
Lithium Enhances Hippocampal Glucose Metabolism in an In Vitro Mice Model of Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
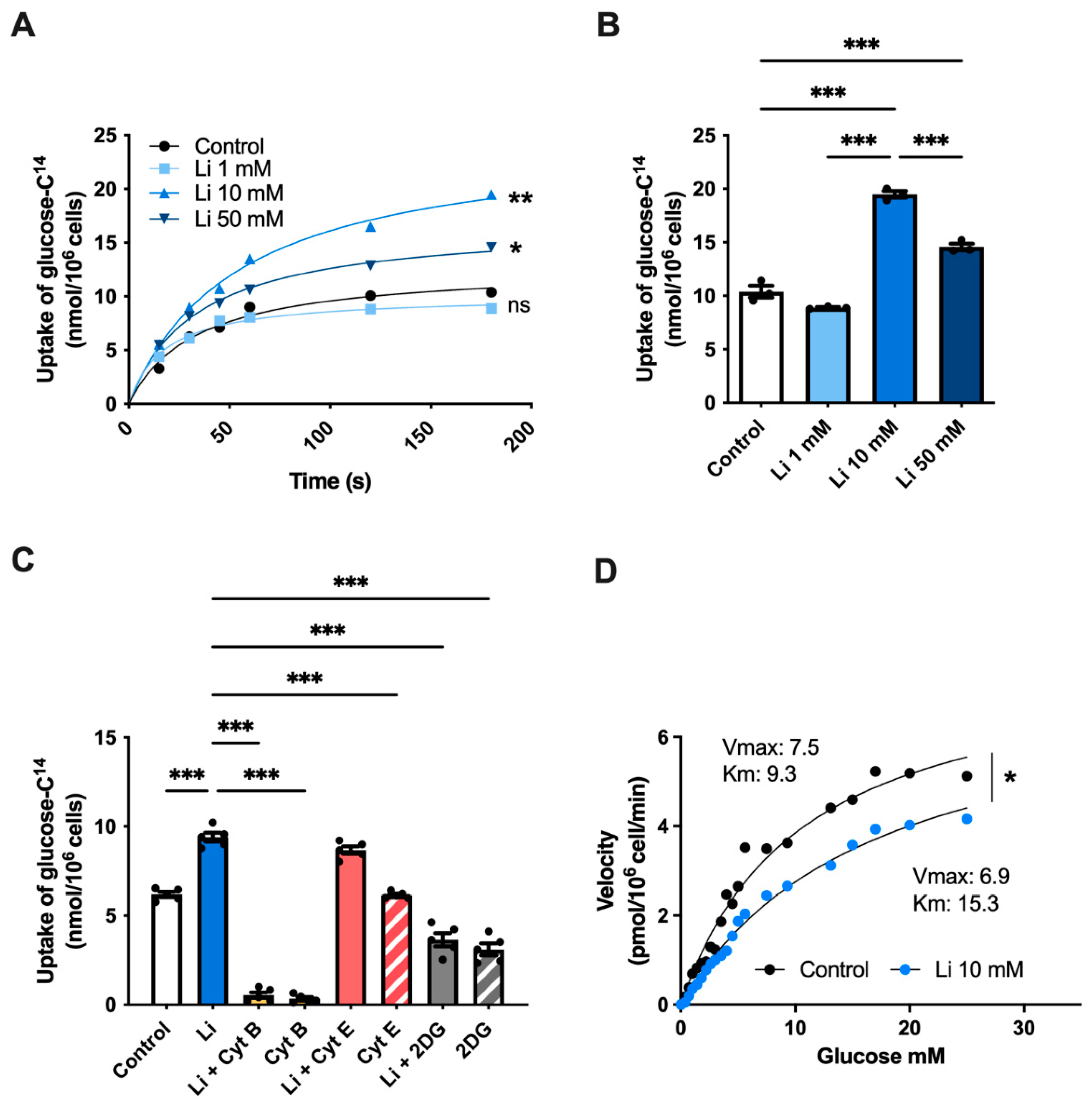
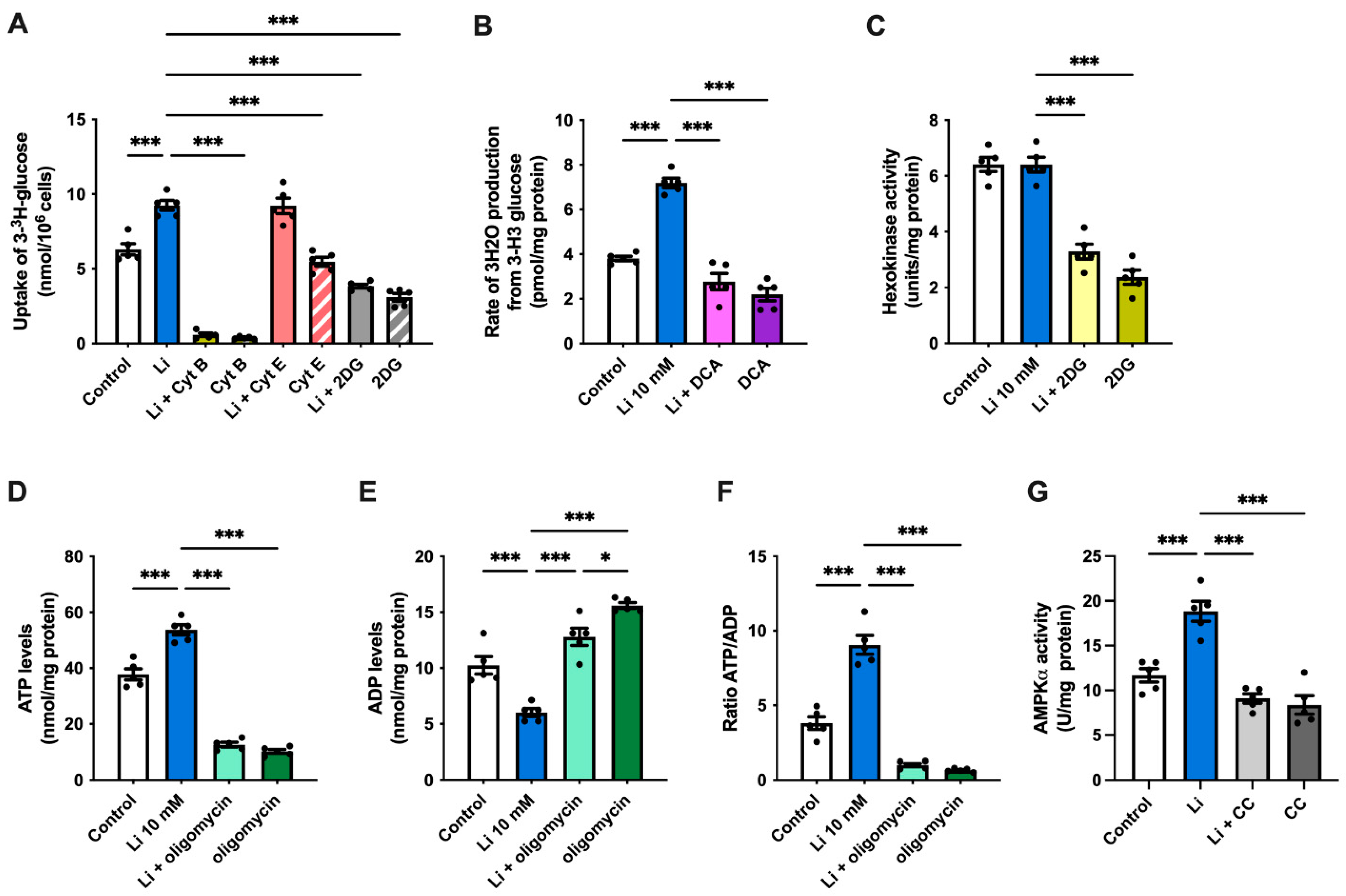
2.1. Lithium Stimulates Glucose Uptake in Hippocampal Neurons
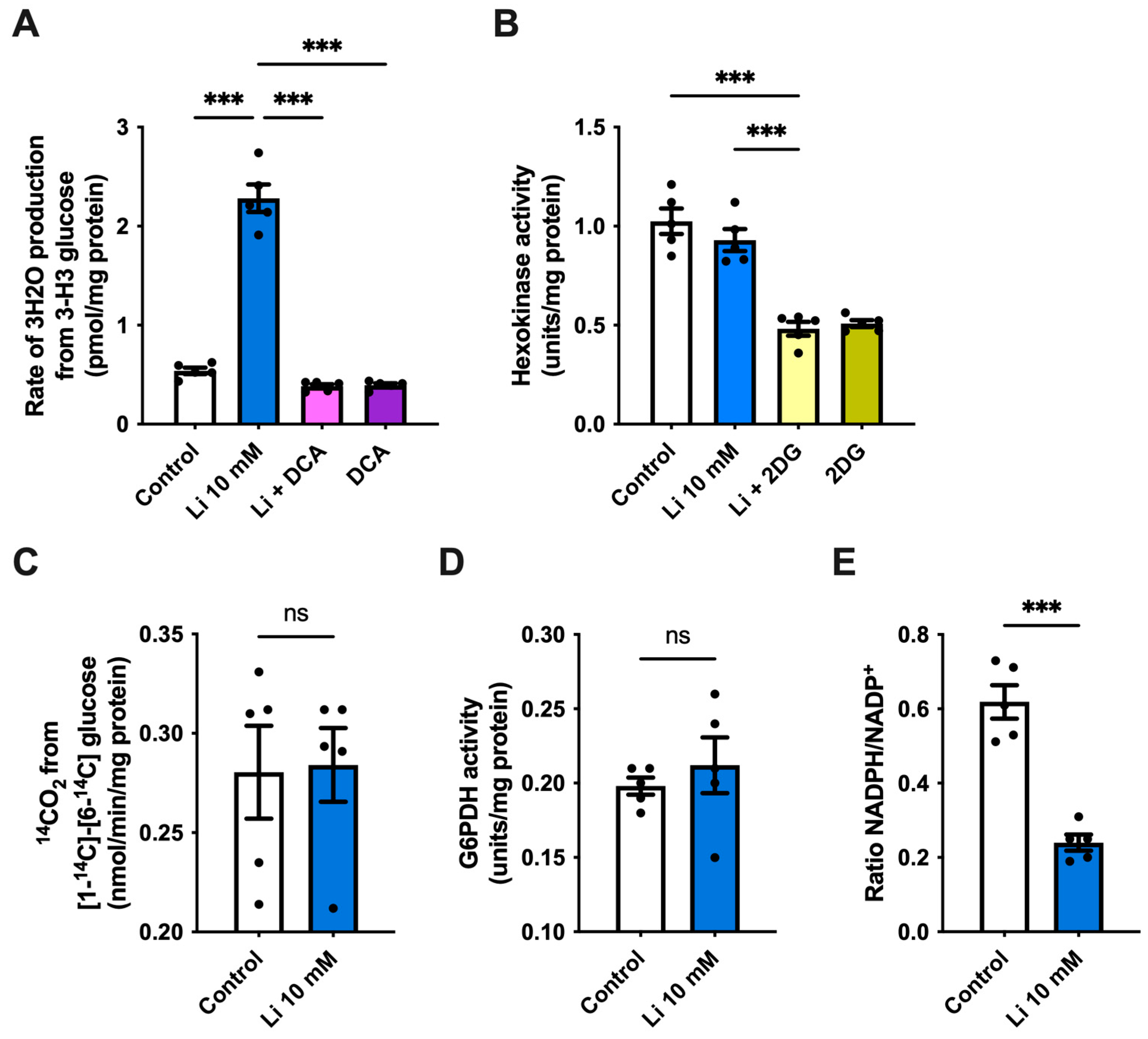
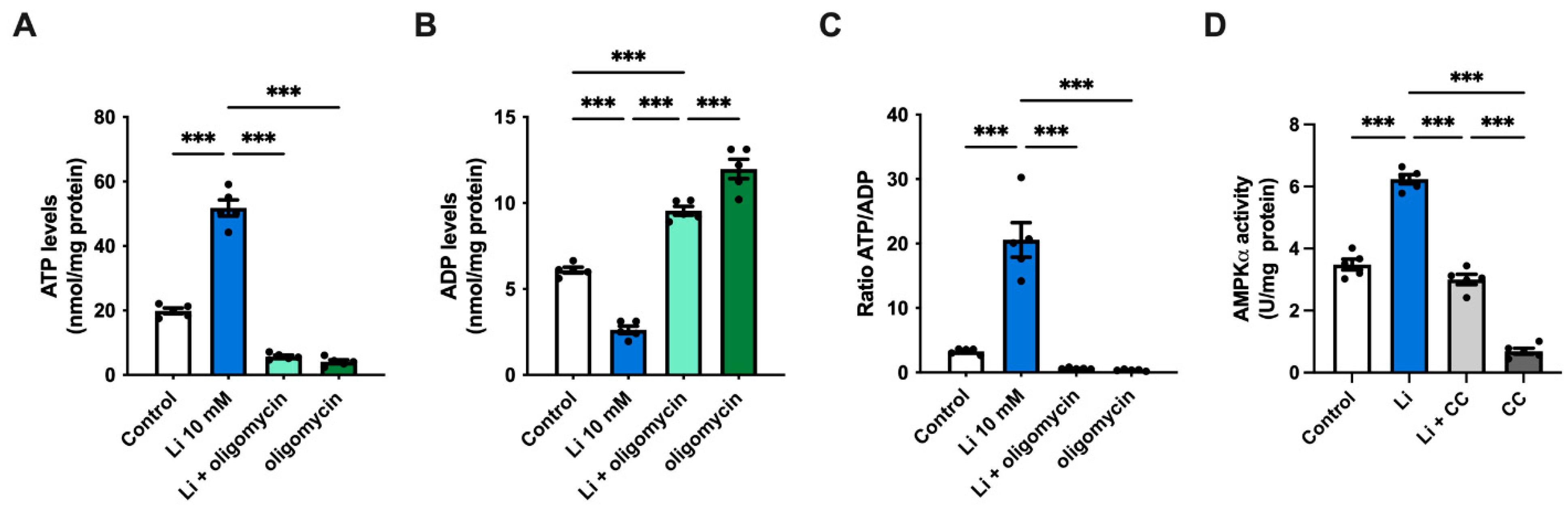
2.2. Lithium Promotes Glycolysis and ATP Synthesis in Hippocampal Neurons
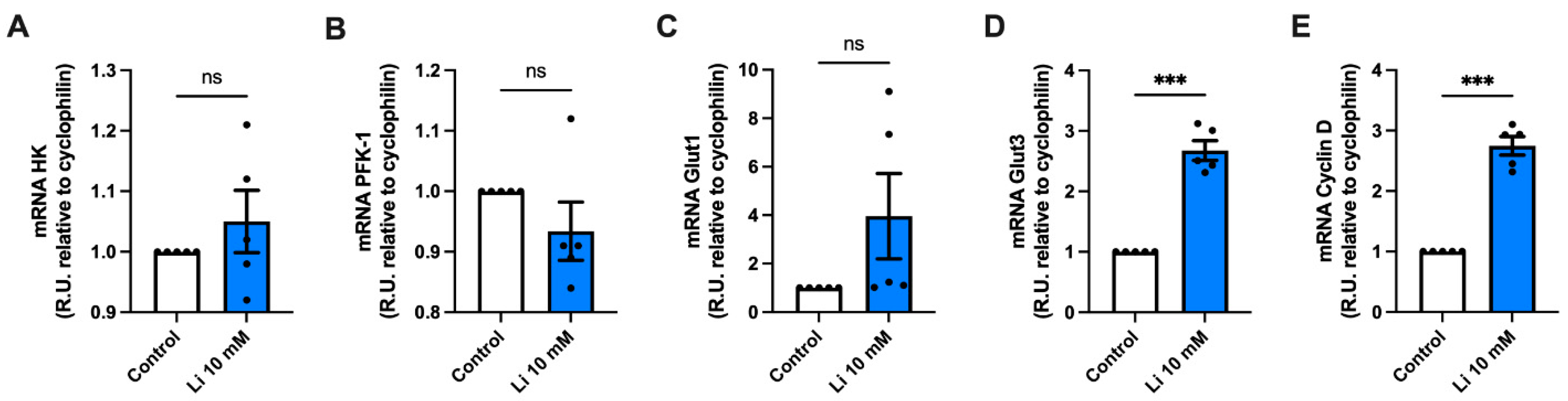
2.3. Lithium Alters the Genetic Expression of Metabolic Genes
2.4. Lithium Enhances Glycolysis and AMPK Activity in Hippocampal Slices of APP/PS1 Mice
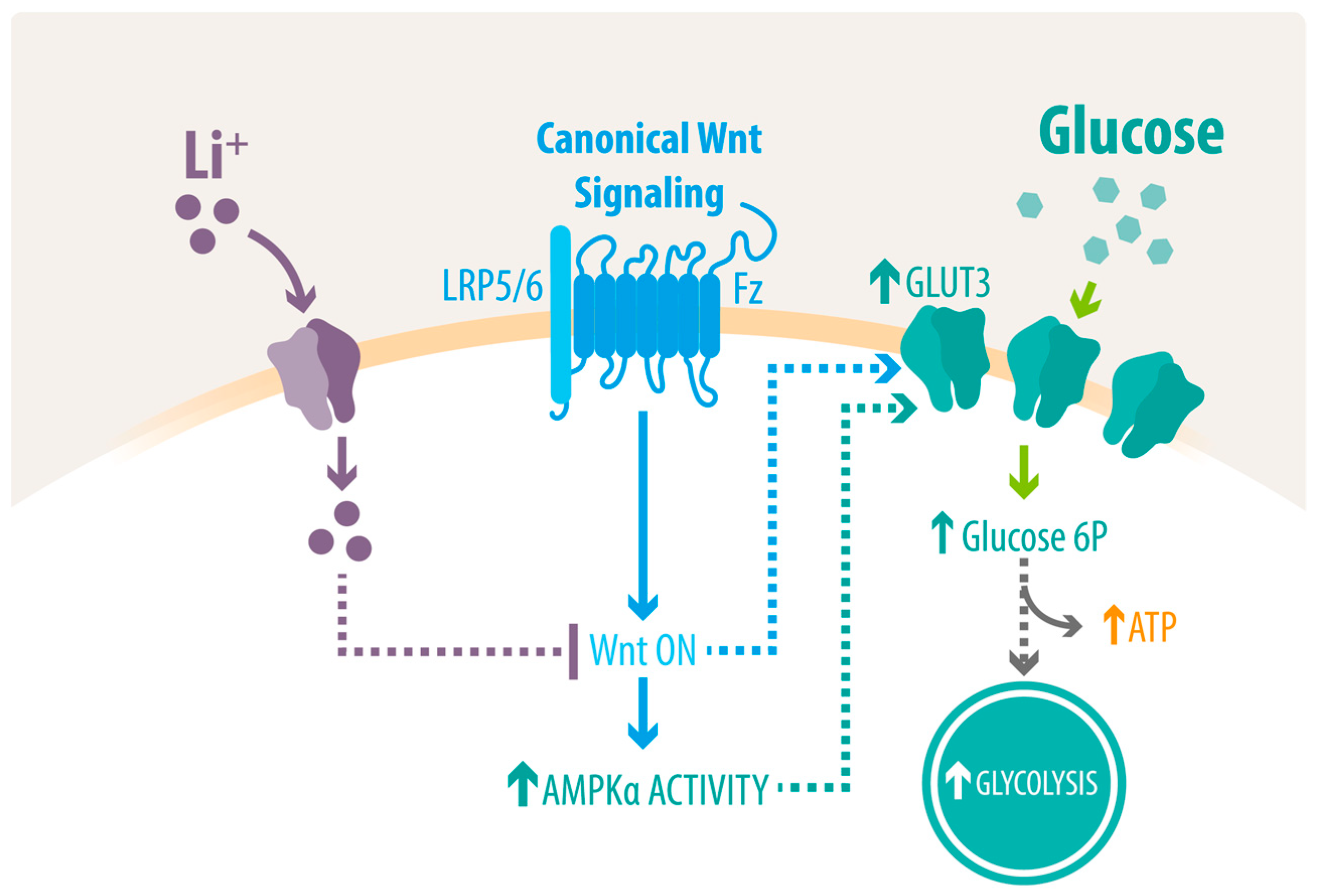
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Primary Hippocampal Neuronal Cultures
4.3. Hippocampal Slices Preparation
4.4. Quantitative Real-Time PCR (qRT-PCR)
4.5. Glucose Uptake Analysis
4.6. Determination of the Glycolytic Rate
4.7. Hexokinase (HK) Activity
4.8. Quantification of ATP, ADP and NADPH/NADP+
4.9. AMPKα Activity
4.10. Pentose Phosphate Pathway Measurements
4.11. Determination of G6PDH Activity
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease: Mechanism of disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease: FDG-PET studies in MCI and AD. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 486–510. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosconi, L.; De Santi, S.; Li, J.; Tsui, W.H.; Li, Y.; Boppana, M.; Laska, E.; Rusinek, H.; de Leon, M.J. Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol. Aging 2008, 29, 676–692. [Google Scholar] [CrossRef] [Green Version]
- Gordon, B.A.; Blazey, T.M.; Su, Y.; Hari-Raj, A.; Dincer, A.; Flores, S.; Christensen, J.; McDade, E.; Wang, G.; Xiong, C.; et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: A longitudinal study. Lancet Neurol. 2018, 17, 241–250. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L. Glucose metabolism in normal aging and Alzheimer’s disease: Methodological and physiological considerations for PET studies. Clin. Transl. Imaging 2013, 1, 217–233. [Google Scholar] [CrossRef] [Green Version]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.-C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease–Associated Aβ oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef]
- Niccoli, T.; Cabecinha, M.; Tillmann, A.; Kerr, F.; Wong, C.T.; Cardenes, D.; Vincent, A.J.; Bettedi, L.; Li, L.; Grönke, S.; et al. Increased Glucose Transport into Neurons Rescues Aβ Toxicity in Drosophila. Curr. Biol. 2016, 26, 2291–2300. [Google Scholar] [CrossRef] [Green Version]
- Gejl, M.; Brock, B.; Egefjord, L.; Vang, K.; Rungby, J.; Gjedde, A. Blood-Brain Glucose Transfer in Alzheimer’s disease: Effect of GLP-1 Analog Treatment. Sci. Rep. 2017, 7, 17490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cisternas, P.; Zolezzi, J.M.; Martinez, M.; Torres, V.I.; Wong, G.W.; Inestrosa, N.C. Wnt-induced activation of glucose metabolism mediates the in vivo neuroprotective roles of Wnt signaling in Alzheimer disease. J. Neurochem. 