
Inhibition of Glutamine Uptake Resensitizes Paclitaxel Resistance in SKOV3-TR Ovarian Cancer Cell via mTORC1/S6K Signaling Pathway

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
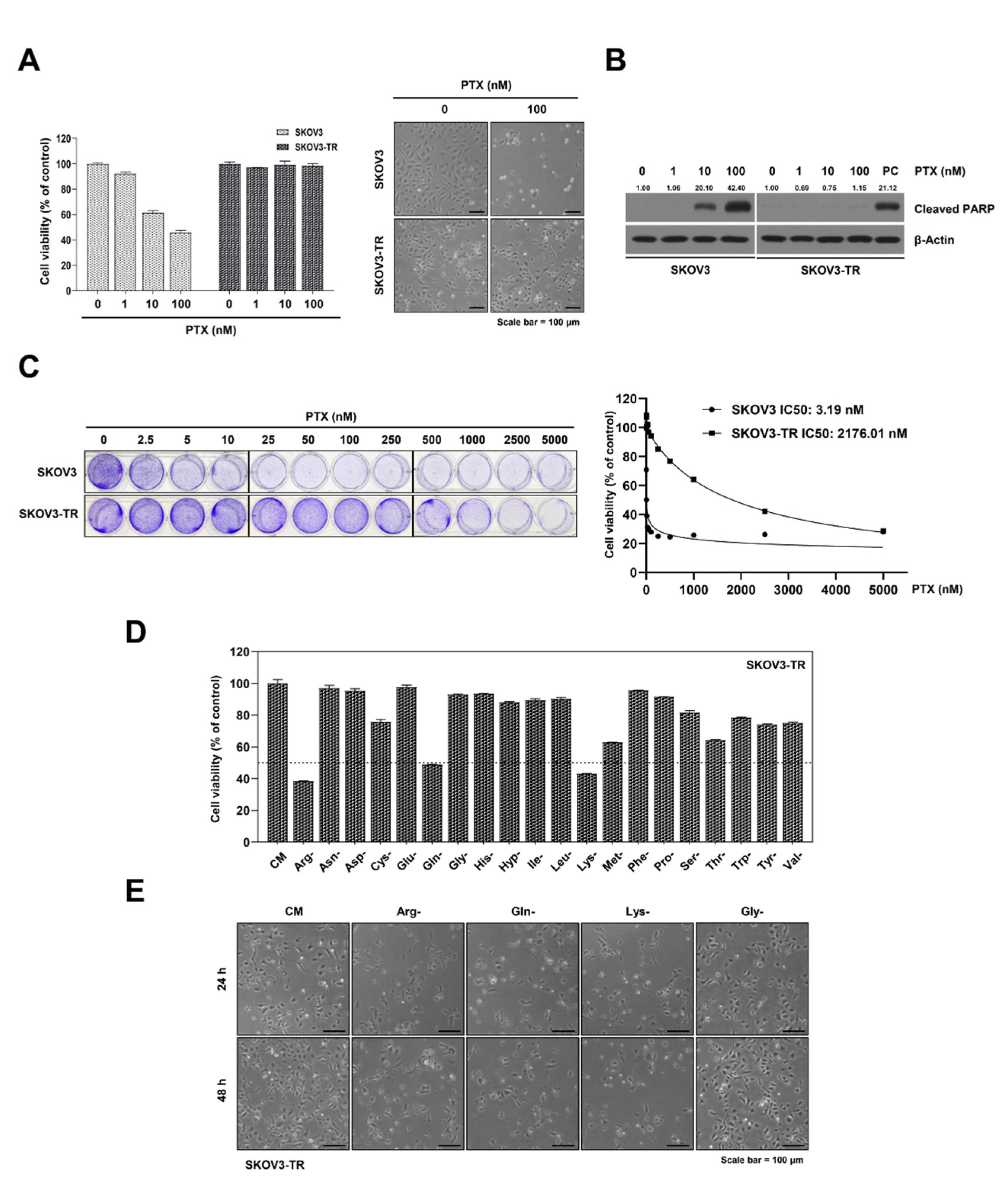
2.1. The Cell Viabilities in Individual Amino Acid-Deprived Conditions in SKOV3-TR Cells
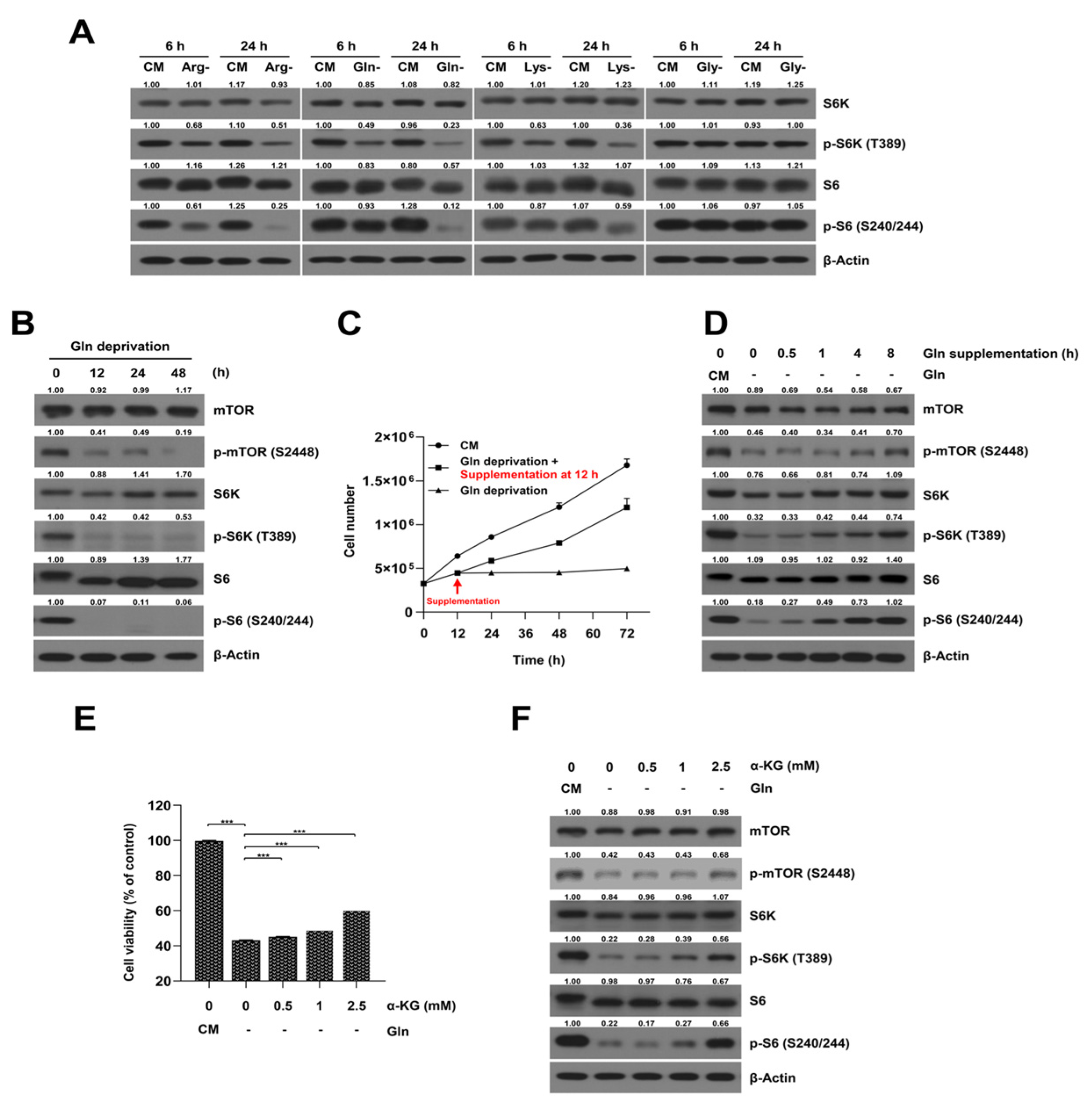
2.2. Glutamine-Deprived Culture Condition Inhibits mTORC1/S6K Signaling Pathway in SKOV3-TR Cells
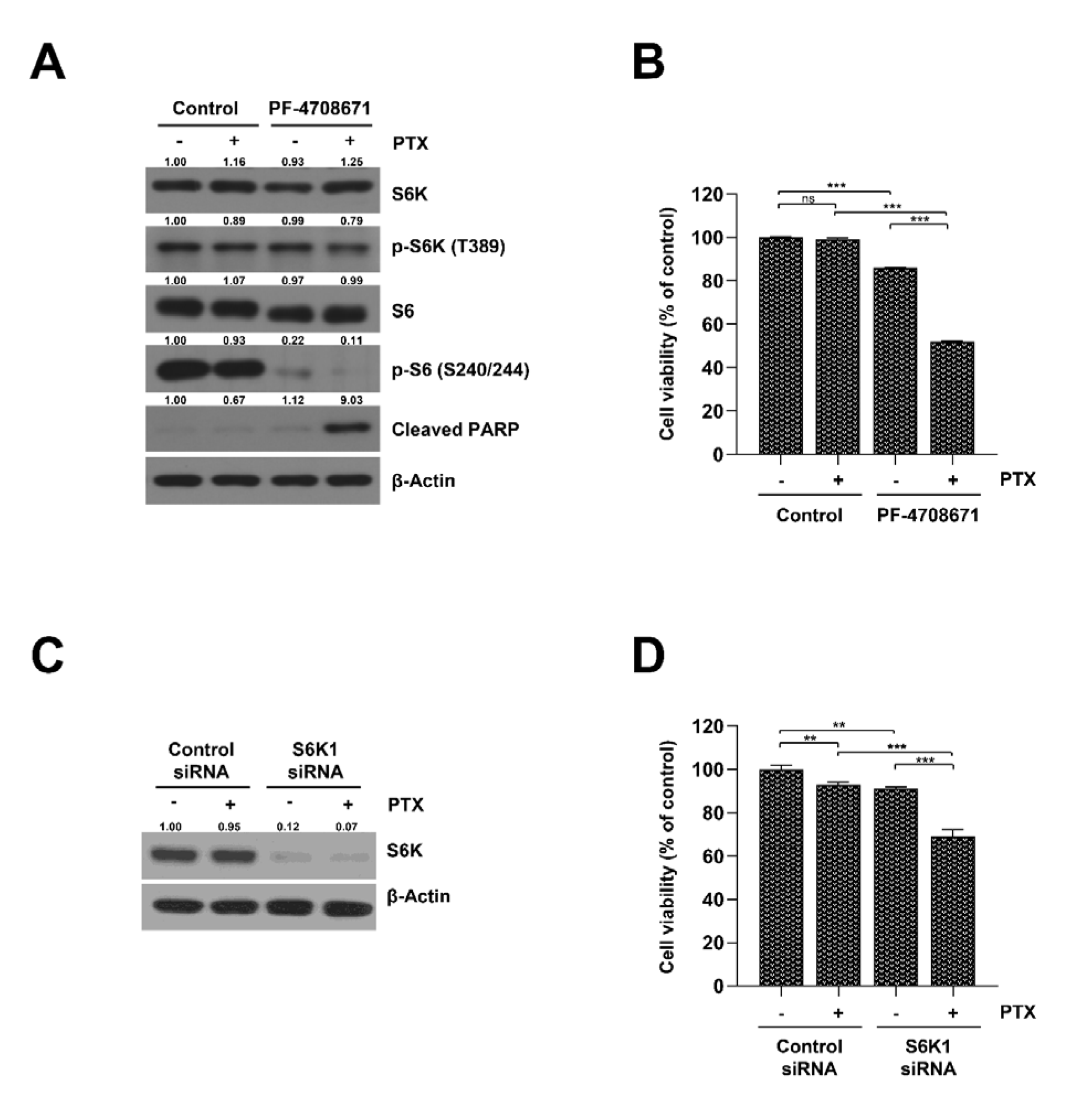
2.3. Inhibition of S6K Resensitizes SKOV3-TR Cells to Paclitaxel
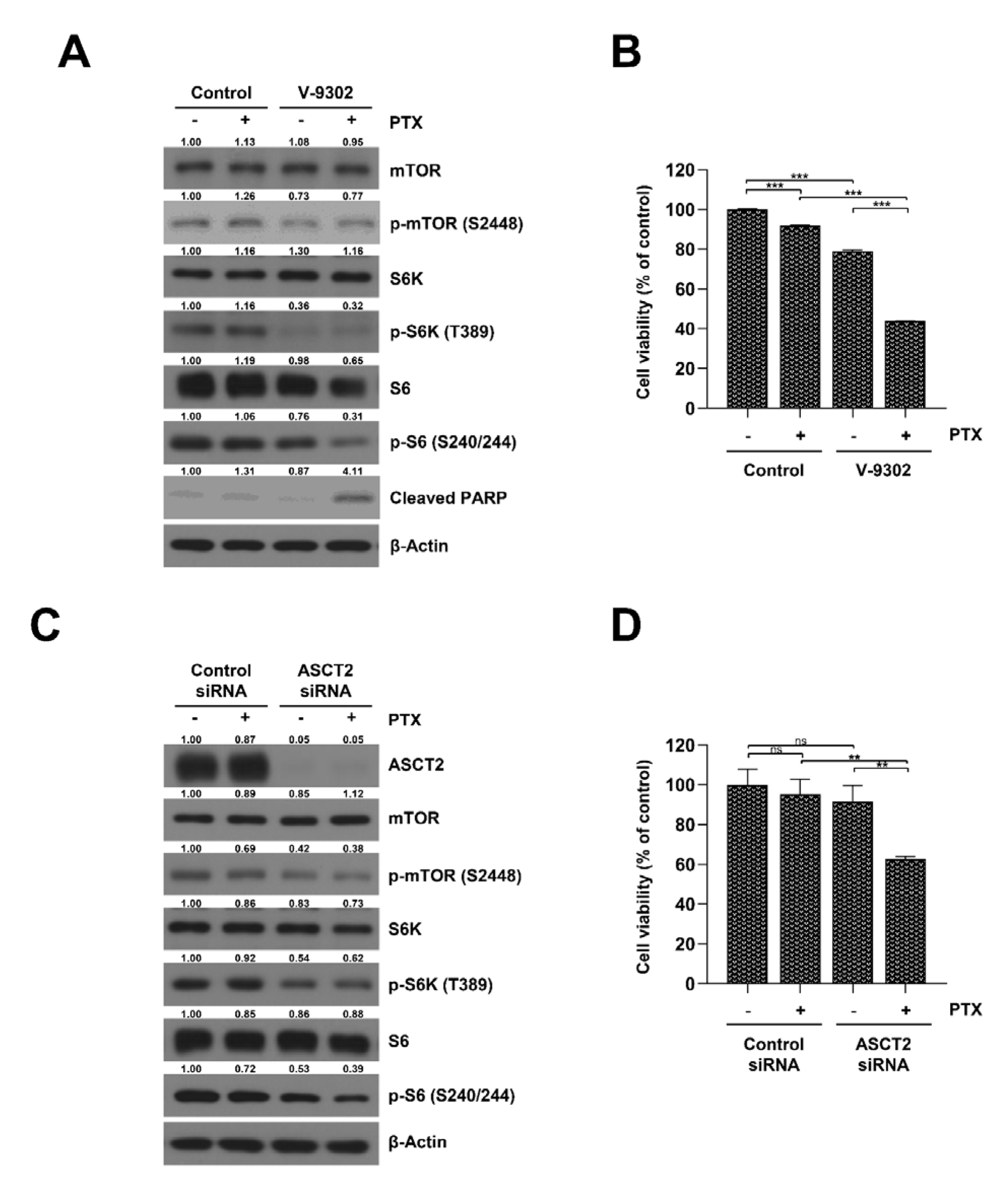
2.4. Inhibition of the Glutamine Transporter ASCT2 Suppresses mTORC1/S6K Signaling and Resensitizes SKOV3-TR Cells to Paclitaxel
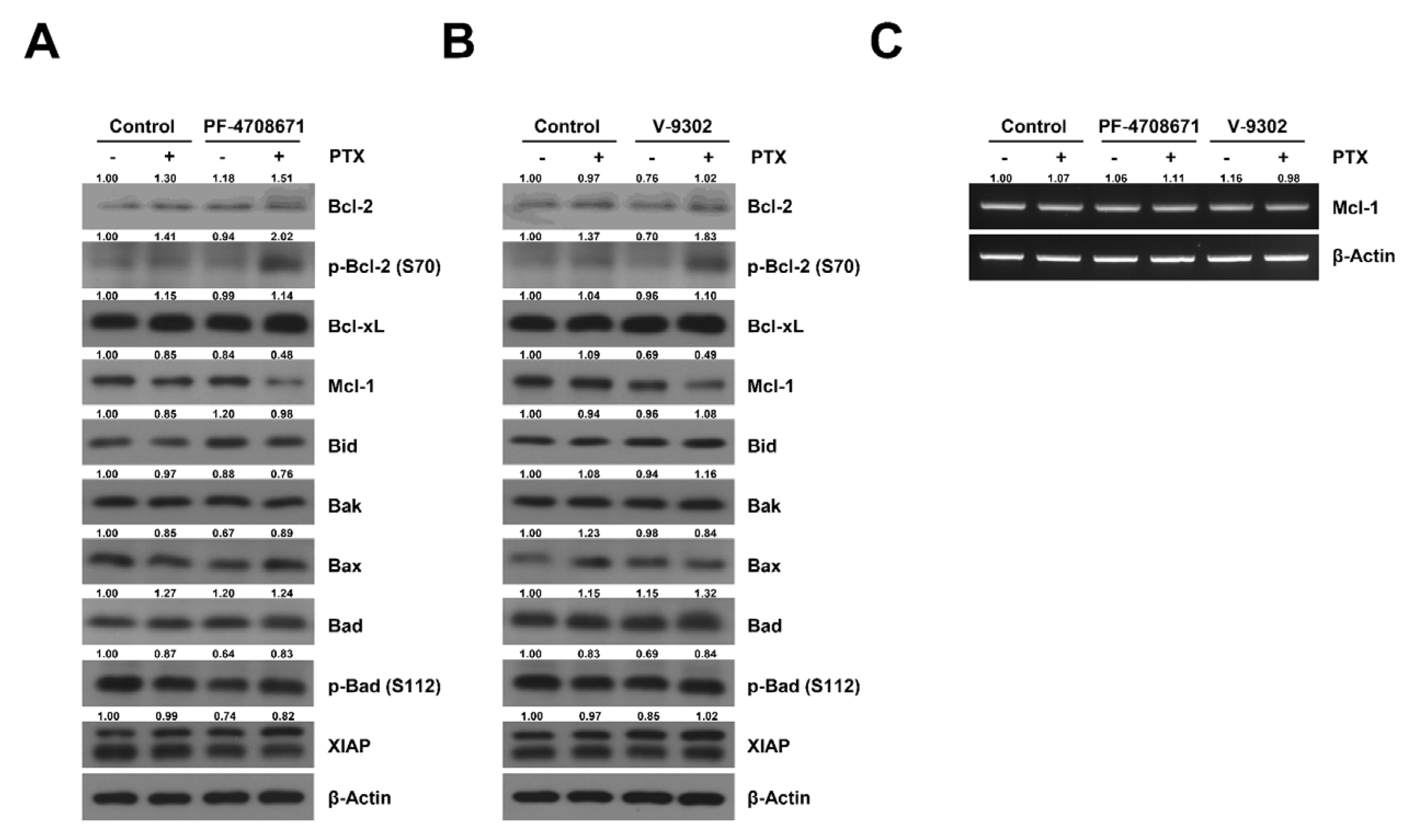
2.5. Cotreatment Paclitaxel with S6K Inhibitor and Glutamine Uptake Inhibitor Induced the Phosphorylation of Bcl-2 and Decreased the Expression of Mcl-1 in SKOV3-TR Cells
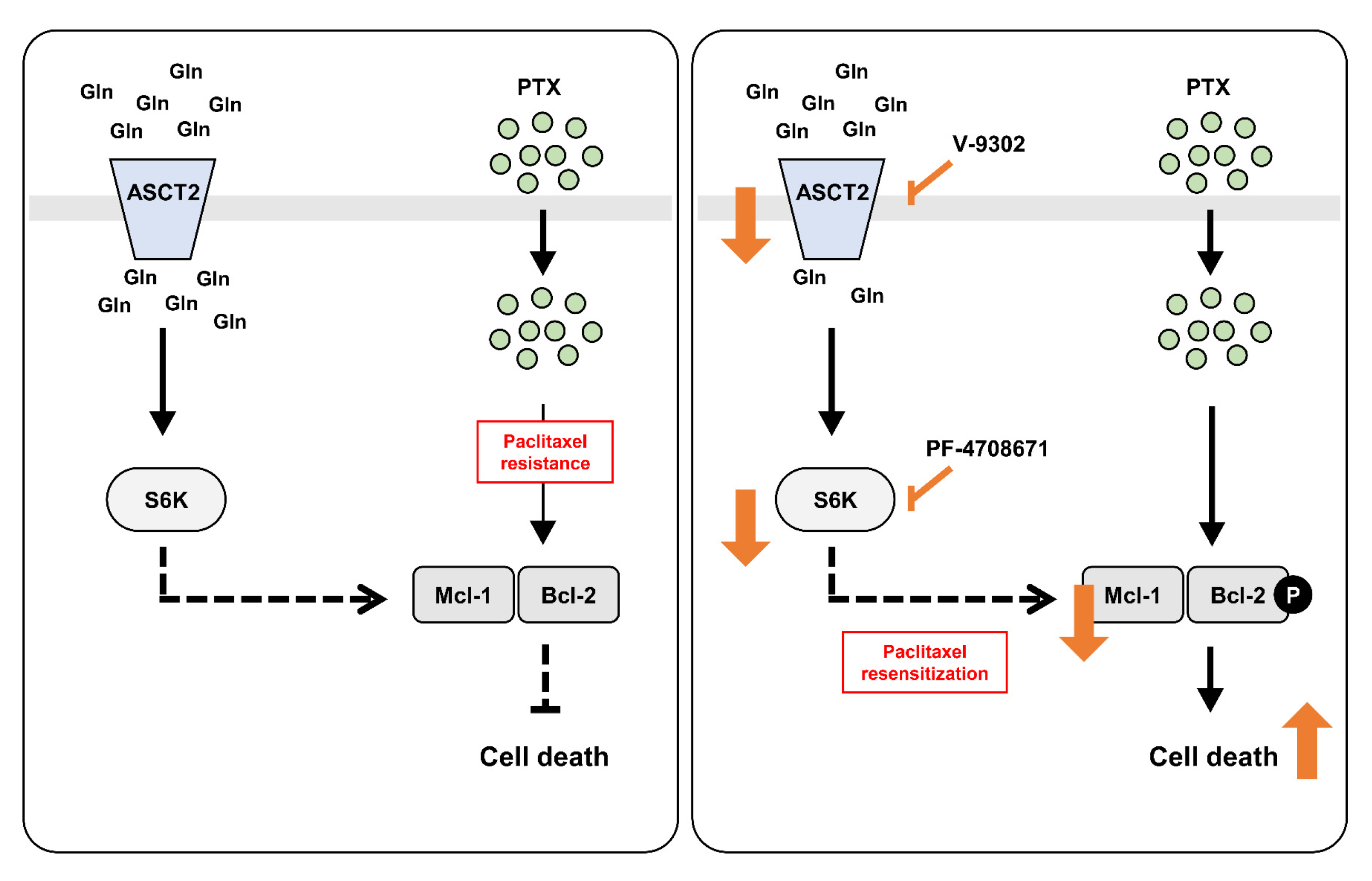
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Measurement of Cell Viability
4.3. Crystal Violet Staining
4.4. Transfection
4.5. Immunoblot Analysis
4.6. RNA Extraction and Reverse Transcription PCR Analysis
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9. [Google Scholar] [PubMed]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Davis, A.; Tinker, A.V.; Friedlander, M. “Platinum resistant” ovarian cancer: What is it, who to treat and how to measure benefit? Gynecol. Oncol. 2014, 133, 624–631. [Google Scholar] [CrossRef]
- Bhalla, K.N. Microtubule-targeted anticancer agents and apoptosis. Oncogene 2003, 22, 9075–9086. [Google Scholar] [CrossRef]
- Kim, A.; Ueda, Y.; Naka, T.; Enomoto, T. Therapeutic strategies in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 2012, 31, 14. [Google Scholar] [CrossRef]
- Agarwal, R.; Kaye, S.B. Ovarian cancer: Strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar]
- Locasale, J.W.; Cantley, L.C. Altered metabolism in cancer. BMC Biol. 2010, 8, 18–26. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Morii, E. Metabolic reprogramming of cancer cells during tumor progression and metastasis. Metabolites 2021, 11, 28. [Google Scholar] [CrossRef]
