
Molecular Dynamics of CYFIP2 Protein and Its R87C Variant Related to Early Infantile Epileptic Encephalopathy

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
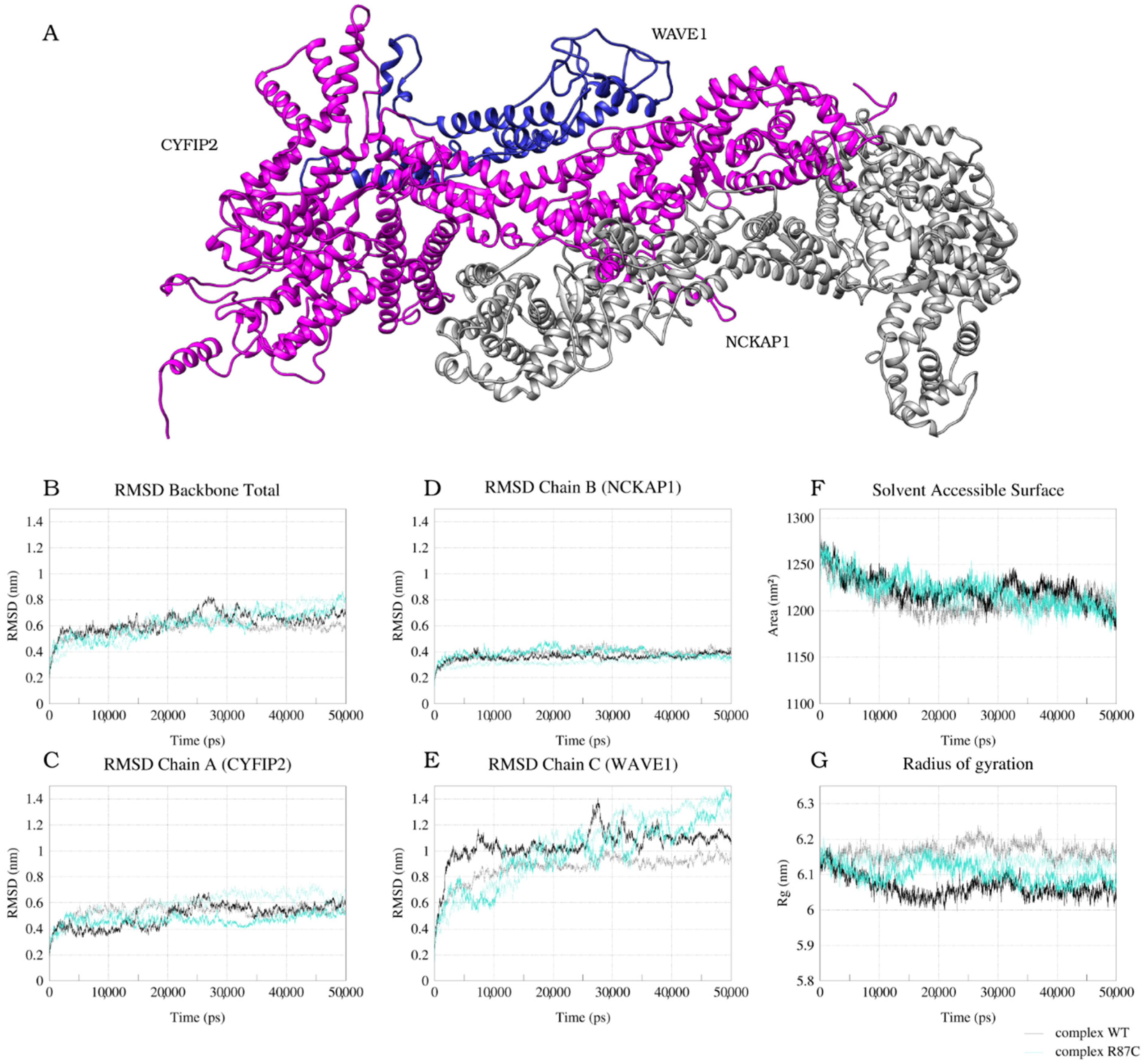
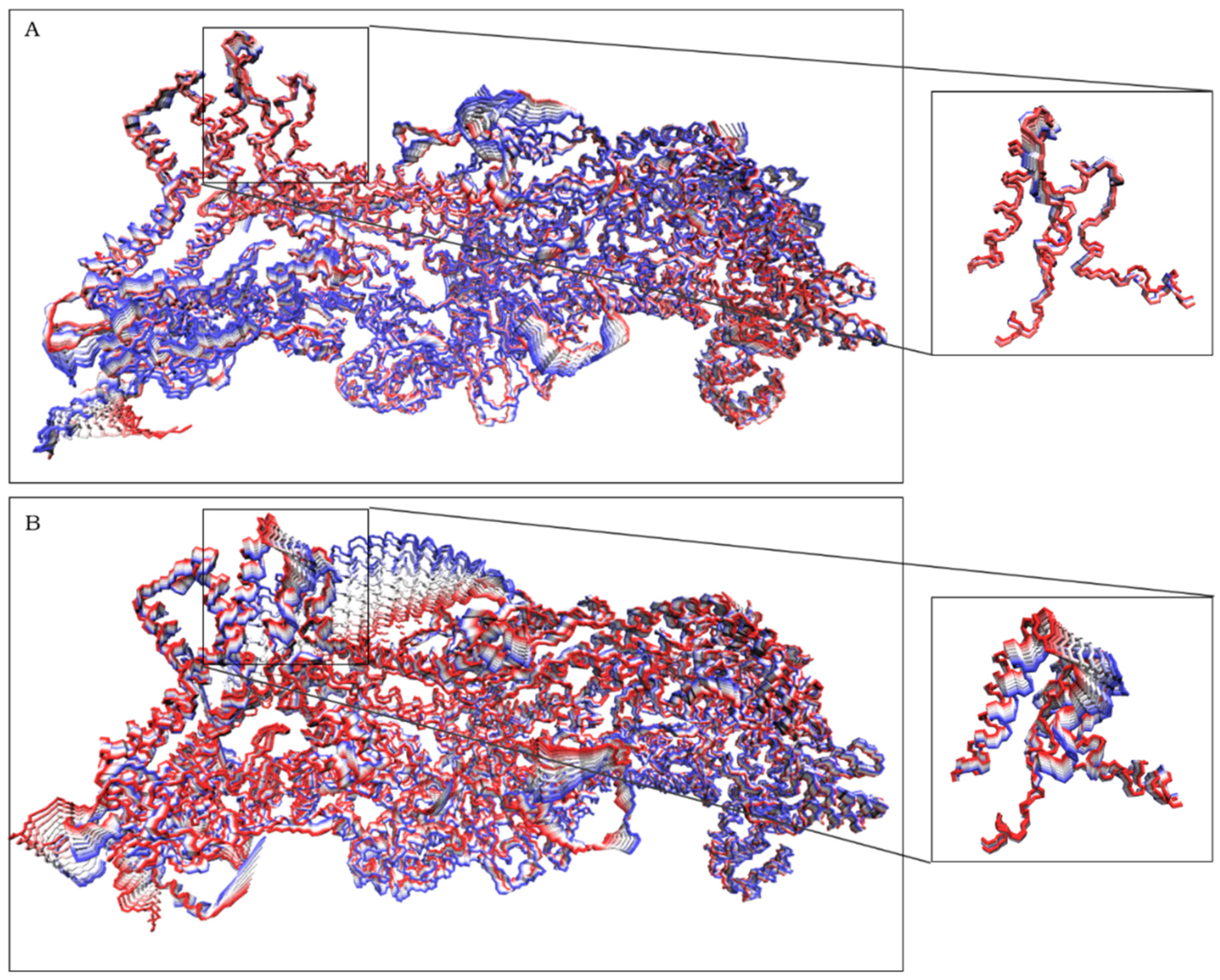
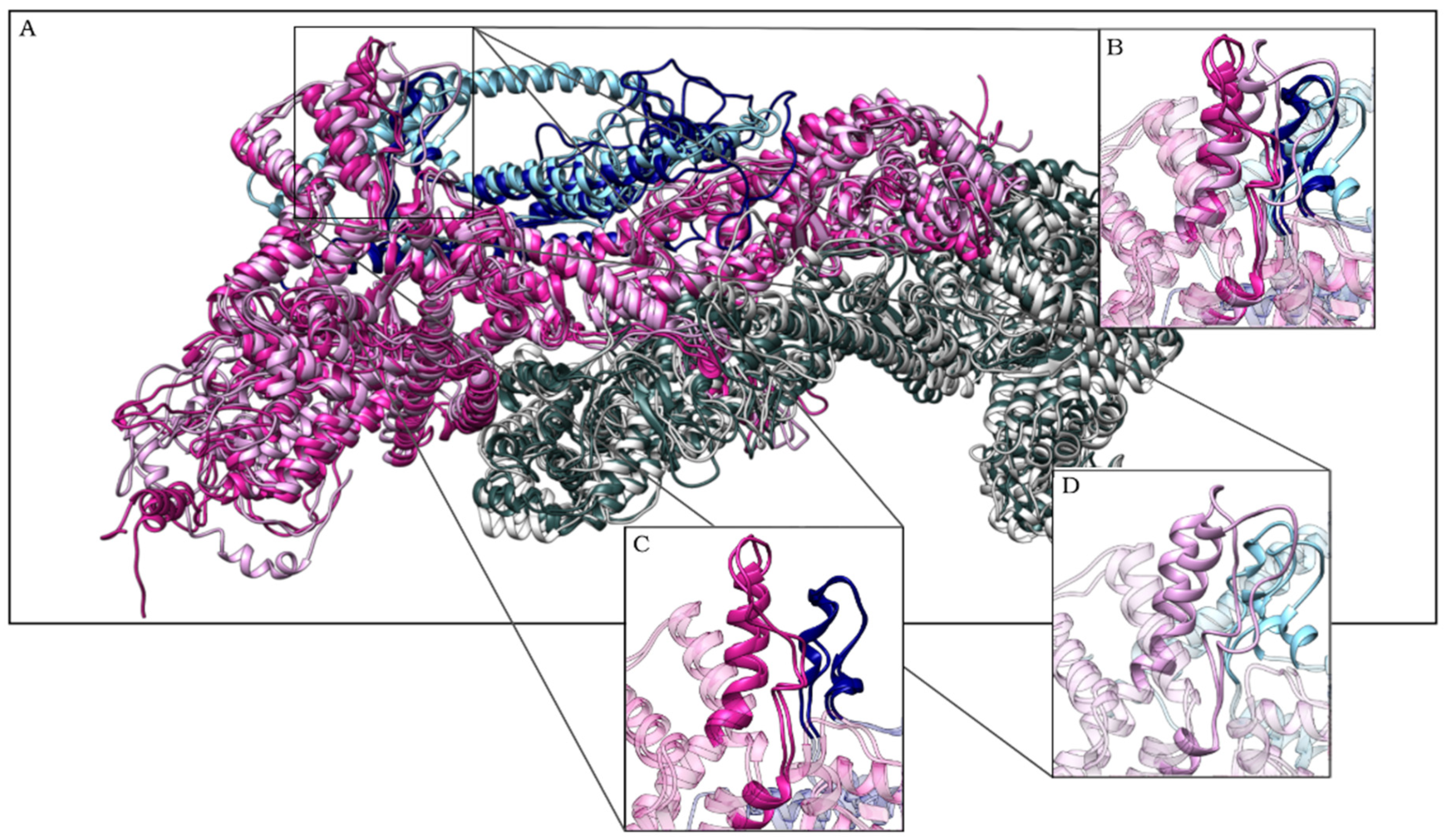
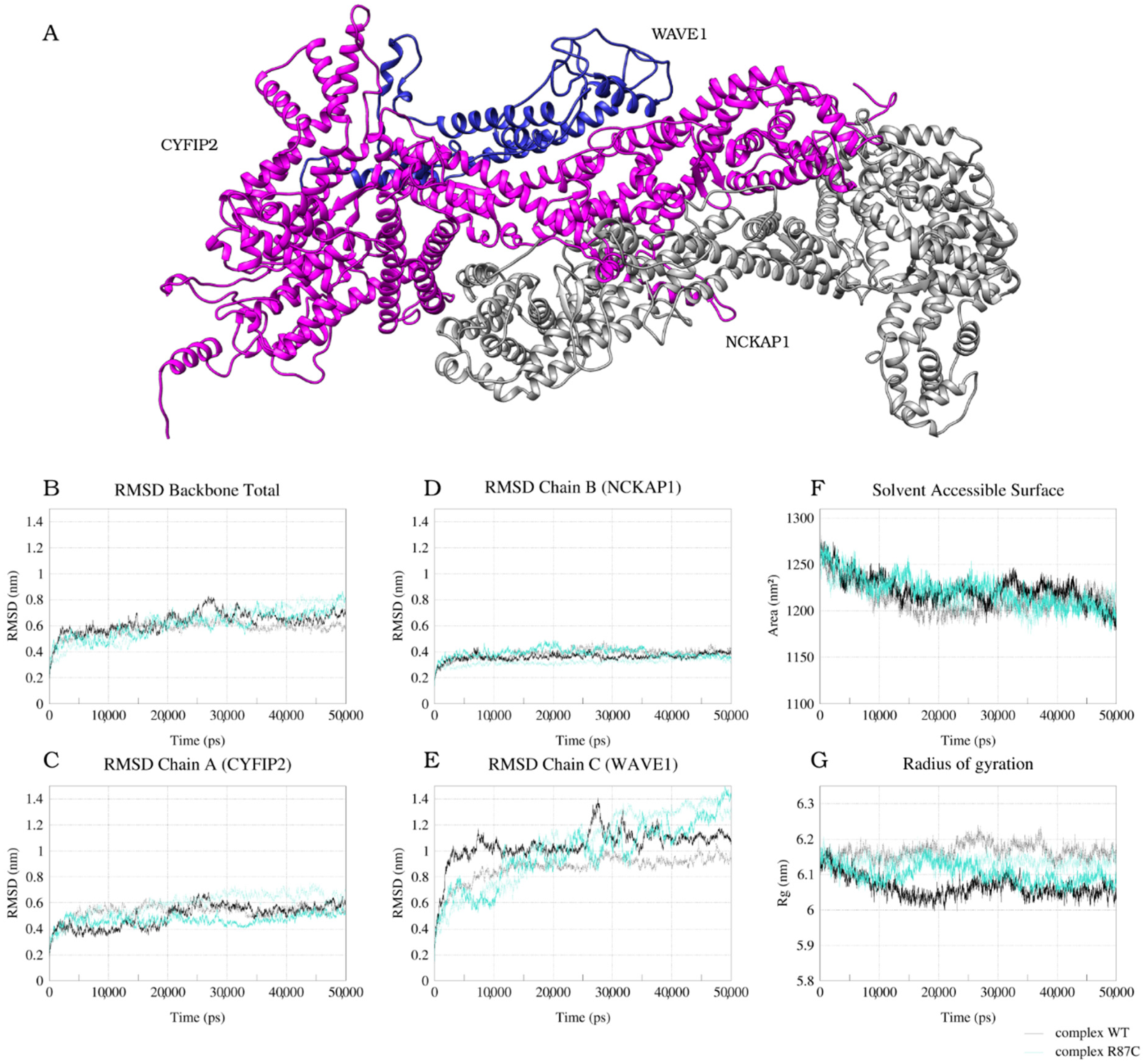
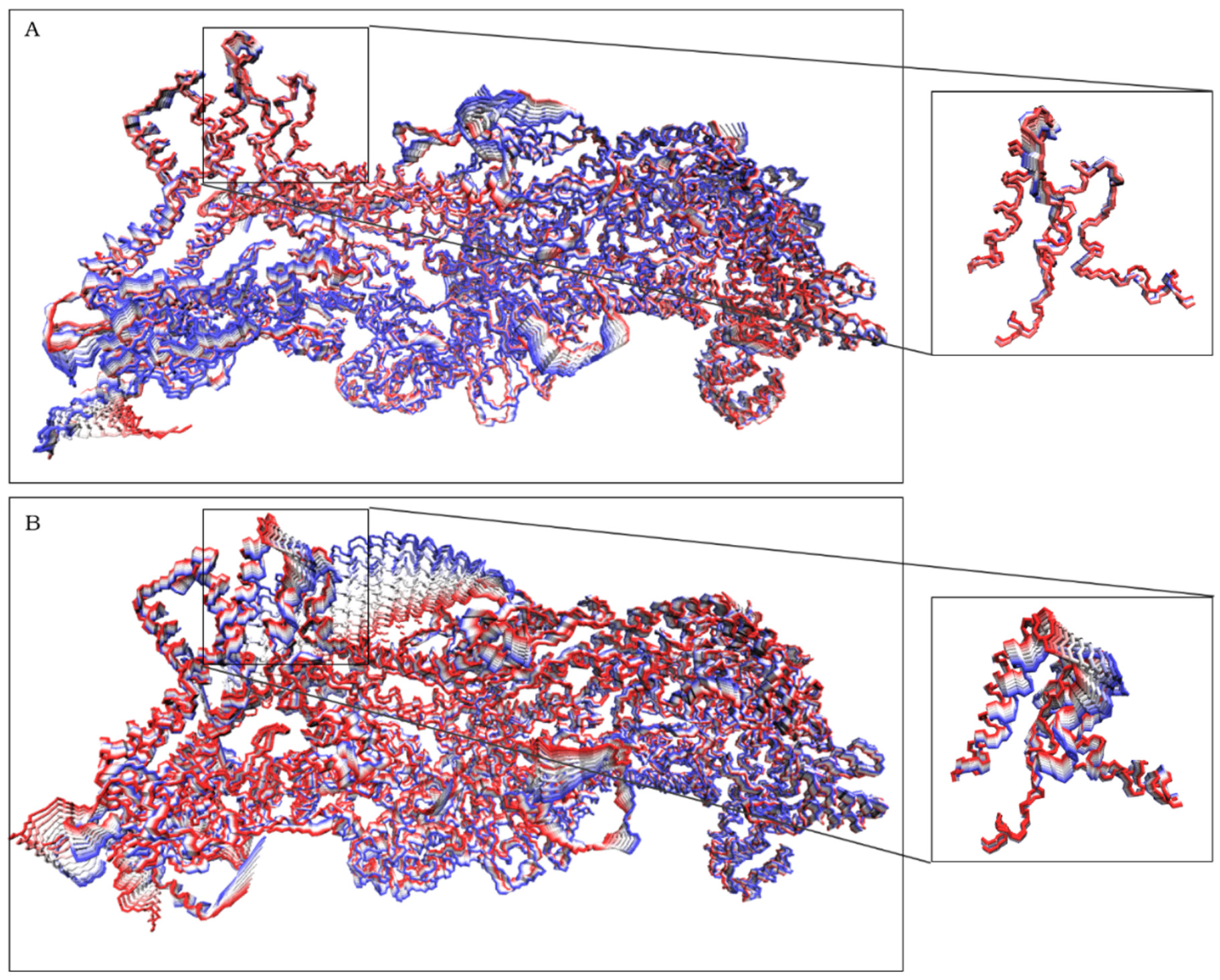
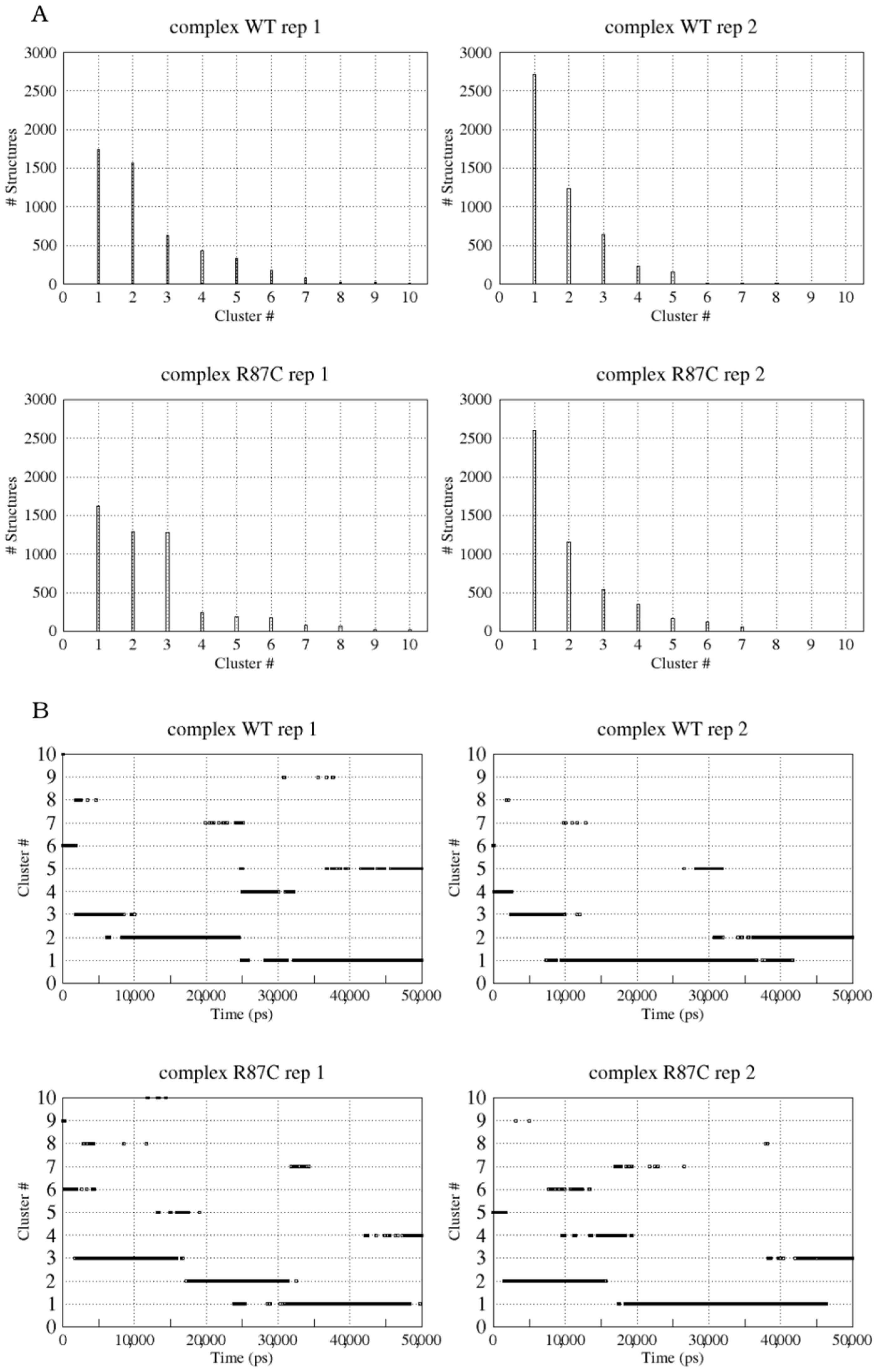
2.1. CYFIP2 WT and CYFIP2 R87C Evolution through the Simulations
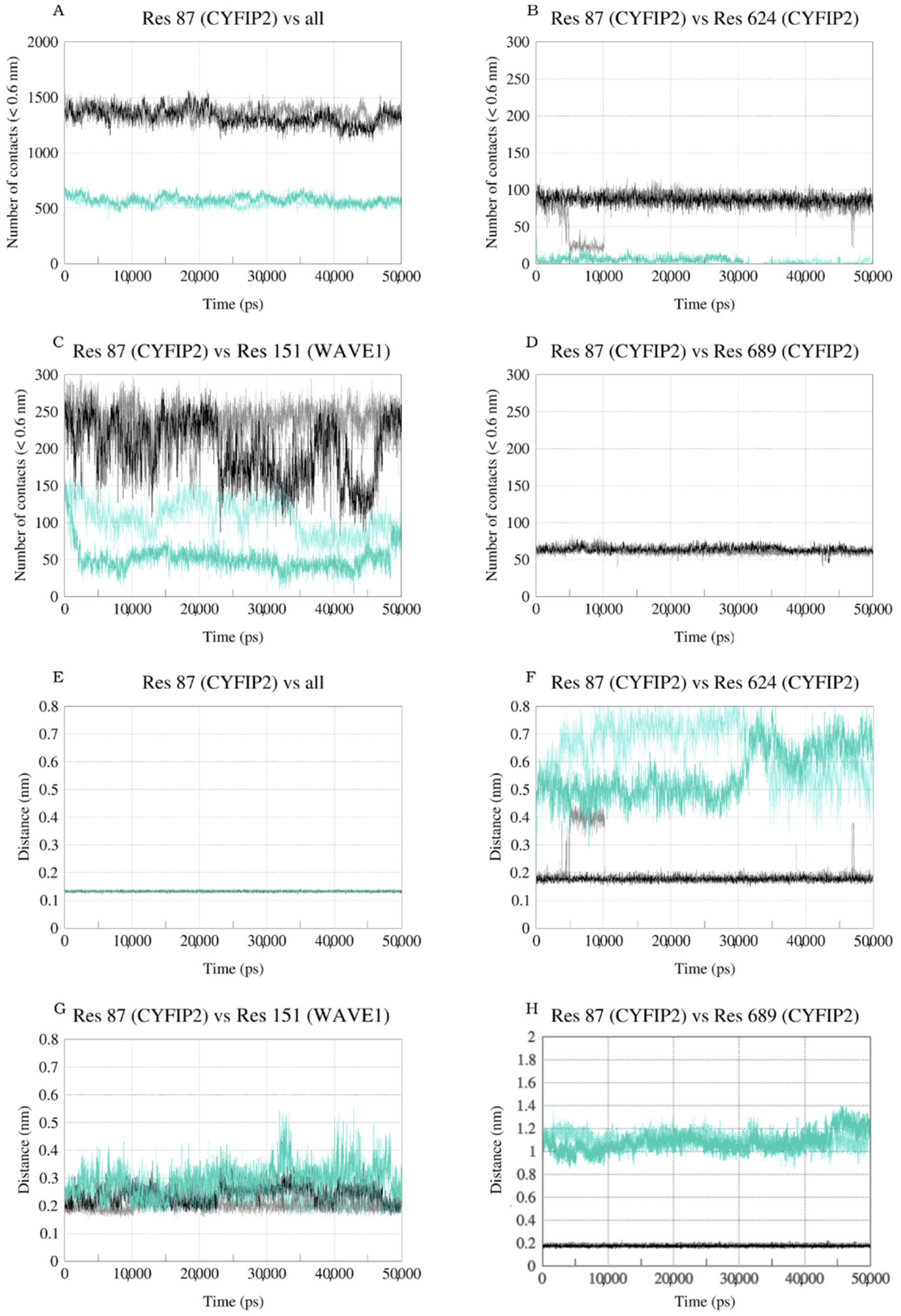
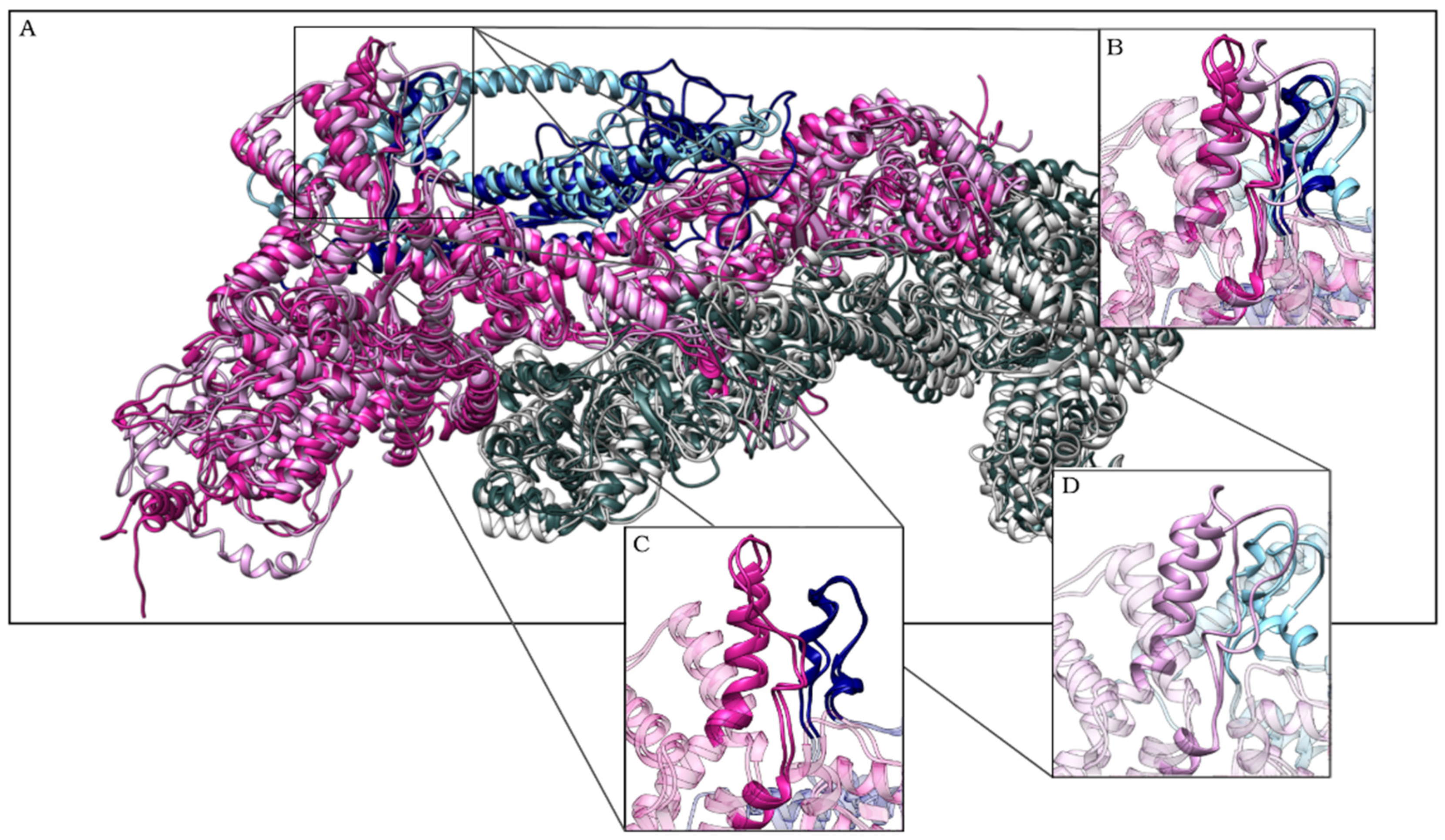
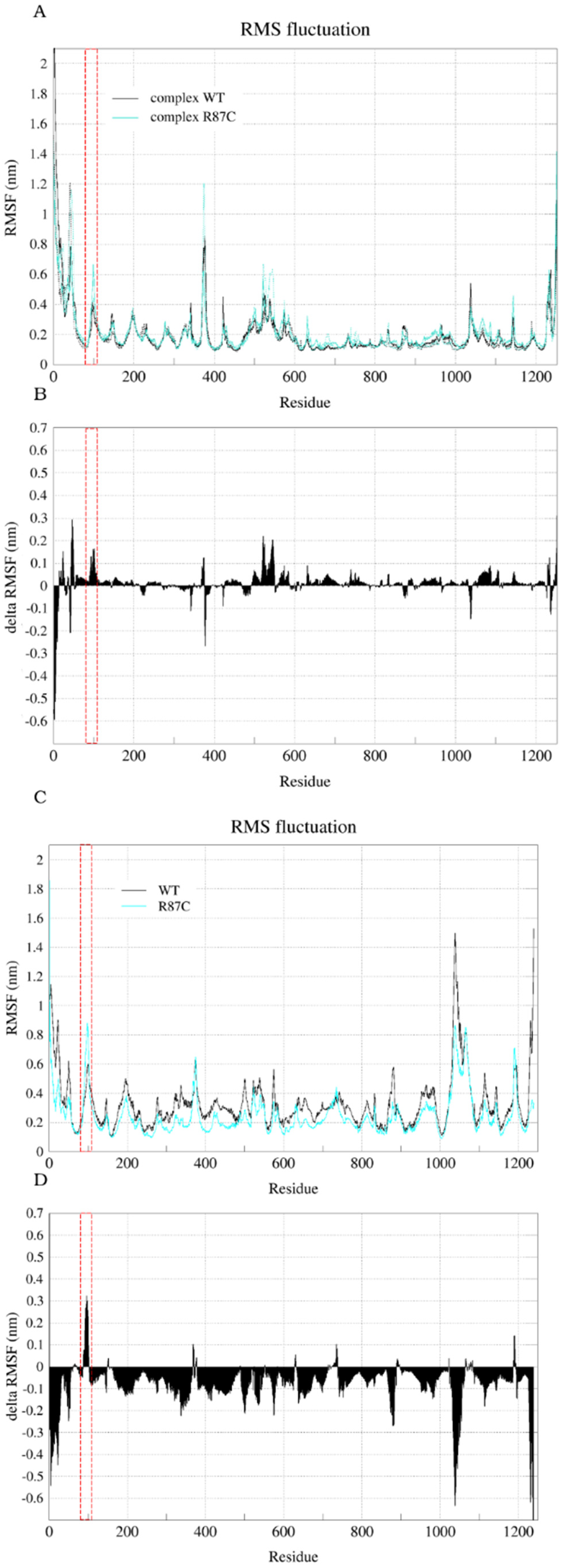
2.2. The Simulation Showed A Flexibilization of the Loop (80–110) in Complex R87C
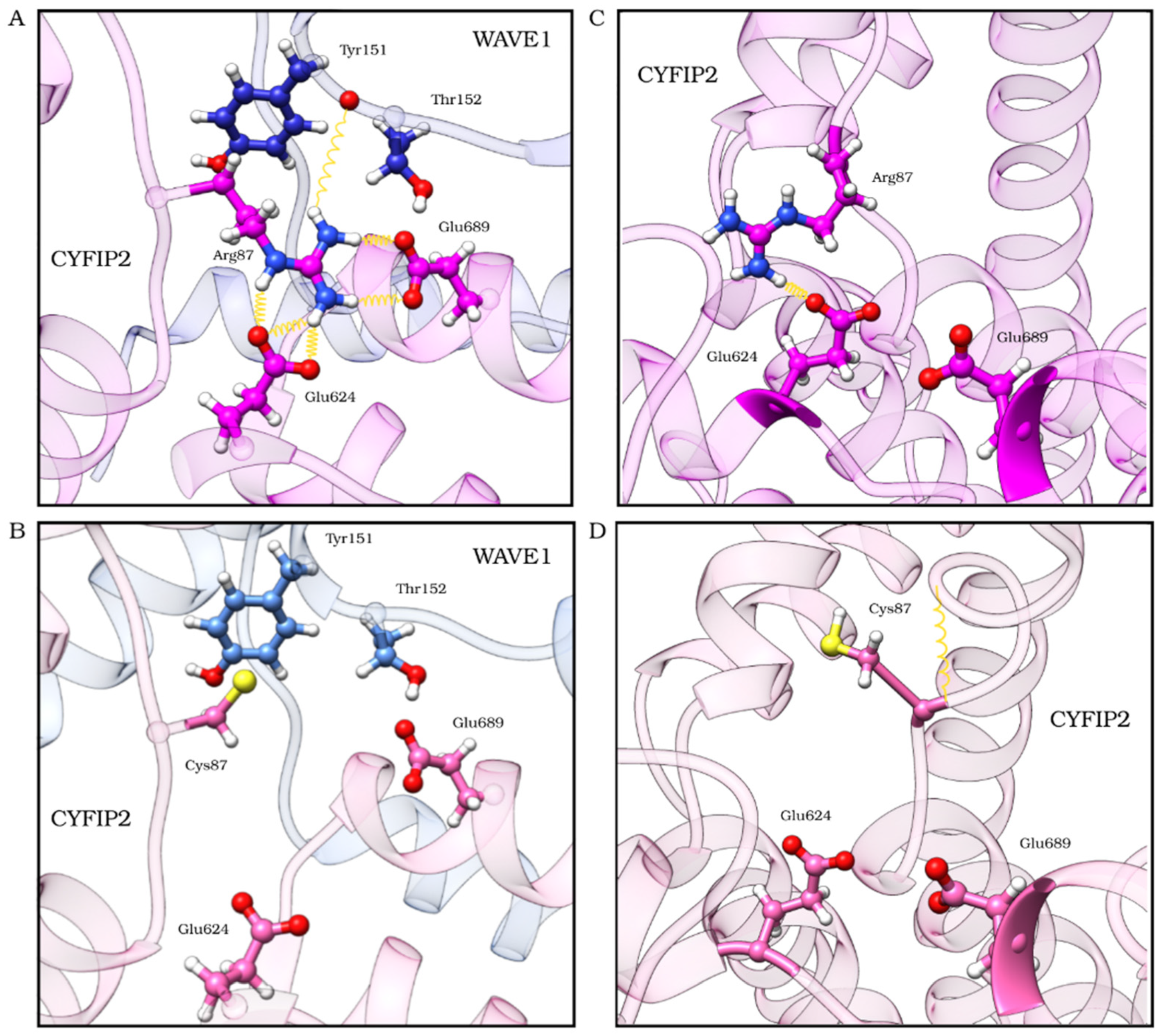
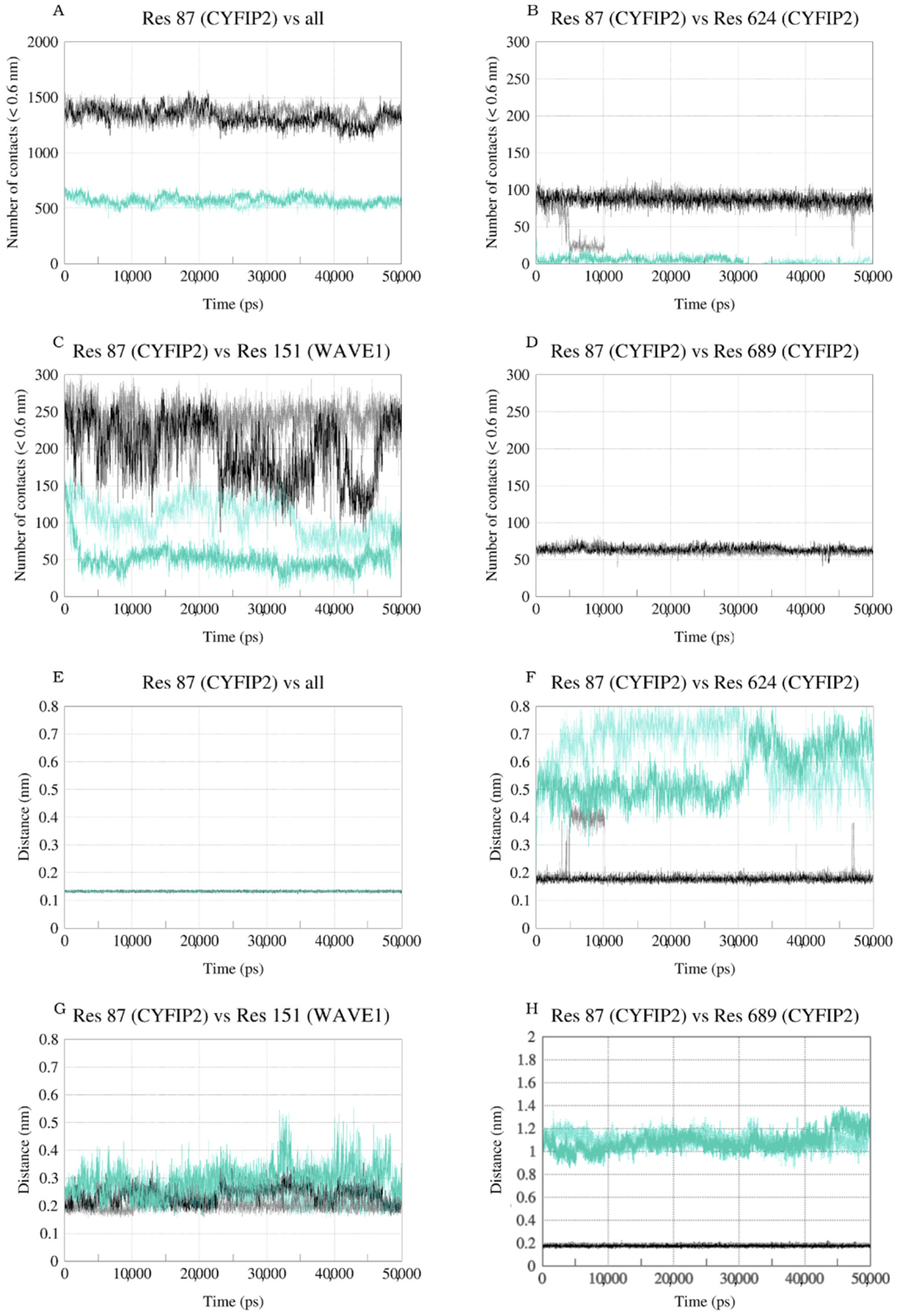
2.3. The R87C Mutation Underlies A Flexibilization of Its Loop Inside CYFIP2 Caused by the Loss of Important Interactions
3. Discussion
4. Materials and Methods
4.1. Tridimensional Models
4.2. Molecular Dynamics of CYFIP2 WT and CYFIP2 R87C
4.3. Molecular Dynamics of the WRC Complex
4.4. Molecular Dynamics Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engel, J. A Proposed Diagnostic Scheme for People with Epileptic Seizures and with Epilepsy: Report of the ILAE Task Force on Classification and Terminology. Epilepsia 2001, 42, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Al Baradie, R. Epileptic Encephalopathies: An Overview. Epilepsy Res. Treat. 2012, 2012, 403592. [Google Scholar] [CrossRef] [PubMed]
