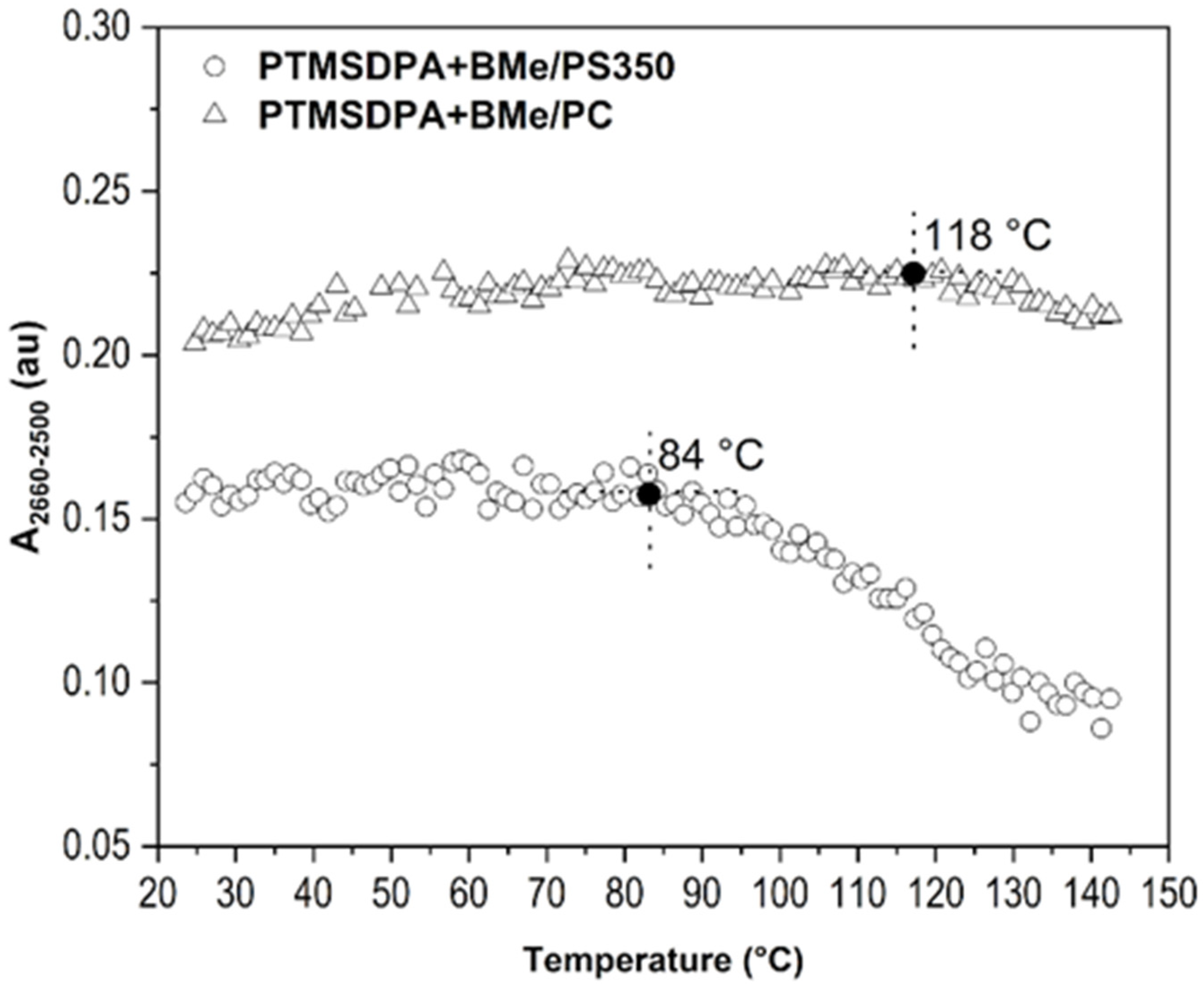{kind=link}
| Thin Film | Solvent | Solution for Spin Coating | Solid Thin Film | Film Thickness (nm) | |
|---|---|---|---|---|---|
| Polymer Weight | Borane Weight | Polymer:Borane Weight Ratio | |||
| per 1 mL of Solvent | |||||
| MEH-PPV+BMePL | chloroform | 3.0 mg | 1.5 mg | 2:1 | 60 ± 1 |
| PTMSDPA+BMePL | cyclohexane | 2.0 mg | 1.0 mg | 2:1 | 46 ± 3 |
| PDMSi-MPSi+BMePL | chloroform | 20.0 mg | 10.0 mg | 2:1 | 180 ± 30 |
| PTMSDPA+Bet | cyclohexane | 1.0 mg | 0.6 mg | 2:1 | 113 ± 5 |
| MEH-PPV+Bet | chloroform | 8.0 mg | 4.5 mg | 2:1 | 116 ± 5 |
| PDMSi-MPSi+Bet | chloroform | 23.0 mg | 10.0 mg | 2:1 | 200 ± 40 |
| PDMSi-MPSi+Bet | chloroform | 23.0 mg | 3.3 mg | 7:1 | 200 ± 20 |
| PVK+BMe | chloroform | 10.0 mg | 5.0 mg | 2:1 | 110 ± 5 |
| PS350+B | 1-chlorpentane | 12.5 mg | 1.3 mg | 10:1 | 130 ± 5 |
| toluene | 19.0 mg | 2 mg | 10:1 | 190 ± 40 | |
| PS35+B | toluene | 19.0 mg | 2 mg | 10:1 | 140 ± 20 |
| PS350+BMe | 1-chlorpentane | 12.5 mg | 1.3 mg | 10:1 | 120 ± 5 |
| PS350+BEt | 1-chlorpentane | 12.5 mg | 1.3 mg | 10:1 | 100 ± 5 |
| toluene | 30.0 mg | 6.0 mg | 5:1 | 358 ± 7 | |
