
Deletion of Meg8-DMR Enhances Migration and Invasion of MLTC-1 Depending on the CTCF Binding Sites


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
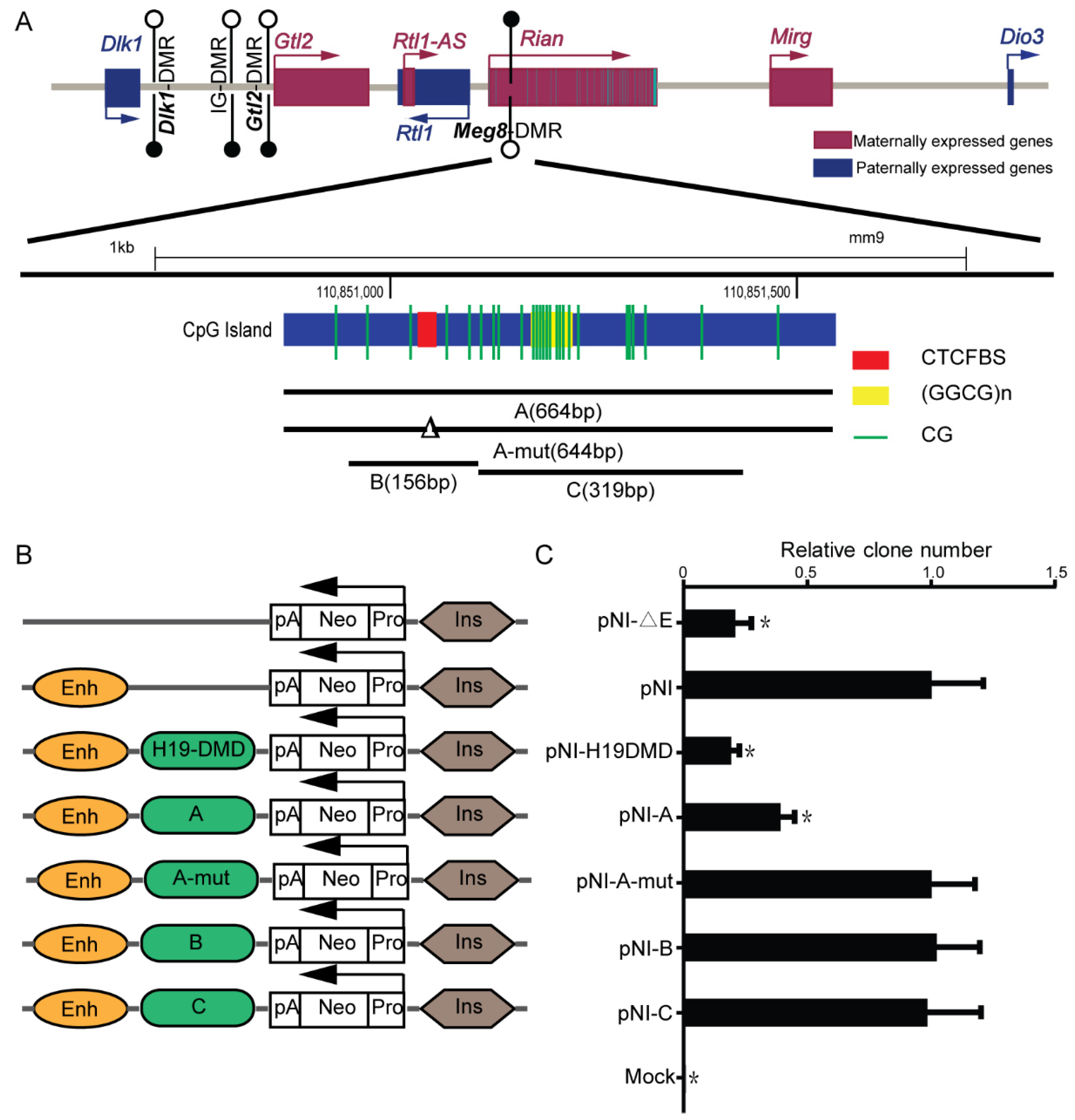
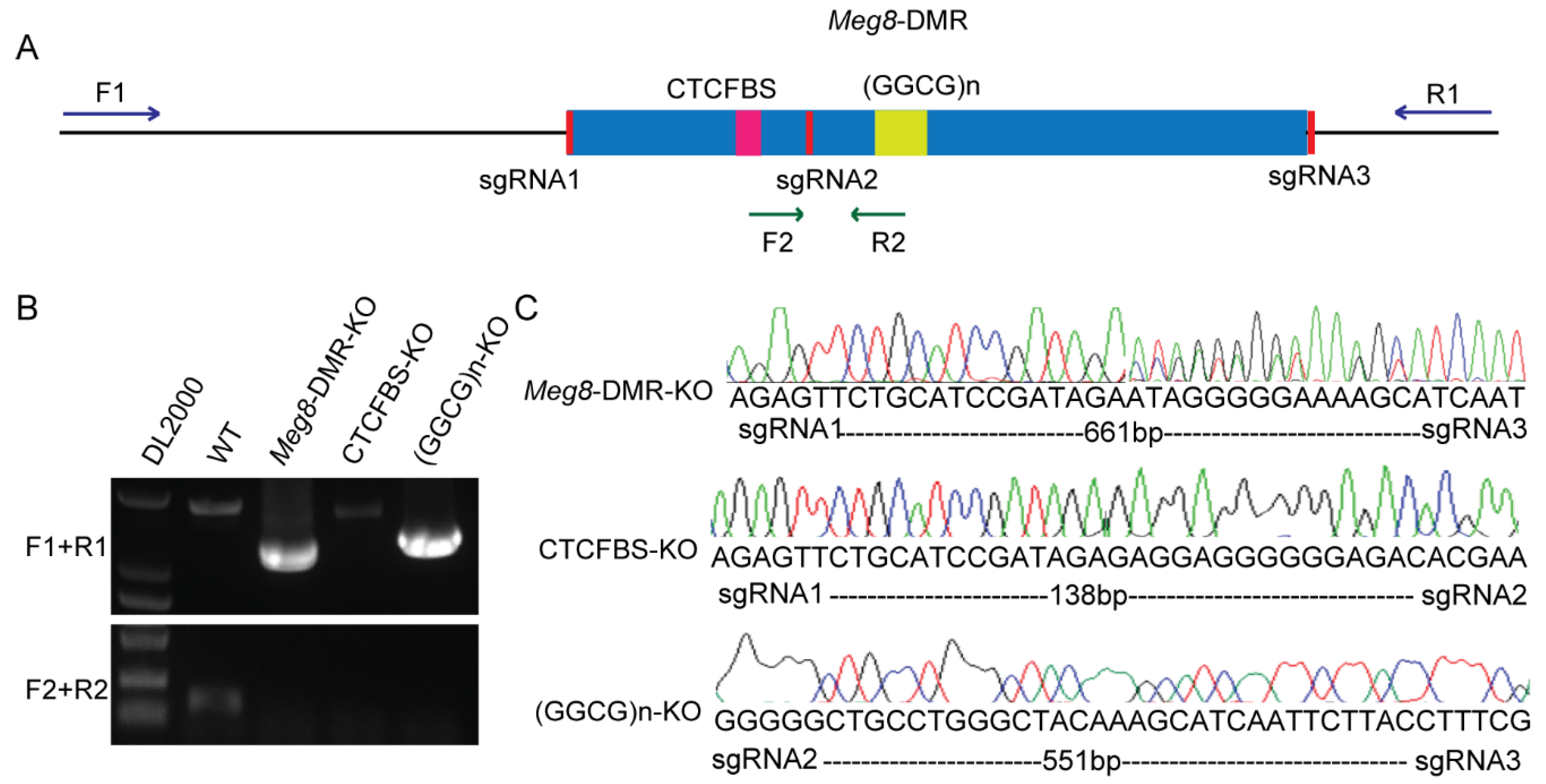
2.1. Meg8-DMR Acts as an Insulator Dependent on the CTCF Binding Sites
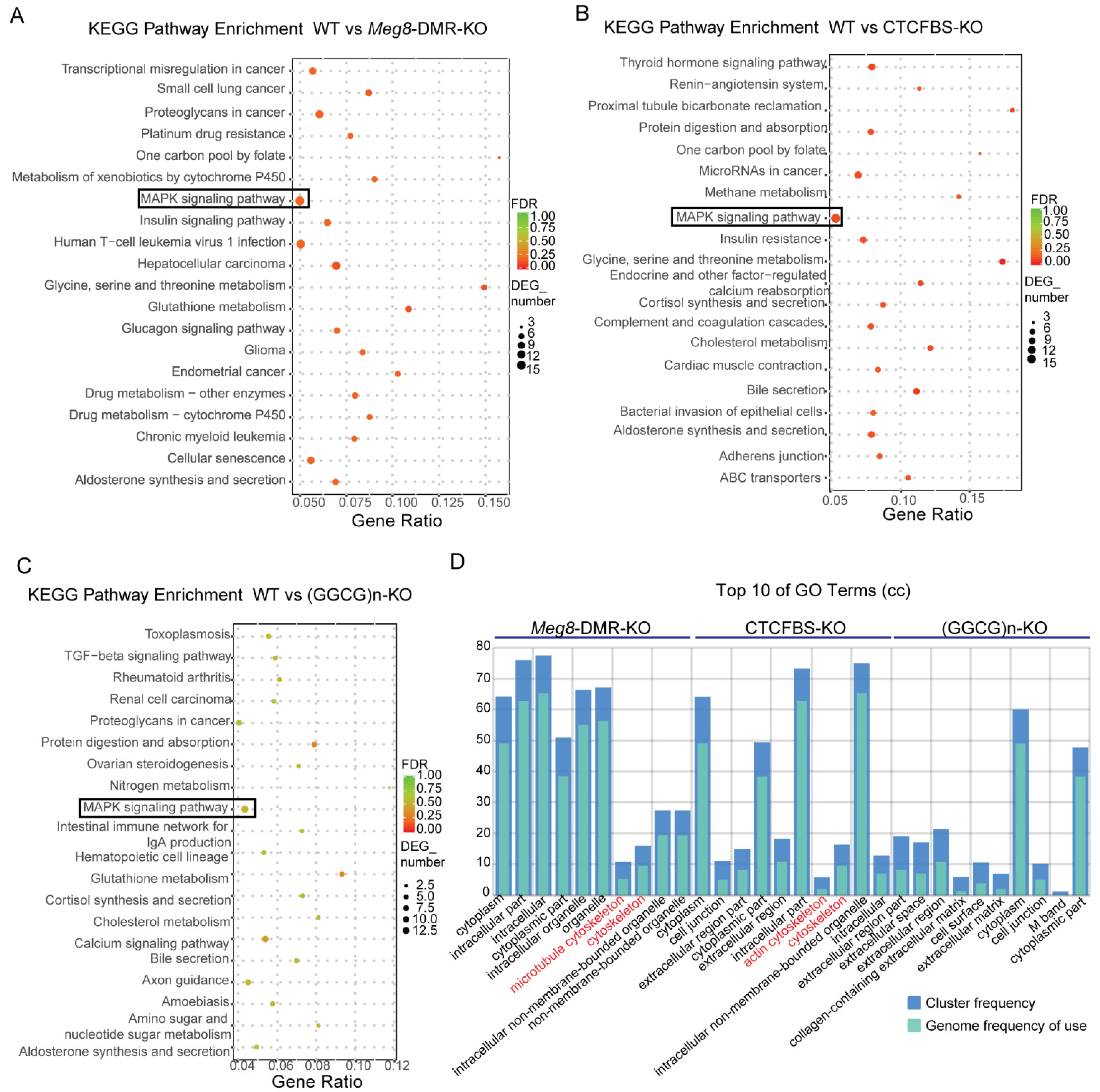
2.2. Deletion of Meg8-DMR Reduced the Expression of Many Genes on Chromosome 12
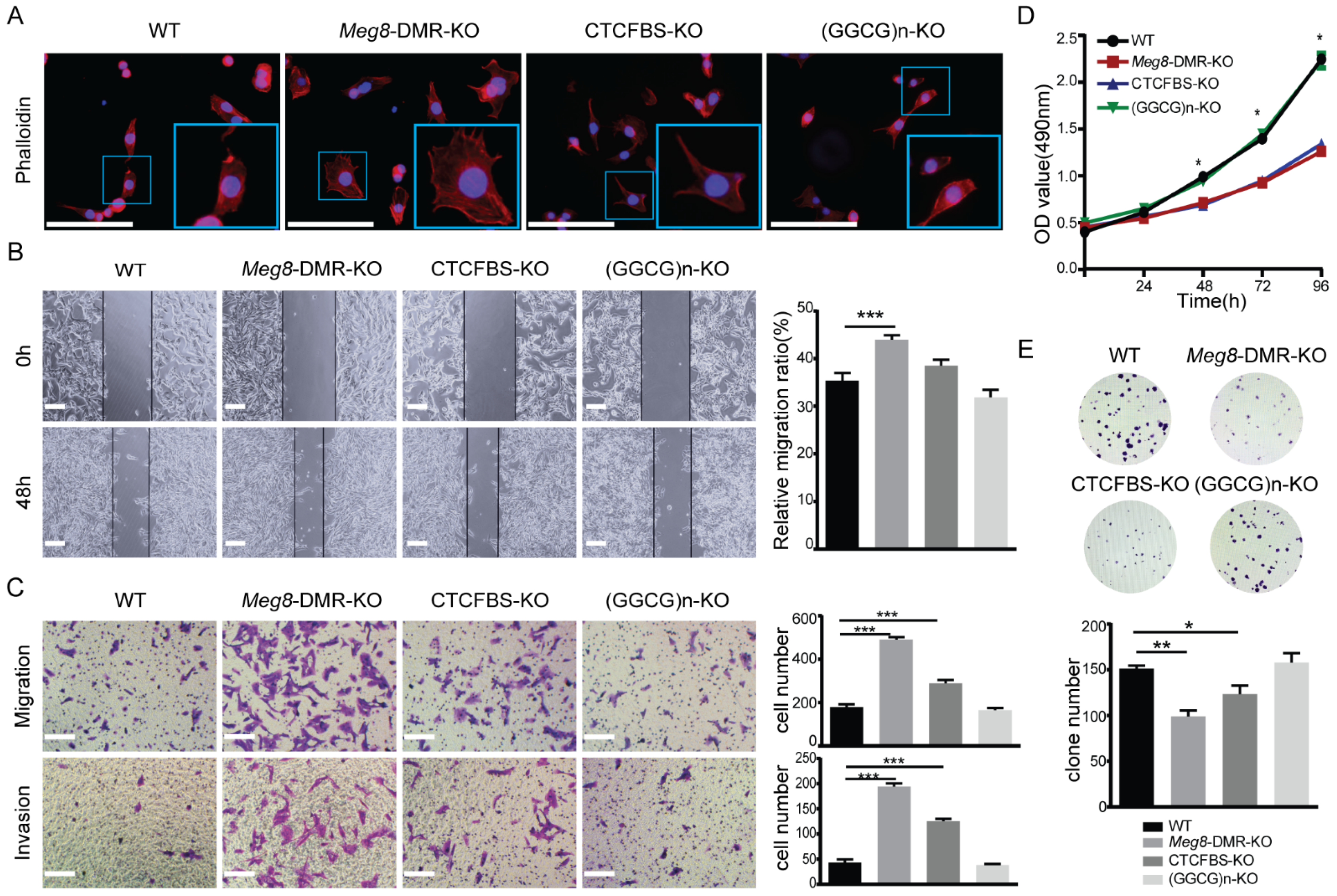
2.3. Meg8-DMR Deletion Enhanced Migration and Invasion of MLTC-1
2.4. Overexpression of Dlk1 in Meg8-DMR-KO Suppressed Cells’ Migration and Invasion by Blocking Notch1-Rhoc-MAPK/ERK
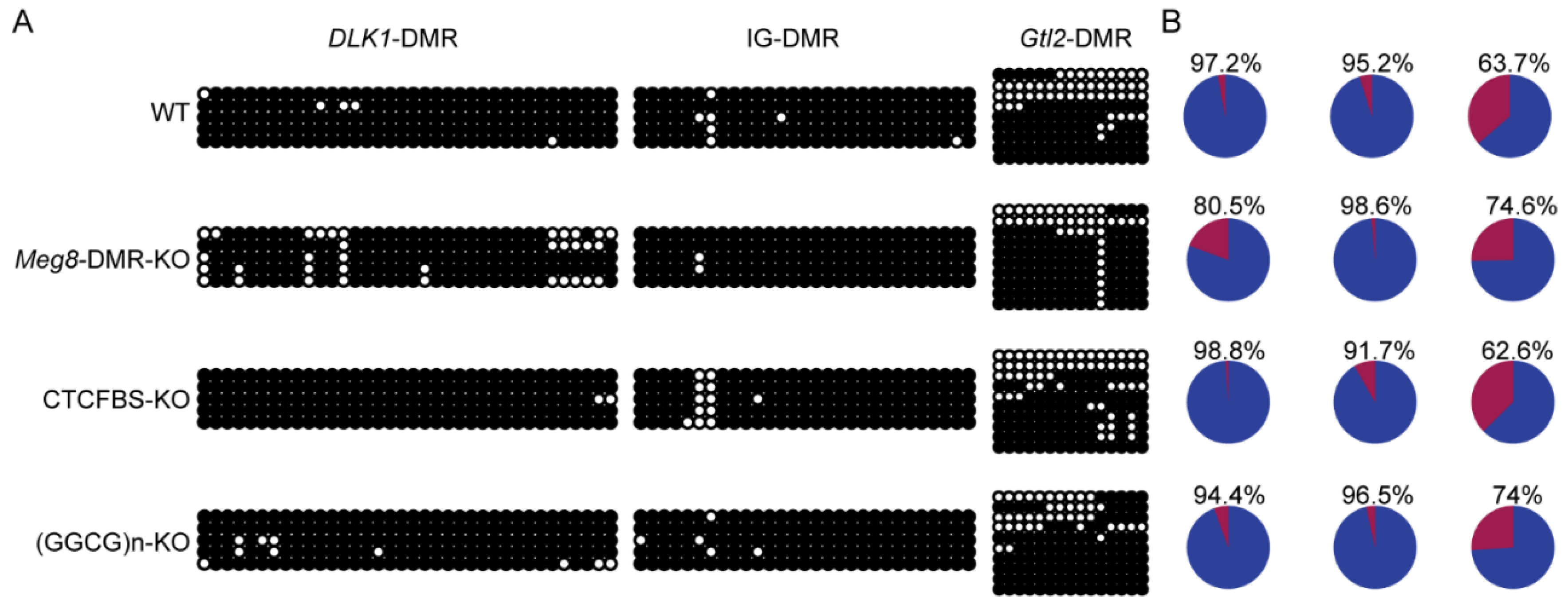
2.5. Absence of Meg8-DMR Has No Impact on the Methylation of Other DMRs in the Dlk1-Dio3 Domain
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. The Enhancer Blocking Assay
4.3. CRISPR/Cas9-Mediated Deletion of Meg8-DMR, CTCF Binding Sites and GGCG Repeats
4.4. DNA Extraction and Methylation Analysis
4.5. RNA Extraction and RNA-Seq Analysis
4.6. qRT-PCR and Strand-Specific RT-PCR
4.7. Immunofluorescence
4.8. Wound Healing, Cell Migration and Invasion
4.9. MTT and Colony Formation Assays
4.10. Western Blotting Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferguson-Smith, A.C. Genomic imprinting: The emergence of an epigenetic paradigm. Nat. Rev. Genet. 2011, 12, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Ferguson-Smith, A.C.; Bourc’his, D. The discovery and importance of genomic imprinting. eLife 2018, 7, e42368. [Google Scholar] [CrossRef] [PubMed]
- Barlow, D.P.; Bartolomei, M.S. Genomic Imprinting in Mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a018382. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G. Genomic imprinting disorders in humans: A mini-review. J. Assist. Reprod. Genet. 2009, 26, 477–486. [Google Scholar] [CrossRef]
- Lin, S.; Ferguson-Smith, A.C.; Schultz, R.M.; Bartolomei, M.S. Nonallelic Transcriptional Roles of CTCF and Cohesins at Imprinted Loci. Mol. Cell. Biol. 2011, 31, 3094–3104. [Google Scholar] [CrossRef]
- Cleaton, M.A.; Edwards, C.A.; Ferguson-Smith, A.C. Phenotypic Outcomes of Imprinted Gene Models in Mice: Elucidation of Pre- and Postnatal Functions of Imprinted Genes. Annu. Rev. Genom. Hum. Genet. 2014, 15, 93–126. [Google Scholar] [CrossRef]