- Vettore, L.; Westbrook, R.L.; Tennant, D.A. New aspects of amino acid metabolism in cancer. Br. J. Cancer 2020, 122, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Labuschagne, C.F.; Van Den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef]
- Maddocks, O.D.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; van den Broek, N.J.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef]
- Yue, M.; Jiang, J.; Gao, P.; Liu, H.; Qing, G. Oncogenic MYC activates a feedforward regulatory loop promoting essential amino acid metabolism and tumorigenesis. Cell Rep. 2017, 21, 3819–3832. [Google Scholar] [CrossRef] [PubMed]
- Elorza, A.; Soro-Arnáiz, I.; Meléndez-Rodríguez, F.; Rodríguez-Vaello, V.; Marsboom, G.; de Cárcer, G.; Acosta-Iborra, B.; Albacete-Albacete, L.; Ordóñez, A.; Serrano-Oviedo, L. HIF2α acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol. Cell 2012, 48, 681–691. [Google Scholar] [CrossRef]
- Grzes, K.M.; Swamy, M.; Hukelmann, J.L.; Emslie, E.; Sinclair, L.V.; Cantrell, D.A. Control of amino acid transport coordinates metabolic reprogramming in T-cell malignancy. Leukemia 2017, 31, 2771–2779. [Google Scholar] [CrossRef]
- Dillon, B.J.; Prieto, V.G.; Curley, S.A.; Ensor, C.M.; Holtsberg, F.W.; Bomalaski, J.S.; Clark, M.A. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers: A method for identifying cancers sensitive to arginine deprivation. Cancer 2004, 100, 826–833. [Google Scholar] [CrossRef]
- Yoon, J.-K.; Frankel, A.E.; Feun, L.G.; Ekmekcioglu, S.; Kim, K.B. Arginine deprivation therapy for malignant melanoma. Clin. Pharmacol. Adv. Appl. 2013, 5, 11. [Google Scholar]
- Delage, B.; Fennell, D.A.; Nicholson, L.; McNeish, I.; Lemoine, N.R.; Crook, T.; Szlosarek, P.W. Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int. J. Cancer 2010, 126, 2762–2772. [Google Scholar] [CrossRef]
- Li, T.; Le, A. Glutamine metabolism in cancer. Heterog. Cancer Metab. 2018, 16, 13–32. [Google Scholar]
- Jin, L.; Alesi, G.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef]