- Lux, A.L. Latest American and European Updates on Infantile Spasms. Curr. Neurol. Neurosci. Rep. 2013, 13, 334. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.L.; Chen, S.; Sivarajah, V.; Stephens, D.; Cortez, M.A. Latitudinal Differences on the Global Epidemiology of Infantile Spasms: Systematic Review and Meta-Analysis. Orphanet J. Rare Dis. 2018, 13, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavone, P.; Polizzi, A.; Marino, S.D.; Corsello, G.; Falsaperla, R.; Marino, S.; Ruggieri, M. West Syndrome: A Comprehensive Review. Neurol. Sci. 2020, 41, 3547–3562. [Google Scholar] [CrossRef]
- Magalhães, P.H.M.; Moraes, H.T.; Athie, M.C.P.; Secolin, R.; Lopes-Cendes, I. New Avenues in Molecular Genetics for the Diagnosis and Application of Therapeutics to the Epilepsies. Epilepsy Behav. 2019, 121, 106428. [Google Scholar] [CrossRef]
- Nakashima, M.; Kato, M.; Aoto, K.; Shiina, M.; Belal, H.; Mukaida, S.; Kumada, S.; Sato, A.; Zerem, A.; Lerman-Sagie, T.; et al. De Novo Hotspot Variants in CYFIP2 Cause Early-Onset Epileptic Encephalopathy. Ann. Neurol. 2018, 83, 794–806. [Google Scholar] [CrossRef]