2019, 149, 54–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens, J.; Wang, Q.W.; Kim, Y.; Yu, D.X.; Pham, S.; Yang, B.; Zheng, Y.; Diffenderfer, K.E.; Zhang, J.; Soltani, S.; et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 2015, 527, 95–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhi, G.S.; Tanious, M.; Das, P.; Coulston, C.M.; Berk, M. Potential mechanisms of action of lithium in bipolar disorder: Current understanding. CNS Drugs 2013, 27, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Baastrup, P.C.; Poulsen, J.C.; Schou, M.; Thomsen, K.; Amdisen, A. Prophylactic lithium: Double blind discontinuation in manic-depressive and recurrent-depressive disorders. Lancet 1970, 296, 326–330. [Google Scholar] [CrossRef]
- Haussmann, R.; Noppes, F.; Brandt, M.D.; Bauer, M.; Donix, M. Lithium: A therapeutic option in Alzheimer’s disease and its prodromal stages? Neurosci. Lett. 2021, 760, 136044. [Google Scholar] [CrossRef]
- Andrade Nunes, M.; Araujo Viel, T.; Sousa Buck, H. Microdose Lithium Treatment Stabilized Cognitive Impairment in Patients with Alzheimer’s Disease. Curr. Alzheimer Res. 2013, 10, 104–107. [Google Scholar]
- Macdonald, A.; Briggs, K.; Poppe, M.; Higgins, A.; Velayudhan, L.; Lovestone, S. A feasibility and tolerability study of lithium in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2008, 23, 704–711. [Google Scholar] [CrossRef]
- Matsunaga, S.; Kishi, T.; Annas, P.; Basun, H.; Hampel, H.; Iwata, N. Lithium as a Treatment for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2015, 48, 403–410. [Google Scholar] [CrossRef]
- Hong, M.; Chen, D.C.R.; Klein, P.S.; Lee, V.M.Y. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J. Biol. Chem. 1997, 272, 25326–25332. [Google Scholar] [CrossRef] [Green Version]
- Asano, R.; Hamano, T.; Yen, S.; Sasaki, H.; Yamaguchi, T.; Endo, Y.; Enomoto, S.; Shirafuji, N.; Ikawa, M.; Yamamura, O.; et al. Lithium chloride inhibits phosphorylation of tau protein. Alzheimer’s Dement. 2021, 17, 54807. [Google Scholar] [CrossRef]
- Wilson, E.N.; Do Carmo, S.; Welikovitch, L.A.; Hall, H.; Aguilar, L.F.; Foret, M.K.; Iulita, M.F.; Jia, D.T.; Marks, A.R.; Allard, S.; et al. NP03, a Microdose Lithium Formulation, Blunts Early Amyloid Post-Plaque Neuropathology in McGill-R-Thy1-APP Alzheimer-Like Transgenic Rats. J. Alzheimer’s Dis. 2020, 73, 723–739. [Google Scholar] [CrossRef]
- Toledo, E.M.; Inestrosa, N.C. Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1ΔE9 mouse model of Alzheimer’s disease. Mol. Psychiatry 2010, 15, 272–285. [Google Scholar] [CrossRef]
- Noble, W.; Planel, E.; Zehr, C.; Olm, V.; Meyerson, J.; Suleman, F.; Gaynor, K.; Wang, L.; LaFrancois, J.; Feinstein, B.; et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 6990–6995. [Google Scholar] [CrossRef] [Green Version]
- Cristobal, J.; Garcia-Garcia, L.; Delgado, M.; Pozo, M.; Medina, M. A longitudinal FDG-PET study of transgenic mice overexpressing GSK- 3β in the brain. Curr. Alzheimer Res. 2014, 11, 175–181. [Google Scholar] [CrossRef]