- da Rocha, S.T.; Edwards, C.A.; Ito, M.; Ogata, T.; Ferguson-Smith, A.C. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008, 24, 306–316. [Google Scholar] [CrossRef]
- Hagan, J.P.; O’Neill, B.L.; Stewart, C.L.; Kozlov, S.V.; Croce, C.M. At Least Ten Genes Define the Imprinted Dlk1-Dio3 Cluster on Mouse Chromosome 12qF1. PLoS ONE 2009, 4, e4352. [Google Scholar] [CrossRef]
- Seitz, H.; Royo, H.; Bortolin, M.L.; Lin, S.P.; Ferguson-Smith, A.C.; Cavaillé, J. A Large Imprinted microRNA Gene Cluster at the Mouse Dlk1-Gtl2 Domain. Genome Res. 2004, 14, 1741–1748. [Google Scholar] [CrossRef]
- Lin, S.P.; Youngson, N.; Takada, S.; Seitz, H.; Reik, W.; Paulsen, M.; Cavaille, J.; Ferguson-Smith, A.C. Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12. Nat. Genet. 2003, 35, 97–102. [Google Scholar] [CrossRef]
- Nowak, K.; Stein, G.; Powell, E.; He, L.M.; Naik, S.; Morris, J.; Marlow, S.; Davis, T.L. Establishment of paternal allele-specific DNA methylation at the imprinted mouseGtl2locus. Epigenetics 2011, 6, 1012–1020. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gagne, A.; Hochman, A.; Qureshi, M.; Tong, C.; Arbon, J.; McDaniel, K.; Davis, T.L. Analysis of DNA methylation acquisition at the imprinted Dlk1 locus reveals asymmetry at CpG dyads. Epigenetics Chromatin 2014, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.L.; Wu, Y.Q.; Kwabi-Addo, B.; Coveler, K.J.; Reid Sutton, V.; Shaffer, L.G. Allele-specific methylation of a functional CTCF binding site upstream of MEG3 in the human imprinted domain of 14q32. Chromosom. Res. 2005, 13, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Llères, D.; Moindrot, B.; Pathak, R.; Piras, V.; Matelot, M.; Pignard, B.; Marchand, A.; Poncelet, M.; Perrin, A.; Tellier, V.; et al. CTCF modulates allele-specific sub-TAD organization and imprinted gene activity at the mouse Dlk1-Dio3 and Igf2-H19 domains. Genome Biol. 2019, 20, 272. [Google Scholar] [CrossRef] [PubMed]
- Merkenschlager, M.; Nora, E.P. CTCF and Cohesin in Genome Folding and Transcriptional Gene Regulation. Annu. Rev. Genom. Hum. Genet. 2016, 17, 17–43. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, D.; Feil, R. Differential 3D chromatin organization and gene activity in genomic imprinting. Curr. Opin. Genet. Dev. 2020, 61, 17–24. [Google Scholar] [CrossRef]