- Lebensohn, A.M.; Kirschner, M.W. Activation of the WAVE Complex by Coincident Signals Controls Actin Assembly. Mol. Cell 2009, 36, 512–524. [Google Scholar] [CrossRef] [Green Version]
- Innocenti, M.; Zucconi, A.; Disanza, A.; Frittoli, E.; Areces, L.B.; Steffen, A.; Stradal, T.E.B.; Di Fiore, P.P.; Carlier, M.F.; Scita, G. Abi1 Is Essential for the Formation and Activation of a WAVE2 Signalling Complex. Nat. Cell Biol. 2004, 6, 319–327. [Google Scholar] [CrossRef]
- Eden, S.; Rohatgi, R.; Podtelejnikov, A.V.; Mann, M.; Kirschner, M.W. Mechanism of Regulation of WAVE1-Induced Actin Nucleation by Rac1 and Nck. Nature 2002, 418, 790–793. [Google Scholar] [CrossRef]
- Gautreau, A.; Ho, H.Y.H.; Li, J.; Steen, H.; Gygi, S.P.; Kirschner, M.W. Purification and Architecture of the Ubiquitous Wave Complex. Proc. Natl. Acad. Sci. USA 2004, 101, 4379–4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derivery, E.; Lombard, B.; Loew, D.; Gautreau, A. The Wave Complex Is Intrinsically Inactive. Cell Motil. Cytoskelet. 2009, 66, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Borek, D.; Padrick, S.B.; Gomez, T.S.; Metlagel, Z.; Ismail, A.M.; Umetani, J.; Billadeau, D.D.; Otwinowski, Z.; Rosen, M.K. Structure and Control of the Actin Regulatory WAVE Complex. Nature 2010, 468, 533–538. [Google Scholar] [CrossRef]