- Hao, Y.; Samuels, Y.; Li, Q.; Krokowski, D.; Guan, B.-J.; Wang, C.; Jin, Z.; Dong, B.; Cao, B.; Feng, X. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat. Commun. 2016, 7, 11971. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Hernandez-Davies, J.E.; Tran, T.Q.; Reid, M.A.; Rosales, K.R.; Lowman, X.H.; Pan, M.; Moriceau, G.; Yang, Y.; Wu, J.; Lo, R.S. Vemurafenib resistance reprograms melanoma cells towards glutamine dependence. J. Transl. Med. 2015, 13, 210. [Google Scholar] [CrossRef]
- Qing, G.; Li, B.; Vu, A.; Skuli, N.; Walton, Z.E.; Liu, X.; Mayes, P.A.; Wise, D.R.; Thompson, C.B.; Maris, J.M. ATF4 regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell 2012, 22, 631–644. [Google Scholar] [CrossRef]
- Zhao, X.; Petrashen, A.P.; Sanders, J.A.; Peterson, A.L.; Sedivy, J.M. SLC1A5 glutamine transporter is a target of MYC and mediates reduced mTORC1 signaling and increased fatty acid oxidation in long-lived Myc hypomorphic mice. Aging Cell 2019, 18, e12947. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.-Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Christie, G.R.; Hajduch, E.; Hundal, H.S.; Proud, C.G.; Taylor, P.M. Intracellular sensing of amino acids in Xenopus laevis oocytes stimulates p70 S6 kinase in a target of rapamycin-dependent manner. J. Biol. Chem. 2002, 277, 9952–9957. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef]
- Vellai, T. How the Amino acid Leucine Activates the Key Cell-Growth Regulator mTOR; Nature Publishing Group: London, UK, 2021. [Google Scholar]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Ip, C.K.; Wong, A.S. Exploiting p70 S6 kinase as a target for ovarian cancer. Expert Opin. Ther. Targets 2012, 16, 619–630. [Google Scholar] [CrossRef]
- Tavares, M.R.; Pavan, I.C.; Amaral, C.L.; Meneguello, L.; Luchessi, A.D.; Simabuco, F.M. The S6K protein family in health and disease. Life Sci. 2015, 131, 1–10. [Google Scholar] [CrossRef]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef]
- Tsun, Z.-Y.; Possemato, R. Amino acid management in cancer. Semin. Cell Dev. Biol. 2015, 43, 22–32. [Google Scholar] [CrossRef]