- Hollander, E.; Buchsbaum, M.S.; Haznedar, M.M.; Berenguer, J.; Berlin, H.A.; Chaplin, W.; Goodman, C.R.; LiCalzi, E.M.; Newmark, R.; Pallanti, S. FDG-PET study in pathological gamblers: 1. Lithium increases orbitofrontal, dorsolateral and cingulate metabolism. Neuropsychobiology 2008, 58, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Forlenza, O.V.; Coutinho, A.M.N.; Aprahamian, I.; Prando, S.; Mendes, L.L.; Diniz, B.S.; Gattaz, W.F.; Buchpiguel, C.A. Long-term lithium treatment reduces glucose metabolism in the cerebellum and hippocampus of nondemented older adults: An [18F]FDG-PET study. ACS Chem. Neurosci. 2014, 5, 484–489. [Google Scholar] [CrossRef] [Green Version]
- Dixit, P.K.; Smithberg, M. Toxic Effect of Lithium in Mouse Brain. Proc. Soc. Exp. Biol. Med. 1988, 187, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Xiong, N.; Liu, L.; Huang, J.; Han, C.; Zhang, G.; Li, J.; Xu, X.; Lin, Z.; Wang, T. Lithium protects dopaminergic cells from rotenone toxicity via autophagy enhancement. BMC Neurosci. 2015, 16, 82. [Google Scholar] [CrossRef] [Green Version]
- Stampone, E.; Bencivenga, D.; Barone, C.; Aulitto, A.; Verace, F.; Ragione, F.D.; Borriello, A. High dosage lithium treatment induces DNA damage and p57kip2 decrease. Int. J. Mol. Sci. 2020, 21, 1169. [Google Scholar] [CrossRef] [Green Version]
- Pajak, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-D-Glucose and its analogs: From diagnostic to therapeutic agents. Int. J. Mol. Sci. 2020, 21, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.; Koh, J.; Kim, S.; Kim, K. Effect of lithium on the mechanism of glucose transport in skeletal muscles. J. Nutr. Sci. Vitaminol. 2017, 63, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Tabata, I.; Schluter, J.; Gulve, E.A.; Holloszy, J.O. Lithium increases susceptibility of muscle glucose transport to stimulation by various agents. Diabetes 1994, 43, 903–907. [Google Scholar] [CrossRef]
- Leroy, C.; Pierre, K.; Simpson, I.A.; Pellerin, L.; Vannucci, S.J.; Nehlig, A. Temporal changes in mRNA expression of the brain nutrient transporters in the lithium-pilocarpine model of epilepsy in the immature and adult rat. Neurobiol. Dis. 2011, 43, 588–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, F.; Simpson, I.A. Modulation of expression of glucose transporters GLUT3 and GLUT1 by potassium and N-methyl-d-aspartate in cultured cerebellar granule neurons. Mol. Cell Neurosci. 1994, 5, 369–375. [Google Scholar] [CrossRef]
- Szablewski, L. Glucose Transporters in Brain: In Health and in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 55, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.Y.; Zhao, X.Y.; Huang, Q.L.; Sun, H.Y.; Sun, C.; Yuan, J.; He, C.; Sun, Y.; Huang, X.; Kong, W.; et al. Activation of Wnt/β-catenin signaling by lithium chloride attenuates d-galactose-induced neurodegeneration in the auditory cortex of a rat model of aging. FEBS Open Bio 2017, 7, 759–776. [Google Scholar] [CrossRef] [PubMed]