- Begemann, A.; Sticht, H.; Begtrup, A.; Vitobello, A.; Faivre, L.; Banka, S.; Alhaddad, B.; Asadollahi, R.; Becker, J.; Bierhals, T.; et al. New Insights into the Clinical and Molecular Spectrum of the Novel CYFIP2-Related Neurodevelopmental Disorder and Impairment of the WRC-Mediated Actin Dynamics. Genet. Med. 2020, 23, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Wang, Y.; He, F.; Chen, C.; Wu, L.W.; Yang, L.F.; Ma, Y.P.; Zhang, W.; Shi, Z.Q.; Chen, C.; et al. Novel West Syndrome Candidate Genes in a Chinese Cohort. CNS Neurosci. Ther. 2018, 24, 1196–1206. [Google Scholar] [CrossRef]
- Zhong, M.; Liao, S.; Li, T.; Wu, P.; Wang, Y.; Wu, F.; Li, X.; Hong, S.; Yan, L.; Jiang, L. Early Diagnosis Improving the Outcome of an Infant with Epileptic Encephalopathy with Cytoplasmic FMRP Interacting Protein 2 Mutation: Case Report and Literature Review. Medicine (Baltimore) 2019, 98, e17749. [Google Scholar] [CrossRef] [PubMed]
- Zweier, M.; Begemann, A.; McWalter, K.; Cho, M.T.; Abela, L.; Banka, S.; Behring, B.; Berger, A.; Brown, C.W.; Carneiro, M.; et al. Spatially Clustering de Novo Variants in CYFIP2, Encoding the Cytoplasmic FMRP Interacting Protein 2, Cause Intellectual Disability and Seizures. Eur. J. Hum. Genet. 2018, 27, 747–759. [Google Scholar] [CrossRef] [Green Version]
- Schaks, M.; Reinke, M.; Witke, W.; Rottner, K. Molecular Dissection of Neurodevelopmental Disorder-Causing Mutations in CYFIP2. Cells 2020, 9, 1355. [Google Scholar] [CrossRef]