- Shuvayeva, G.; Bobak, Y.; Igumentseva, N.; Titone, R.; Morani, F.; Stasyk, O.; Isidoro, C. Single amino acid arginine deprivation triggers prosurvival autophagic response in ovarian carcinoma SKOV3. BioMed Res. Int. 2014, 2014, 505041. [Google Scholar] [CrossRef]
- Durán, R.V.; Oppliger, W.; Robitaille, A.M.; Heiserich, L.; Skendaj, R.; Gottlieb, E.; Hall, M.N. Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 2012, 47, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Zhen, M.-C.; Wang, F.-Q.; Wu, S.-F.; Zhao, Y.-L.; Liu, P.-G.; Yin, Z.-Y. Identification of mTOR as a primary resistance factor of the IAP antagonist AT406 in hepatocellular carcinoma cells. Oncotarget 2017, 8, 9466. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, W.; Chang, J.; Liu, M.; Yuan, J.; Zhang, J.; Qin, J.; Xia, X.; Wang, Y. Quantitative proteomics profiling reveals activation of mTOR pathway in trastuzumab resistance. Oncotarget 2017, 8, 45793. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H.-C.; Han, J.-M. Amino Acid Metabolism in Cancer Drug Resistance. Cells 2022, 11, 140. [Google Scholar] [CrossRef] [PubMed]
- Faried, L.S.; Faried, A.; Kanuma, T.; Nakazato, T.; Tamura, T.; Kuwano, H.; Minegishi, T. Inhibition of the mammalian target of rapamycin (mTOR) by rapamycin increases chemosensitivity of CaSki cells to paclitaxel. Eur. J. Cancer 2006, 42, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Cheaib, B.; Auguste, A.; Leary, A. The PI3K/Akt/mTOR pathway in ovarian cancer: Therapeutic opportunities and challenges. Chin. J. Cancer 2015, 34, 4–16. [Google Scholar] [CrossRef]
- Hernández-Prat, A.; Rodriguez-Vida, A.; Juanpere-Rodero, N.; Arpi, O.; Menéndez, S.; Soria-Jiménez, L.; Martínez, A.; Iarchouk, N.; Rojo, F.; Albanell, J. Novel Oral mTORC1/2 Inhibitor TAK-228 Has Synergistic Antitumor Effects When Combined with Paclitaxel or PI3Kα Inhibitor TAK-117 in Preclinical Bladder Cancer ModelsmTOR Inhibitor TAK-228 in Preclinical Bladder Cancer Models. Mol. Cancer Res. 2019, 17, 1931–1944. [Google Scholar] [CrossRef]
- Dai, H.; Ding, H.; Meng, X.W.; Lee, S.-H.; Schneider, P.A.; Kaufmann, S.H. Contribution of Bcl-2 Phosphorylation to Bak Binding and Drug ResistanceBcl-2 Phosphorylation, Bak Binding, and Paclitaxel Resistance. Cancer Res. 2013, 73, 6998–7008. [Google Scholar] [CrossRef]