- Hedgepeth, C.M.; Conrad, L.J.; Zhang, J.; Huang, H.C.; Lee, V.M.Y.; Klein, P.S. Activation of the Wnt signaling pathway: A molecular mechanism for lithium action. Dev. Biol. 1997, 185, 82–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clément-Lacroix, P.; Ai, M.; Morvan, F.; Roman-Roman, S.; Vayssière, B.; Belleville, C.; Estrera, K.; Warman, M.L.; Baron, R.; Rawadi, G. Lrp5-independent activation of Wnt signaling by lithium chloride increases bone formation and bone mass in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 17406–17411. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L.; Mistur, R.; Switalski, R.; Tsui, W.H.; Glodzik, L.; Li, Y.; Pirraglia, E.; De Santi, S.; Reisberg, B.; Wisniewski, T.; et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Dukart, J.; Kherif, F.; Mueller, K.; Adaszewski, S.; Schroeter, M.L.; Frackowiak, R.S.J.; Draganski, B. Generative FDG-PET and MRI Model of Aging and Disease Progression in Alzheimer’s Disease. PLoS Comput. Biol. 2013, 9, e1002987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, G.W.; Ercoli, L.M.; Silverman, D.H.S.; Huang, S.C.; Komo, S.; Bookheimer, S.Y.; Lavretsky, H.; Miller, K.; Siddarth, P.; Rasgon, N.L.; et al. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 6037–6042. [Google Scholar] [CrossRef] [Green Version]
- Toppala, S.; Ekblad, L.L.; Viitanen, M.; Rinne, J.O.; Jula, A. Oral glucose tolerance test predicts episodic memory decline: A 10-year population-based follow-up study. Diabetes Care 2021, 44, 2435–2437. [Google Scholar] [CrossRef]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculoneuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cisternas, P.; Oliva, C.A.; Torres, V.I.; Barrera, D.P.; Inestrosa, N.C. Presymptomatic treatment with andrographolide improves brain metabolic markers and cognitive behavior in a model of early-onset alzheimer’s disease. Front. Cell Neurosci. 2019, 13, 295. [Google Scholar] [CrossRef]
- Gejl, M.; Gjedde, A.; Egefjord, L.; Møller, A.; Hansen, S.B.; Vang, K.; Rodell, A.; Brændgaard, H.; Gottrup, H.; Schacht, A.; et al. In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: Randomized, placebo-controlled, double-blind clinical trial. Front. Aging Neurosci. 2016, 8, 108. [Google Scholar] [CrossRef]
- Gherardelli, C.; Cisternas, P.; Gutiérrez, J.; Martinez, M.; Inestrosa, N.C. Andrographolide restores glucose uptake in rat hippocampal neurons. J. Neurochem. 2020, 8, 108. [Google Scholar] [CrossRef]
- Gherardelli, C.; Cisternas, P.; Vera-Salazar, R.F.; Mendez-Orellana, C.; Inestrosa, N.C. Age- and Sex-Associated Glucose Metabolism Decline in a Mouse Model of Alzheimer’s Disease. J. Alzheimer Dis. 2022, 87, 901–917. [Google Scholar] [CrossRef]
- Shorter, E. The history of lithium therapy. Bipolar Disord. 2009, 11, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Plenge, P. Lithium effects on rat brain glucose metabolism in vivo. Effects after administration of lithium by various routes. Psychopharmacology 1982, 77, 348–355. [Google Scholar] [CrossRef]