- Zhang, Y.; Lee, Y.; Han, K. Neuronal Function and Dysfunction of CYFIP2: From Actin Dynamics to Early Infantile Epileptic Encephalopathy. BMB Rep. 2019, 52, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Zhang, Y.; Kang, H.; Bang, G.; Kim, Y.; Kang, H.R.; Ma, R.; Jin, C.; Kim, J.Y.; Han, K. Epilepsy- and Intellectual Disability-Associated CYFIP2 Interacts with Both Actin Regulators and RNA-Binding Proteins in the Neonatal Mouse Forebrain. Biochem. Biophys. Res. Commun. 2020, 529, 1–6. [Google Scholar] [CrossRef]
- Vidal, R.; Ma, Y.; Sastry, S.S. Principal Component Analysis. In Generalized Principal Component Analysis; Interdisciplinary Applied Mathematics; Springer: New York, NY, USA, 2016; Volume 40. [Google Scholar] [CrossRef]
- Jäntschi, L. The Eigenproblem Translated for Alignment of Molecules. Symmetry 2019, 11, 1027. [Google Scholar] [CrossRef] [Green Version]
- Ding, B.; Yang, S.; Schaks, M.; Liu, Y.; Brown, A.; Rottner, K.; Chowdhury, S.; Chen, B. Structures Reveal a Key Mechanism of WAVE Regulatory Complex Activation by Rac1 GTPase. BioRxiv 2022. [Google Scholar] [CrossRef]