- Jones, N.A.; Turner, J.; McIlwrath, A.J.; Brown, R.; Dive, C. Cisplatin-and paclitaxel-induced apoptosis of ovarian carcinoma cells and the relationship between bax and bak up-regulation and the functional status of p53. Mol. Pharmacol. 1998, 53, 819–826. [Google Scholar]
- Beale, P.; Rogers, P.; Boxall, F.; Sharp, S.; Kelland, L. BCL-2 family protein expression and platinum drug resistance in ovarian carcinoma. Br. J. Cancer 2000, 82, 436–440. [Google Scholar] [CrossRef]
- Blagosklonny, M. Unwinding the loop of Bcl-2 phosphorylation. Leukemia 2001, 15, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Goberdhan, D.C.; Wilson, C.; Harris, A.L. Amino acid sensing by mTORC1: Intracellular transporters mark the spot. Cell Metab. 2016, 23, 580–589. [Google Scholar] [CrossRef]
- Lukasheva, E.V.; Babayeva, G.; Karshieva, S.S.; Zhdanov, D.D.; Pokrovsky, V.S. L-Lysine α-Oxidase: Enzyme with Anticancer Properties. Pharmaceuticals 2021, 14, 1070. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Chandel, N.S. Mitochondrial metabolism and cancer. Ann. N. Y. Acad. Sci. 2009, 1177, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yi, D.; Hou, Y.; Ding, B.; Li, K.; Li, B.; Zhu, H.; Liu, Y.; Wu, G. Dietary supplementation with α-ketoglutarate activates mTOR signaling and enhances energy status in skeletal muscle of lipopolysaccharide-challenged piglets. J. Nutr. 2016, 146, 1514–1520. [Google Scholar] [CrossRef]
- Yuan, L.; Sheng, X.; Willson, A.K.; Roque, D.R.; Stine, J.E.; Guo, H.; Jones, H.M.; Zhou, C.; Bae-Jump, V.L. Glutamine promotes ovarian cancer cell proliferation through the mTOR/S6 pathway. Endocr. Relat. Cancer 2015, 22, 577–591. [Google Scholar] [CrossRef]
- Schulte, M.L.; Fu, A.; Zhao, P.; Li, J.; Geng, L.; Smith, S.T.; Kondo, J.; Coffey, R.J.; Johnson, M.O.; Rathmell, J.C. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat. Med. 2018, 24, 194–202. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Babu, E.; Ramachandran, S.; Ganapathy, V. Amino acid transporters in cancer and their relevance to “glutamine addiction”: Novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015, 75, 1782–1788. [Google Scholar] [CrossRef]