- Prickett, A.R.; Barkas, N.; McCole, R.B.; Hughes, S.; Amante, S.M.; Schulz, R.; Oakey, R.J. Genome-wide and parental allele-specific analysis of CTCF and cohesin DNA binding in mouse brain reveals a tissue-specific binding pattern and an association with imprinted differentially methylated regions. Genome Res. 2013, 23, 1624–1635. [Google Scholar] [CrossRef]
- Bell, A.C.; Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 2000, 405, 482–485. [Google Scholar] [CrossRef]
- Schoenherr, C.J.; Levorse, J.M.; Tilghman, S.M. CTCF maintains differential methylation at the Igf2/H19 locus. Nat. Genet. 2003, 33, 66–69. [Google Scholar] [CrossRef]
- Ideraabdullah, F.Y.; Thorvaldsen, J.L.; Myers, J.A.; Bartolomei, M.S. Tissue-specific insulator function at H19/Igf2 revealed by deletions at the imprinting control region. Hum. Mol. Genet. 2014, 23, 6246–6259. [Google Scholar] [CrossRef]
- Zeng, T.B.; He, H.J.; Han, Z.B.; Zhang, F.W.; Huang, Z.J.; Liu, Q.; Cui, W.; Wu, Q. DNA methylation dynamics of a maternally methylated DMR in the mouseDlk1-Dio3domain. FEBS Lett. 2014, 588, 4665–4671. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.A.; Ferguson-Smith, A.C. Mechanisms regulating imprinted genes in clusters. Curr. Opin. Cell Biol. 2007, 19, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.C.; West, A.G.; Felsenfeld, G. The Protein CTCF Is Required for the Enhancer Blocking Activity of Vertebrate Insulators. Cell 1999, 98, 387–396. [Google Scholar] [CrossRef]
- Cui, S.; Wu, Q.; Liu, M.; Su, M.; Liu, S.; Shao, L.; Han, X.; He, H. EphA2 super-enhancer promotes tumor progression by recruiting FOSL2 and TCF7L2 to activate the target gene EphA2. Cell Death Dis. 2021, 12, 264. [Google Scholar] [CrossRef] [PubMed]
- Pittaway, J.F.H.; Lipsos, C.; Mariniello, K.; Guasti, L. The role of delta-like non-canonical Notch ligand 1 (DLK1) in cancer. Endocr. Relat. Cancer 2021, 28, R271–R287. [Google Scholar] [CrossRef]
- Perramón, M.; Jiménez, W. Pituitary Tumor-Transforming Gene 1/Delta like Non-Canonical Notch Ligand 1 Signaling in Chronic Liver Diseases. Int. J. Mol. Sci. 2022, 23, 6897. [Google Scholar] [CrossRef]
- Baladrón, V.; Ruiz-Hidalgo, M.J.; Nueda, M.L.; Díaz-Guerra, M.J.; García-Ramírez, J.J.; Bonvini, E.; Gubina, E.; Laborda, J. dlk acts as a negative regulator of Notch1 activation through interactions with specific EGF-like repeats. Exp. Cell Res. 2005, 303, 343–359. [Google Scholar] [CrossRef]