| 30.0 mg | 1.0 mg | 30:1 | 308 ± 4 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capkova, T.; Hanulikova, B.; Sevcik, J.; Urbanek, P.; Antos, J.; Urbanek, M.; Kuritka, I. Incorporation of the New anti-Octadecaborane Laser Dyes into Thin Polymer Films: A Temperature-Dependent Photoluminescence and Infrared Spectroscopy Study. Int. J. Mol. Sci. 2022, 23, 8832. https://doi.org/10.3390/ijms23158832
Capkova T, Hanulikova B, Sevcik J, Urbanek P, Antos J, Urbanek M, Kuritka I. Incorporation of the New anti-Octadecaborane Laser Dyes into Thin Polymer Films: A Temperature-Dependent Photoluminescence and Infrared Spectroscopy Study. International Journal of Molecular Sciences. 2022; 23(15):8832. https://doi.org/10.3390/ijms23158832
Chicago/Turabian StyleCapkova, Tereza, Barbora Hanulikova, Jakub Sevcik, Pavel Urbanek, Jan Antos, Michal Urbanek, and Ivo Kuritka. 2022. "Incorporation of the New anti-Octadecaborane Laser Dyes into Thin Polymer Films: A Temperature-Dependent Photoluminescence and Infrared Spectroscopy Study" International Journal of Molecular Sciences 23, no. 15: 8832. https://doi.org/10.3390/ijms23158832
APA StyleCapkova, T., Hanulikova, B., Sevcik, J., Urbanek, P., Antos, J., Urbanek, M., & Kuritka, I. (2022). Incorporation of the New anti-Octadecaborane Laser Dyes into Thin Polymer Films: A Temperature-Dependent Photoluminescence and Infrared Spectroscopy Study. International Journal of Molecular Sciences, 23(15), 8832. https://doi.org/10.3390/ijms23158832