- Plenge, P. Acute lithium effects on rat brain glucose metabolism in vivo. Int. Pharm. 1976, 11, 84–92. [Google Scholar] [CrossRef]
- Kohno, T.; Shiga, T.; Toyomaki, A.; Kusumi, I.; Matsuyama, T.; Inoue, T.; Katoh, C.; Koyama, T.; Tamaki, N. Effects of lithium on brain glucose metabolism in healthy men. J. Clin. Psychopharmacol. 2007, 27, 698–702. [Google Scholar] [CrossRef]
- Salimi, A.; Gholamifar, E.; Naserzadeh, P.; Hosseini, M.J.; Pourahmad, J. Toxicity of lithium on isolated heart mitochondria and cardiomyocyte: A justification for its cardiotoxic adverse effect. J. Biochem. Mol. Toxicol. 2017, 31, e21836. [Google Scholar] [CrossRef]
- Struewing, I.T.; Barnett, C.D.; Tang, T.; Mao, C.D. Lithium increases PGC-1α expression and mitochondrial biogenesis in primary bovine aortic endothelial cells. FEBS J. 2007, 274, 2749–2765. [Google Scholar] [CrossRef]
- Rizak, J.D. The Inhibition of ATP Production by Lithium: A Preliminary Study in Whole Mitochondria from Rat Brain and a Putative Model for Bipolar Disorder. Ann. Psychiatry Ment. Health 2014, 2, 1014. [Google Scholar]
- Dudev, T.; Grauffel, C.; Lim, C. How native and alien metal cations bind ATP: Implications for lithium as a therapeutic agent. Sci. Rep. 2017, 7, srep42377. [Google Scholar] [CrossRef]
- Briggs, K.T.; Giulian, G.G.; Li, G.; Kao, J.P.Y.; Marino, J.P. A Molecular Model for Lithium’s Bioactive Form. Biophys. J. 2016, 111, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Maurer, I.C.; Schippel, P.; Volz, H.P. Lithium-induced enhancement of mitochondrial oxidative phosphorylation in human brain tissue. Bipolar Disord. 2009, 11, 515–522. [Google Scholar]
- Osete, J.R.; Akkouh, I.A.; de Assis, D.R.; Szabo, A.; Frei, E.; Hughes, T.; Smeland, O.B.; Steen, N.E.; Andreassen, O.A.; Djurovic, S. Lithium increases mitochondrial respiration in iPSC-derived neural precursor cells from lithium responders. Mol. Psychiatry 2021, 26, 6789–6805. [Google Scholar] [CrossRef]
- Rydström, J. Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Haugaard, E.S.; Mickel, R.A.; Haugaard, N. Actions of lithium ions and insulin on glucose utilization, glycogen synthesis and glycogen synthase in the isolated rat diaphragm. Biochem. Pharm. 1974, 23, 1675–1685. [Google Scholar] [CrossRef]
- Cheng, K.; Creacy, S.; Larner, J. “Insulin-like” effects of lithium ion on isolated rat adipocytes I. Stimulation of glycogenesis beyond glucose transport. Mol. Cell Biochem. 1983, 56, 177–182. [Google Scholar] [CrossRef]
- Macko, A.R.; Beneze, A.N.; Teachey, M.K.; Henriksen, E.J. Roles of insulin signalling and p38 MAPK in the activation by lithium of glucose transport in insulin-resistant rat skeletal muscle. Arch. Physiol. Biochem. 2008, 114, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fürnsinn, C.; Noe, C.; Herdlicka, R.; Roden, M.; Nowotny, P.; Leighton, B.; Waldhäusl, W. More marked stimulation by lithium than insulin of the glycogenic pathway in rat skeletal muscle. Am. J. Physiol. 1997, 273, E514–E520. [Google Scholar] [CrossRef]