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci. 2018, 43, 752–789. [Google Scholar] [CrossRef]
- Miller, A.V.; Hicks, M.A.; Nakajima, W.; Richardson, A.C.; Windle, J.J.; Harada, H. Paclitaxel-induced apoptosis is BAK-dependent, but BAX and BIM-independent in breast tumor. PLoS ONE 2013, 8, e60685. [Google Scholar] [CrossRef]
- Srivastava, R.K.; Mi, Q.-S.; Hardwick, J.M.; Longo, D.L. Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc. Natl. Acad. Sci. USA 1999, 96, 3775–3780. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Jena, N.; Croce, C.M. Inactivation of Bcl-2 by phosphorylation. Proc. Natl. Acad. Sci. USA 1995, 92, 4507–4511. [Google Scholar] [CrossRef]
- Ruvolo, P.; Deng, X.; May, W. Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia 2001, 15, 515–522. [Google Scholar] [CrossRef]
- Bassik, M.C.; Scorrano, L.; Oakes, S.A.; Pozzan, T.; Korsmeyer, S.J. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004, 23, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Shuang, W.; Hou, L.; Zhu, Y.; Li, Q.; Hu, W. Mcl-1 stabilization confers resistance to taxol in human gastric cancer. Oncotarget 2017, 8, 82981. [Google Scholar] [CrossRef] [PubMed]
- Park, J.A.; Jin, H.O.; Lee, H.N.; Kim, J.H.; Park, I.C.; Noh, W.C.; Chang, Y.H.; Hong, Y.J.; Kim, K.C.; Lee, J.K. S6K1 inhibition enhances the apoptotic cell death of breast cancer cells in response to Bcl-2/Bcl-xL inhibition by the downregulation of survivin. Oncol. Lett. 2015, 10, 829–834. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Harada, H.; Andersen, J.S.; Mann, M.; Terada, N.; Korsmeyer, S.J. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc. Natl. Acad. Sci. USA 2001, 98, 9666–9670. [Google Scholar] [CrossRef]
- Ferlini, C.; Raspaglio, G.; Mozzetti, S.; Distefano, M.; Filippetti, F.; Martinelli, E.; Ferrandina, G.; Gallo, D.; Ranelletti, F.O.; Scambia, G. Bcl-2 down-regulation is a novel mechanism of paclitaxel resistance. Mol. Pharmacol. 2003, 64, 51–58. [Google Scholar] [CrossRef]