- Traustadóttir, G.; Jensen, C.H.; Thomassen, M.; Beck, H.C.; Mortensen, S.B.; Laborda, J.; Baladrón, V.; Sheikh, S.P.; Andersen, D.C. Evidence of non-canonical NOTCH signaling: Delta-like 1 homolog (DLK1) directly interacts with the NOTCH1 receptor in mammals. Cell. Signal. 2016, 28, 246–254. [Google Scholar] [CrossRef]
- Lou, Y.; Jiang, Y.; Liang, Z.; Liu, B.; Li, T.; Zhang, D. Role of RhoC in cancer cell migration. Cancer Cell Int. 2021, 21, 527. [Google Scholar] [CrossRef]
- Takada, S.; Paulsen, M.; Tevendale, M.; Tsai, C.E.; Kelsey, G.; Cattanach, B.M.; Ferguson-Smith, A.C. Epigenetic analysis of the Dlk1-Gtl2 imprinted domain on mouse chromosome 12: Implications for imprinting control from comparison with Igf2-H19. Hum. Mol. Genet. 2002, 11, 77–86. [Google Scholar] [CrossRef]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C.; the Erice Imprinting Group. Genomic Imprinting and Physiological Processes in Mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Georgiades, P.; Watkins, M.; Surani, M.A.; Ferguson-Smith, A.C. Parental origin-specific developmental defects in mice with uniparental disomy for chromosome 12. Development 2000, 127, 4719–4728. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Kumagai, T.; Kawahara, M.; Ogawa, H.; Hiura, H.; Obata, Y.; Takano, R.; Kono, T. Regulated expression of two sets of paternally imprinted genes is necessary for mouse parthenogenetic development to term. Reproduction 2006, 131, 481–488. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Monk, D.; Mackay, D.J.G.; Eggermann, T.; Maher, E.R.; Riccio, A. Genomic imprinting disorders: Lessons on how genome, epigenome and environment interact. Nat. Rev. Genet. 2019, 20, 235–248. [Google Scholar] [CrossRef]
- Hark, A.T.; Schoenherr, C.J.; Katz, D.J.; Ingram, R.S.; Levorse, J.M.; Tilghman, S.M. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 2000, 405, 486–489. [Google Scholar] [CrossRef]
- Thorvaldsen, J.L.; Duran, K.L.; Bartolomei, M.S. Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev. 1998, 12, 3693–3702. [Google Scholar] [CrossRef]
- Yoon, B.; Herman, H.; Hu, B.; Park, Y.J.; Lindroth, A.; Bell, A.; West, A.G.; Chang, Y.; Stablewski, A.; Piel, J.C.; et al. Rasgrf1 Imprinting Is Regulated by a CTCF-Dependent Methylation-Sensitive Enhancer Blocker. Mol. Cell. Biol. 2005, 25, 11184–11190. [Google Scholar] [CrossRef]