- Bai, S.; Pan, S.; Zhang, K.; Ding, X.; Wang, J.; Zeng, Q.; Xuan, Y.; Su, Z. Long-term effect of dietary overload lithium on the glucose metabolism in broiler chickens. Environ. Toxicol. Pharm. 2017, 54, 191–198. [Google Scholar] [CrossRef]
- Klein, P.S.; Melton, D.A. A molecular mechanism for the effect of lithium on development. Proc. Natl. Acad. Sci. USA 1996, 93, 8455–8459. [Google Scholar] [CrossRef] [Green Version]
- Bosch, F.; Gomez-Foix, A.M.; Arino, J.; Guinovart, J.J. Effects of lithium ions on glycogen synthase and phosphorylase in rat hepatocytes. Biol. Chem. 1986, 261, 16927–16931. [Google Scholar] [CrossRef]
- Summers, S.A.; Kao, A.W.; Kohn, A.D.; Backus, G.S.; Roth, R.A.; Pessin, J.E.; Birnbaum, M.J. The role of glycogen synthase kinase 3β in insulin-stimulated glucose metabolism. J. Biol. Chem. 1999, 274, 17934–17940. [Google Scholar] [CrossRef] [Green Version]
- Jope, R.S. Lithium and GSK-3: One inhibitor, two inhibitory actions, multiple outcomes. Trends Pharm. Sci. 2003, 24, 441–443. [Google Scholar] [CrossRef]
- Forlenza, O.V.; De-Paula, V.J.R.; Diniz, B.S.O. Neuroprotective effects of lithium: Implications for the treatment of Alzheimer’s disease and related neurodegenerative disorders. ACS Chem. Neurosci. 2014, 5, 443–450. [Google Scholar] [CrossRef] [Green Version]
- Chuang, D.-M.; Wang, Z.; Chiu, C.-T. GSK-3 as a Target for Lithium-Induced Neuroprotection Against Excitotoxicity in Neuronal Cultures and Animal Models of Ischemic Stroke. Mol. Neurosci. 2011, 4, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valvezan, A.J.; Klein, P.S. GSK-3 and Wnt signaling in neurogenesis and bipolar disorder. Front. Mol. Neurosci. 2012, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoli, D.; Pollak, M.; Topisirovic, I. The role of GSK3 in metabolic pathway perturbations in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119059. [Google Scholar] [CrossRef]
- Suzuki, T.; Bridges, D.; Nakada, D.; Skiniotis, G.; Morrison, S.J.; Lin, J.; Saltiel, A.R.; Inoki, K. Inhibition of AMPK catabolic action by GSK3. Mol. Cell 2013, 50, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Pan, W. GSK3: A multifaceted kinase in Wnt signaling. Trends Biochem. Sci. 2010, 35, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ríos, J.A.; Godoy, J.A.; Inestrosa, N.C. Wnt3a ligand facilitates autophagy in hippocampal neurons by modulating a novel GSK-3β-AMPK axis. Cell Commun. Signal. 2018, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shan, J.; Chang, W.; Kim, I.; Bao, J.; Lee, H.J.; Zhang, X.; Samuel, V.T.; Shulman, G.I.; Liu, D.; et al. Chemical and genetic evidence for the involvement of Wnt antagonist Dickkopf2 in regulation of glucose metabolism. Proc. Natl. Acad. Sci. USA 2012, 109, 11402–11407. [Google Scholar] [CrossRef] [Green Version]
- Daneman, R.; Agalliu, D.; Zhou, L.; Kuhnert, F.; Kuo, C.J.; Barres, B.A. Wnt/β-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 641–646. [Google Scholar] [CrossRef] [Green Version]