- Chong, S.J.F.; Iskandar, K.; Lai, J.X.H.; Qu, J.; Raman, D.; Valentin, R.; Herbaux, C.; Collins, M.; Low, I.C.C.; Loh, T. Serine-70 phosphorylated Bcl-2 prevents oxidative stress-induced DNA damage by modulating the mitochondrial redox metabolism. Nucleic Acids Res. 2020, 48, 12727–12745. [Google Scholar] [CrossRef]
- Mojsa, B.; Lassot, I.; Desagher, S. Mcl-1 ubiquitination: Unique regulation of an essential survival protein. Cells 2014, 3, 418–437. [Google Scholar] [CrossRef]
- Harley, M.E.; Allan, L.A.; Sanderson, H.S.; Clarke, P.R. Phosphorylation of Mcl-1 by CDK1–cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. EMBO J. 2010, 29, 2407–2420. [Google Scholar] [CrossRef] [PubMed]
- Senichkin, V.V.; Streletskaia, A.Y.; Gorbunova, A.S.; Zhivotovsky, B.; Kopeina, G.S. Saga of Mcl-1: Regulation from transcription to degradation. Cell Death Differ. 2020, 27, 405–419. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.; Yue, P.; Deng, X.; Khuri, F.R.; Sun, S.-Y. mTOR complex 2 stabilizes Mcl-1 protein by suppressing its glycogen synthase kinase 3-dependent and SCF-FBXW7-mediated degradation. Mol. Cell. Biol. 2015, 35, 2344–2355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, R.; Shuai, Y.; Huang, Y.; Jin, R.; Wang, X.; Luo, J. ASCT2 (SLC1A5)-dependent glutamine uptake is involved in the progression of head and neck squamous cell carcinoma. Br. J. Cancer 2020, 122, 82–93. [Google Scholar] [CrossRef]
- Dohl, J.; Passos, M.E.P.; Foldi, J.; Chen, Y.; Pithon-Curi, T.; Curi, R.; Gorjao, R.; Deuster, P.A.; Yu, T. Glutamine depletion disrupts mitochondrial integrity and impairs C2C12 myoblast proliferation, differentiation, and the heat-shock response. Nutr. Res. 2020, 84, 42–52. [Google Scholar] [CrossRef]
- Park, H.Y.; Kim, M.-J.; Kim, Y.J.; Lee, S.; Jin, J.; Lee, S.; Choi, Y.-K.; Park, K.-G. V-9302 inhibits proliferation and migration of VSMCs, and reduces neointima formation in mice after carotid artery ligation. Biochem. Biophys. Res. Commun. 2021, 560, 45–51. [Google Scholar] [CrossRef]
- Ratre, Y.K.; Mehta, A.; Sharma, R.; Soni, V.K.; Shukla, D.; Tripathi, V.N.; Vishvakarma, N.K. Glutamine metabolism in liver cancer: Role in progression and potential therapeutic targeting. In Theranostics and Precision Medicine for the Management of Hepatocellular Carcinoma; Elsevier: Amsterdam, The Netherlands, 2022; pp. 199–217. [Google Scholar]