- Schaks, M.; Singh, S.P.; Kage, F.; Thomason, P.; Klünemann, T.; Steffen, A.; Blankenfeldt, W.; Stradal, T.E.; Insall, R.H.; Rottner, K. Distinct Interaction Sites of Rac GTPase with WAVE Regulatory Complex Have Non-Redundant Functions in Vivo. Curr. Biol. 2018, 28, 3674–3684.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, M.J.; Schlessman, J.L.; Sue, G.R.; Bertrand García-Moreno, E. Arginine Residues at Internal Positions in a Protein Are Always Charged. Proc. Natl. Acad. Sci. USA 2011, 108, 18954–18959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saller, E.; Tom, E.; Brunori, M.; Otter, M.; Estreicher, A.; Mack, D.H.; Iggo, R. Increased Apoptosis Induction by 121F Mutant P53. EMBO J. 1999, 18, 4424–4437. [Google Scholar] [CrossRef] [Green Version]
- Schenck, A.; Bardoni, B.; Moro, A.; Bagni, C.; Mandel, J.L. A Highly Conserved Protein Family Interacting with the Fragile X Mental Retardation Protein (FMRP) and Displaying Selective Interactions with FMRP-Related Proteins FXR1P and FXR2P. Proc. Natl. Acad. Sci. USA 2001, 98, 8844–8849. [Google Scholar] [CrossRef] [Green Version]
- Di Marino, D.; Chillemi, G.; De Rubeis, S.; Tramontano, A.; Achsel, T.; Bagni, C. MD and Docking Studies Reveal That the Functional Switch of CYFIP1 Is Mediated by a Butterfly-like Motion Supplementary. J. Chem. Theory Comput. 2015, 11, 3401–3410. [Google Scholar] [CrossRef]