- Crippa, S.; Ancey, P.; Vazquez, J.; Angelino, P.; Rougemont, A.; Guettier, C.; Zoete, V.; Delorenzi, M.; Michielin, O.; Meylan, E. Mutant CTNNB 1 and histological heterogeneity define metabolic subtypes of hepatoblastoma. EMBO Mol. Med. 2017, 9, 1589–1604. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Toledo, E.M. The role of Wnt signaling in neuronal dysfunction in Alzheimer’s Disease. Mol. Neurodegener. 2008, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Ewers, M.; Bürger, K.; Annas, P.; Mörtberg, A.; Bogstedt, A.; Frölich, L.; Schröder, J.; Schönknecht, P.; Riepe, M.W.; et al. Lithium trial in Alzheimer’s disease: A randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 2009, 70, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Fjell, A.M.; Walhovd, K.B.; Fennema-Notestine, C.; McEvoy, L.K.; Hagler, D.J.; Holland, D.; Brewer, J.B.; Dale, A.M. CSF biomarkers in prediction of cerebral and clinical change in mild cognitive impairment and Alzheimer’s disease. J. Neurosci. 2010, 30, 2088–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forlenza, O.V.; Diniz, B.S.; Radanovic, M.; Santos, F.S.; Talib, L.L.; Gattaz, W.F. Disease-modifying properties of long-term lithium treatment for amnestic mild cognitive impairment: Randomised controlled trial. Br. J. Psychiatry 2011, 198, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Montaño, J.R.; Moreno, F.J.; Avila, J.; Díaz-Nido, J. Lithium inhibits Alzheimer’s disease-like tau protein phosphorylation in neurons. FEBS Lett. 1997, 411, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Motoi, Y.; Ishiguro, K.; Kambe, T.; Matsumoto, S.E.; Itaya, M.; Kunichika, M.; Mori, H.; Shinohara, A.; Chiba, M.; et al. Long-term oral lithium treatment attenuates motor disturbance in tauopathy model mice: Implications of autophagy promotion. Neurobiol. Dis. 2012, 46, 101–108. [Google Scholar] [CrossRef]
- Nakashima, H.; Ishihara, T.; Suguimoto, P.; Yokota, O.; Oshima, E.; Kugo, A.; Terada, S.; Hamamura, T.; Trojanowski, J.Q.; Lee, V.M.Y.; et al. Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol. 2005, 110, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Drummond, E.; Wisniewski, T. Alzheimer’s Disease: Experimental Models and Reality. Acta Neuropathol. 2018, 133, 155–175. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2016, 18, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanzi, R.E. The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef]
- Cisternas, P.; Silva-Alvarez, C.; Martínez, F.; Fernandez, E.; Ferrada, L.; Oyarce, K.; Salazar, K.; Bolaños, J.P.; Nualart, F. The oxidized form of vitamin C, dehydroascorbic acid, regulates neuronal energy metabolism. J. Neurochem. 2014, 129, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef]