- Fitzpatrick, G.V.; Pugacheva, E.M.; Shin, J.-Y.; Abdullaev, Z.; Yang, Y.; Khatod, K.; Lobanenkov, V.V.; Higgins, M.J. Allele-Specific Binding of CTCF to the Multipartite Imprinting Control Region KvDMR1. Mol. Cell. Biol. 2007, 27, 2636–2647. [Google Scholar] [CrossRef]
- Shiura, H.; Nakamura, K.; Hikichi, T.; Hino, T.; Oda, K.; Suzuki-Migishima, R.; Kohda, T.; Kaneko-Ishino, T.; Ishino, F. Paternal deletion of Meg1/Grb10 DMR causes maternalization of the Meg1/Grb10 cluster in mouse proximal Chromosome 11 leading to severe pre- and postnatal growth retardation. Hum. Mol. Genet. 2009, 18, 1424–1438. [Google Scholar] [CrossRef]
- Hutter, B.; Helms, V.; Paulsen, M. Tandem repeats in the CpG islands of imprinted genes. Genomics 2006, 88, 323–332. [Google Scholar] [CrossRef]
- Beygo, J.; Citro, V.; Sparago, A.; De Crescenzo, A.; Cerrato, F.; Heitmann, M.; Rademacher, K.; Guala, A.; Enklaar, T.; Anichini, C.; et al. The molecular function and clinical phenotype of partial deletions of the IGF2/H19 imprinting control region depends on the spatial arrangement of the remaining CTCF-binding sites. Hum. Mol. Genet. 2013, 22, 544–557. [Google Scholar] [CrossRef] [PubMed]
- Sparago, A.; Cerrato, F.; Riccio, A. Is ZFP57 binding to H19/IGF2:IG-DMR affected in Silver-Russell syndrome? Clin. Epigenetics 2018, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- De Crescenzo, A.; Coppola, F.; Falco, P.; Bernardo, I.; Ausanio, G.; Cerrato, F.; Falco, L.; Riccio, A. A novel microdeletion in the IGF2/H19 imprinting centre region defines a recurrent mutation mechanism in familial Beckwith–Wiedemann syndrome. Eur. J. Med Genet. 2011, 54, e451–e454. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Hara, S.; Kato, T.; Tamano, M.; Muramatsu, A.; Asahara, H.; Takada, S. A tandem repeat array in IG-DMR is essential for imprinting of paternal allele at the Dlk1–Dio3 domain during embryonic development. Hum. Mol. Genet. 2018, 27, 3283–3292. [Google Scholar] [CrossRef]
- Soares, M.L.; Edwards, C.A.; Dearden, F.L.; Ferrón, S.R.; Curran, S.; Corish, J.A.; Rancourt, R.C.; Allen, S.E.; Charalambous, M.; Ferguson-Smith, M.A.; et al. Targeted deletion of a 170-kb cluster of LINE-1 repeats and implications for regional control. Genome Res. 2018, 28, 345–356. [Google Scholar] [CrossRef]