- Edwards, D.N.; Ngwa, V.M.; Raybuck, A.L.; Wang, S.; Hwang, Y.; Kim, L.C.; Cho, S.H.; Paik, Y.; Wang, Q.; Zhang, S. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J. Clin. Investig. 2021, 131, e140100. [Google Scholar] [CrossRef]
- Halder, J.; Landen, C.N., Jr.; Lutgendorf, S.K.; Li, Y.; Jennings, N.B.; Fan, D.; Nelkin, G.M.; Schmandt, R.; Schaller, M.D.; Sood, A.K. Focal adhesion kinase silencing augments docetaxel-mediated apoptosis in ovarian cancer cells. Clin. Cancer Res. 2005, 11, 8829–8836. [Google Scholar] [CrossRef]
- AAT Bioquest Inc. Quest Graph™ IC50 Calculator. 2020. Available online: https://www.aatbio.com/tools/ic50-calculator (accessed on 30 July 2022).






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, G.; Jang, S.-K.; Kim, Y.J.; Jin, H.-O.; Bae, S.; Hong, J.; Park, I.-C.; Lee, J.H. Inhibition of Glutamine Uptake Resensitizes Paclitaxel Resistance in SKOV3-TR Ovarian Cancer Cell via mTORC1/S6K Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 8761. https://doi.org/10.3390/ijms23158761
Kim G, Jang S-K, Kim YJ, Jin H-O, Bae S, Hong J, Park I-C, Lee JH. Inhibition of Glutamine Uptake Resensitizes Paclitaxel Resistance in SKOV3-TR Ovarian Cancer Cell via mTORC1/S6K Signaling Pathway. International Journal of Molecular Sciences. 2022; 23(15):8761. https://doi.org/10.3390/ijms23158761
Chicago/Turabian StyleKim, Gyeongmi, Se-Kyeong Jang, Yu Jin Kim, Hyeon-Ok Jin, Seunghee Bae, Jungil Hong, In-Chul Park, and Jae Ho Lee. 2022. "Inhibition of Glutamine Uptake Resensitizes Paclitaxel Resistance in SKOV3-TR Ovarian Cancer Cell via mTORC1/S6K Signaling Pathway" International Journal of Molecular Sciences 23, no. 15: 8761. https://doi.org/10.3390/ijms23158761
APA StyleKim, G., Jang, S.-K., Kim, Y. J., Jin, H.-O., Bae, S., Hong, J., Park, I.-C., & Lee, J. H. (2022). Inhibition of Glutamine Uptake Resensitizes Paclitaxel Resistance in SKOV3-TR Ovarian Cancer Cell via mTORC1/S6K Signaling Pathway. International Journal of Molecular Sciences, 23(15), 8761. https://doi.org/10.3390/ijms23158761