- Cerpa, W.; Farias, G.G.; Godoy, J.A.; Fuenzalida, M.; Bonansco, C.; Inestrosa, N.C. Wnt-5a occludes Abeta oligomer-induced depression of glutamatergic transmission in hippocampal neurons. Mol. Neurodegener. 2010, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Cisternas, P.; Salazar, P.; Silva-álvarez, C.; Barros, L.F. Activation of Wnt signaling in cortical neurons enhances glucose utilization through glycolysis. J. Biol. Chem. 2016, 291, 25950–25964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, M.; Torres, V.I.; Vio, C.P.; Inestrosa, N.C. Canonical Wnt Signaling Modulates the Expression of Pre- and Postsynaptic Components in Different Temporal Patterns. Mol. Neurobiol. 2020, 57, 1389–1404. [Google Scholar] [CrossRef]
- Sun, S.; Hu, F.; Wu, J.; Zhang, S. Cannabidiol attenuates OGD/R-induced damage by enhancing mitochondrial bioenergetics and modulating glucose metabolism via pentose-phosphate pathway in hippocampal neurons. Redox Biol. 2017, 11, 577–585. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Inestrosa, N.C. Wnt signaling loss accelerates the appearance of neuropathological hallmarks of Alzheimer’s disease in J20-APP transgenic and wild-type mice. J. Neurochem. 2018, 144, 443–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cisternas, P.; Gherardelli, C.; Salazar, P.; Inestrosa, N.C. Disruption of Glucose Metabolism in Aged Octodon degus: A Sporadic Model of Alzheimer’s Disease. Front. Integr. Neurosci. 2021, 15, 733007. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Rojas, C.; Schüller, A.; Lindsay, C.B.; Ureta, R.C.; Mejias-reyes, C.; Hancke, J.; Melo, F.; Inestrosa, N.C. Andrographolide activates the canonical Wnt signalling pathway by a mechanism that implicates the non-ATP competitive inhibition of GSK-3β: Autoregulation of GSK-3β in vivo. Biochem. J. 2015, 466, 415–430. [Google Scholar] [CrossRef]
- Bolaños, J.P.; Delgado-Esteban, M.; Herrero-Mendez, A.; Fernandez-Fernandez, S.; Almeida, A. Regulation of glycolysis and pentose-phosphate pathway by nitric oxide: Impact on neuronal survival. Biochim. Biophys. Acta 2008, 1777, 789–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.S.; Chen, Q. Purification and kinetic characterization of 6-phosphogluconate dehydrogenase from Schizosaccharomyces pombe. Biochem. Cell Biol. 1998, 76, 107–113. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gherardelli, C.; Cisternas, P.; Inestrosa, N.C. Lithium Enhances Hippocampal Glucose Metabolism in an In Vitro Mice Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 8733. https://doi.org/10.3390/ijms23158733
Gherardelli C, Cisternas P, Inestrosa NC. Lithium Enhances Hippocampal Glucose Metabolism in an In Vitro Mice Model of Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(15):8733. https://doi.org/10.3390/ijms23158733
Chicago/Turabian StyleGherardelli, Camila, Pedro Cisternas, and Nibaldo C. Inestrosa. 2022. "Lithium Enhances Hippocampal Glucose Metabolism in an In Vitro Mice Model of Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 15: 8733. https://doi.org/10.3390/ijms23158733
APA StyleGherardelli, C., Cisternas, P., & Inestrosa, N. C. (2022). Lithium Enhances Hippocampal Glucose Metabolism in an In Vitro Mice Model of Alzheimer’s Disease. International Journal of Molecular Sciences, 23(15), 8733. https://doi.org/10.3390/ijms23158733