- Surmacz, B.; Noisa, P.; Risner-Janiczek, J.R.; Hui, K.; Ungless, M.; Cui, W.; Li, M. DLK1 Promotes Neurogenesis of Human and Mouse Pluripotent Stem Cell-Derived Neural Progenitors Via Modulating Notch and BMP Signalling. Stem Cell Rev. Rep. 2012, 8, 459–471. [Google Scholar] [CrossRef]
- Rodríguez, P.; Higueras, M.A.; González-Rajal, A.; Alfranca, A.; Fierro-Fernández, M.; García-Fernández, R.A.; Ruiz-Hidalgo, M.J.; Monsalve, M.; Rodríguez-Pascual, F.; Redondo, J.M.; et al. The non-canonical NOTCH ligand DLK1 exhibits a novel vascular role as a strong inhibitor of angiogenesis. Cardiovasc. Res. 2012, 93, 232–241. [Google Scholar] [CrossRef]
- Müller, D.; Cherukuri, P.; Henningfeld, K.; Poh, C.H.; Wittler, L.; Grote, P.; Schlüter, O.; Schmidt, J.; Laborda, J.; Bauer, S.R.; et al. Dlk1 Promotes a Fast Motor Neuron Biophysical Signature Required for Peak Force Execution. Science 2014, 343, 1264–1266. [Google Scholar] [CrossRef]
- García-Gallastegui, P.; Ibarretxe, G.; Garcia-Ramírez, J.J.; Baladrón, V.; Aurrekoetxea, M.; Nueda, M.L.; Naranjo, A.I.; Santaolalla, F.; Rey, A.S.D.; Laborda, J.; et al. DLK1 regulates branching morphogenesis and parasympathetic innervation of salivary glands through inhibition of NOTCH signalling. Biol. Cell 2014, 106, 237–253. [Google Scholar] [CrossRef]
- Traustadóttir, G.; Lagoni, L.V.; Ankerstjerne, L.B.S.; Bisgaard, H.C.; Jensen, C.H.; Andersen, D.C. The imprinted gene Delta like non-canonical Notch ligand 1 (Dlk1) is conserved in mammals, and serves a growth modulatory role during tissue development and regeneration through Notch dependent and independent mechanisms. Cytokine Growth Factor Rev. 2019, 46, 17–27. [Google Scholar] [CrossRef]
- Srivastava, S.; Ramdass, B.; Nagarajan, S.; Rehman, M.; Mukherjee, G.; Krishna, S. Notch1 regulates the functional contribution of RhoC to cervical carcinoma progression. Br. J. Cancer 2010, 102, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ren, H.; Jiang, J.; Wang, Q.; Wudu, M.; Zhang, Q.; Su, H.; Wang, C.; Jiang, L.; Qiu, X. KIAA 0247 inhibits growth, migration, invasion of non-small-cell lung cancer through regulating the Notch pathway. Cancer Sci. 2018, 109, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Gou, W.F.; Zhao, Y.; Lu, H.; Yang, X.F.; Xiu, Y.L.; Zhao, S.; Liu, J.M.; Zhu, Z.T.; Sun, H.Z.; Liu, Y.P.; et al. The role of RhoC in epithelial-to-mesenchymal transition of ovarian carcinoma cells. BMC Cancer 2014, 14, 477. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Pranatharthi, A.; Ross, C.; Srivastava, S. RhoC: A fascinating journey from a cytoskeletal organizer to a Cancer stem cell therapeutic target. J. Exp. Clin. Cancer Res. 2019, 38, 328. [Google Scholar] [CrossRef]
- van Golen, K.L.; Bao, L.W.; Pan, Q.; Miller, F.R.; Wu, Z.F.; Merajver, S.D. Mitogen activated protein kinase pathway is involved in RhoC GTPase induced motility, invasion and angiogenesis in inflammatory breast cancer. Clin. Exp. Metastasis 2002, 19, 301–311. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, X.; Klibanski, A. MEG3 noncoding RNA: A tumor suppressor. J. Mol. Endocrinol. 2012, 48, R45–R53. [Google Scholar] [CrossRef]
- Fan, G.; Ye, D.; Zhu, S.; Xi, J.; Guo, X.; Qiao, J.; Wu, Y.; Jia, W.; Wang, G.; Fan, G.; et al. RTL1 promotes melanoma proliferation by regulating Wnt/β-catenin signalling. Oncotarget 2017, 8, 106026–106037. [Google Scholar] [CrossRef]
- Sheng, F.; Sun, N.; Ji, Y.; Ma, Y.; Ding, H.; Zhang, Q.; Yang, F.; Li, W. Aberrant expression of imprinted lncRNA MEG8 causes trophoblast dysfunction and abortion. J. Cell. Biochem. 2019, 120, 17378–17390. [Google Scholar] [CrossRef]
- Zhang, B.; Dong, Y.; Zhao, Z. LncRNA MEG8 regulates vascular smooth muscle cell proliferation, migration and apoptosis by targeting PPARα. Biochem. Biophys. Res. Commun. 2019, 510, 171–176. [Google Scholar] [CrossRef]
- Ling, L.; Hu, H.L.; Liu, K.Y.; Ram, Y.I.; Gao, J.L.; Cao, Y.M. Long noncoding RNA MIRG induces osteoclastogenesis and bone resorption in osteoporosis through negative regulation of miR-1897. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 10195–10203. [Google Scholar] [CrossRef]
- Terashima, M.; Ishimura, A.; Wanna-Udom, S.; Suzuki, T. MEG8 long noncoding RNA contributes to epigenetic progression of the epithelial-mesenchymal transition of lung and pancreatic cancer cells. J. Biol. Chem. 2018, 293, 18016–18030. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, L.; Shang, P.; Song, X. LncRNA MEG8 promotes tumor progression of non-small cell lung cancer via regulating miR-107/CDK6 axis. Anti-Cancer Drugs 2020, 31, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Lou, J.; Yan, W.; Li, Q.Y.; Zhu, A.K.; Tan, B.Q.; Dong, R.; Zou, X.Z.; Liu, T. LncRNA MEG8 plays an oncogenic role in hepatocellular carcinoma progression through miR-367-3p/14-3-3ζ/TGFβR1 axis. Neoplasma 2021, 68, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cheunsuchon, P.; Nakayama, Y.; Lawlor, M.W.; Zhong, Y.; Rice, K.A.; Zhang, L.; Zhang, X.; Gordon, F.E.; Lidov, H.G.; et al. Activation of paternally expressed genes and perinatal death caused by deletion of the Gtl2 gene. Development 2010, 137, 2643–2652. [Google Scholar] [CrossRef]
- Zhu, W.; Botticelli, E.M.; Kery, R.E.; Mao, Y.; Wang, X.; Yang, A.; Wang, X.; Zhou, J.; Zhang, X.; Soberman, R.J.; et al. Meg3-DMR, not the Meg3 gene, regulates imprinting of the Dlk1-Dio3 locus. Dev. Biol. 2019, 455, 10–18. [Google Scholar] [CrossRef]
- Barlow, D.P. Genomic Imprinting: A Mammalian Epigenetic Discovery Model. Annu. Rev. Genet. 2011, 45, 379–403. [Google Scholar] [CrossRef]
- Riordan, J.D.; Keng, V.W.; Tschida, B.R.; Scheetz, T.E.; Bell, J.B.; Podetz-Pedersen, K.M.; Moser, C.D.; Copeland, N.G.; Jenkins, N.A.; Roberts, L.R.; et al. Identification of Rtl1, a Retrotransposon-Derived Imprinted Gene, as a Novel Driver of Hepatocarcinogenesis. PLoS Genet. 2013, 9, e1003441. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, X.; He, H.; Shao, L.; Cui, S.; Yu, H.; Zhang, X.; Wu, Q. Deletion of Meg8-DMR Enhances Migration and Invasion of MLTC-1 Depending on the CTCF Binding Sites. Int. J. Mol. Sci. 2022, 23, 8828. https://doi.org/10.3390/ijms23158828
Han X, He H, Shao L, Cui S, Yu H, Zhang X, Wu Q. Deletion of Meg8-DMR Enhances Migration and Invasion of MLTC-1 Depending on the CTCF Binding Sites. International Journal of Molecular Sciences. 2022; 23(15):8828. https://doi.org/10.3390/ijms23158828
Chicago/Turabian StyleHan, Xiao, Hongjuan He, Lan Shao, Shuang Cui, Haoran Yu, Ximeijia Zhang, and Qiong Wu. 2022. "Deletion of Meg8-DMR Enhances Migration and Invasion of MLTC-1 Depending on the CTCF Binding Sites" International Journal of Molecular Sciences 23, no. 15: 8828. https://doi.org/10.3390/ijms23158828
APA StyleHan, X., He, H., Shao, L., Cui, S., Yu, H., Zhang, X., & Wu, Q. (2022). Deletion of Meg8-DMR Enhances Migration and Invasion of MLTC-1 Depending on the CTCF Binding Sites. International Journal of Molecular Sciences, 23(15), 8828. https://doi.org/10.3390/ijms23158828