- Lee, Y.; Zhang, Y.; Ryu, J.R.; Kang, H.R.; Kim, D.; Jin, C.; Kim, Y.; Sun, W.; Han, K. Reduced CYFIP2 Stability by Arg87 Variants Causing Human Neurological Disorders. Ann. Neurol. 2019, 86, 803–805. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at Its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.; Sali, A. Statistical Potential for Assessment and Prediction of Protein Structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and Better Reference Data for Improved All-Atom Structure Validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating PK Prediction and the Preparation of Biomolecular Structures for Atomistic Molecular Modeling and Simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Haug, E.J.; Arora, J.S.; Matsui, K. A Steepest-Descent Method for Optimization of Mechanical Systems. J. Optim. Theory Appl. 1976, 19, 401–424. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; van Gunsteren, W.F.; Mark, A.E. Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Original Residue Numbering | Residue Numbering in Models |
|---|---|---|
| CYFIP2 | 1–1252 | 1–1252 |
| NCKAP1 | 1–1127 | 1253–2379 |
| WAVE1 | WAVE1(1–186)-(GlyGlySer)6-(485–559) | WAVE1(2380–2544)-(GlyGlySer)6-(2563–2622) |
| System | Composition (Residues/Molecules) | ||||||
|---|---|---|---|---|---|---|---|
| Chain A (CYFIP2) | Chain B (NCKAP1) | Chain C (WAVE1) | Na+ | Cl− | Water | Total | |
| Complex WT | 1252 | 1127 | 242 | 21 | - | 398,728 | 401,371 |
| Complex R87C | 1252 | 1127 | 242 | 22 | - | 372,980 | 375,624 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biembengut, Í.V.; Shigunov, P.; Frota, N.F.; Lourenzoni, M.R.; de Souza, T.A.C.B. Molecular Dynamics of CYFIP2 Protein and Its R87C Variant Related to Early Infantile Epileptic Encephalopathy. Int. J. Mol. Sci. 2022, 23, 8708. https://doi.org/10.3390/ijms23158708
Biembengut ÍV, Shigunov P, Frota NF, Lourenzoni MR, de Souza TACB. Molecular Dynamics of CYFIP2 Protein and Its R87C Variant Related to Early Infantile Epileptic Encephalopathy. International Journal of Molecular Sciences. 2022; 23(15):8708. https://doi.org/10.3390/ijms23158708
Chicago/Turabian StyleBiembengut, Ísis V., Patrícia Shigunov, Natalia F. Frota, Marcos R. Lourenzoni, and Tatiana A. C. B. de Souza. 2022. "Molecular Dynamics of CYFIP2 Protein and Its R87C Variant Related to Early Infantile Epileptic Encephalopathy" International Journal of Molecular Sciences 23, no. 15: 8708. https://doi.org/10.3390/ijms23158708
APA StyleBiembengut, Í. V., Shigunov, P., Frota, N. F., Lourenzoni, M. R., & de Souza, T. A. C. B. (2022). Molecular Dynamics of CYFIP2 Protein and Its R87C Variant Related to Early Infantile Epileptic Encephalopathy. International Journal of Molecular Sciences, 23(15), 8708. https://doi.org/10.3390/ijms23158708