
Gene Variants Related to Cardiovascular and Pulmonary Diseases May Correlate with Severe Outcome of COVID-19

 , , , , ,
, , , , ,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
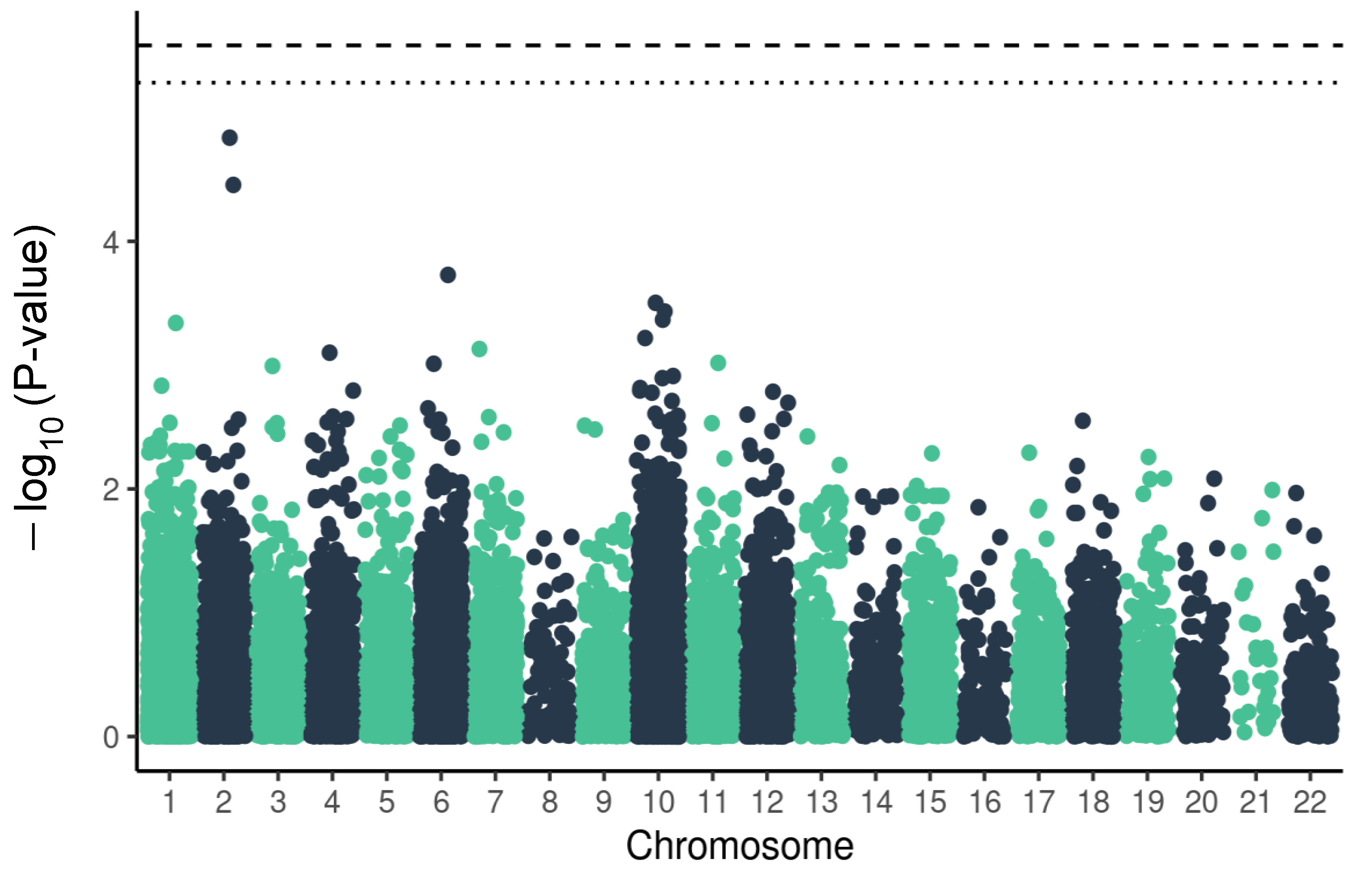
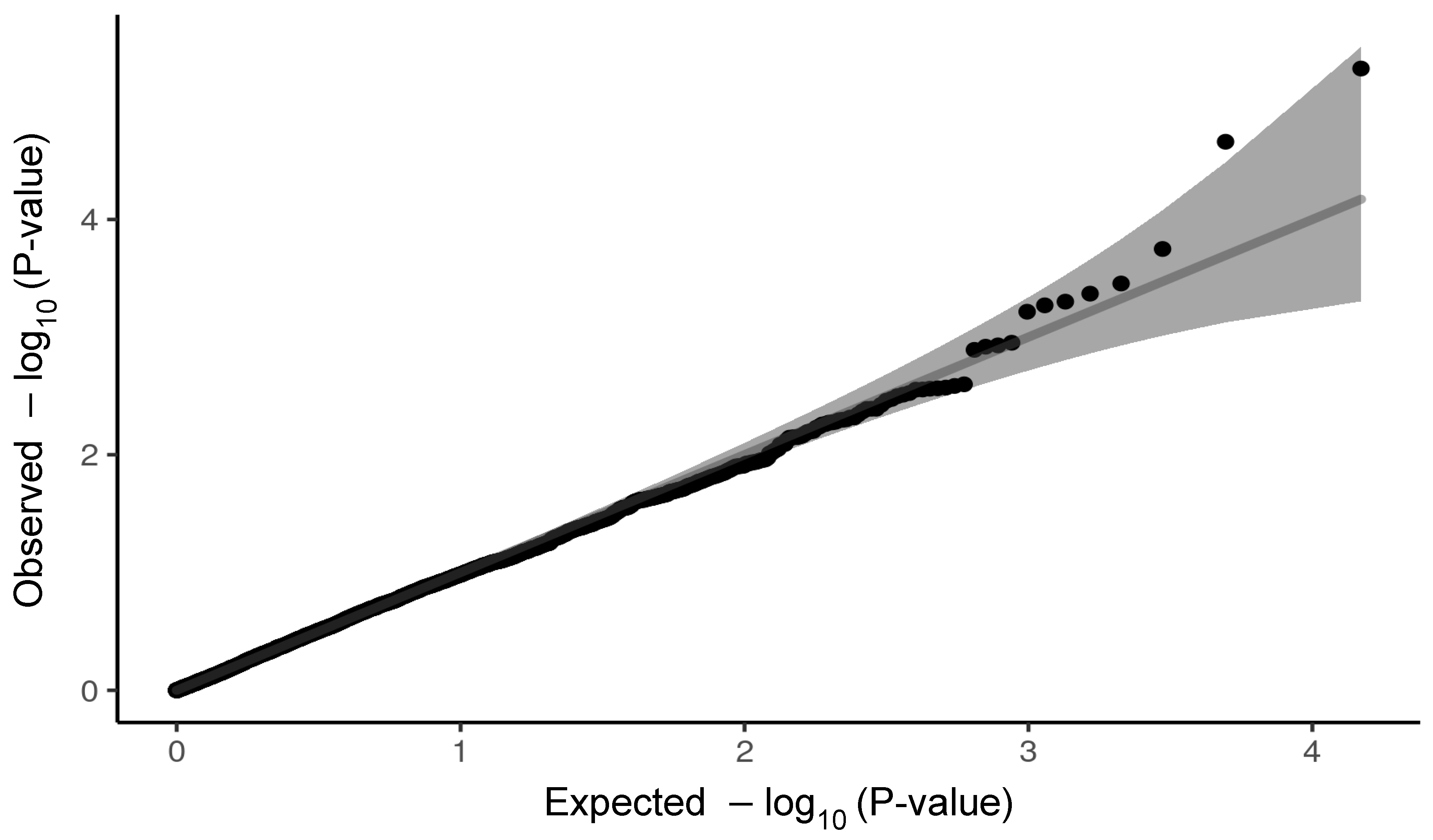
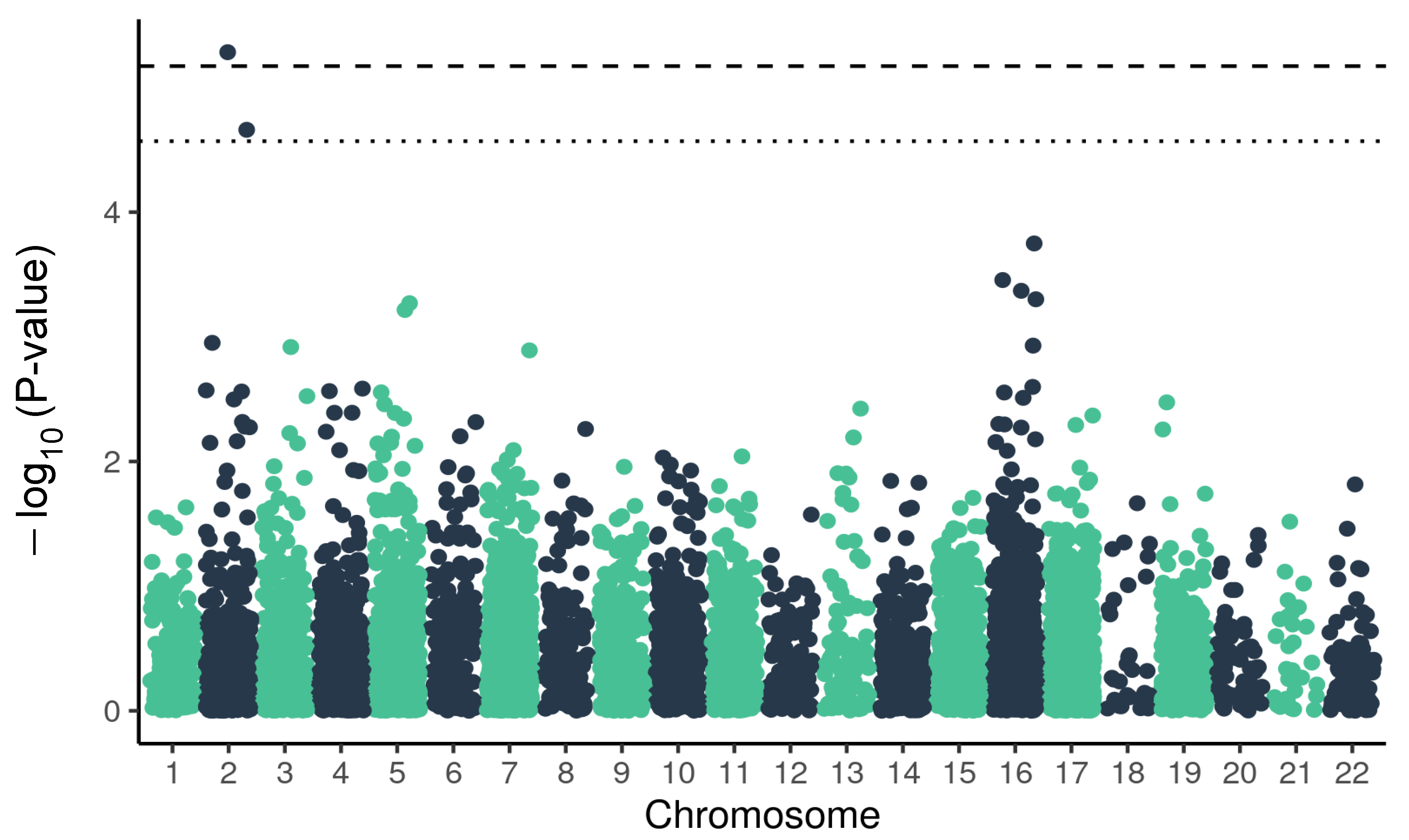
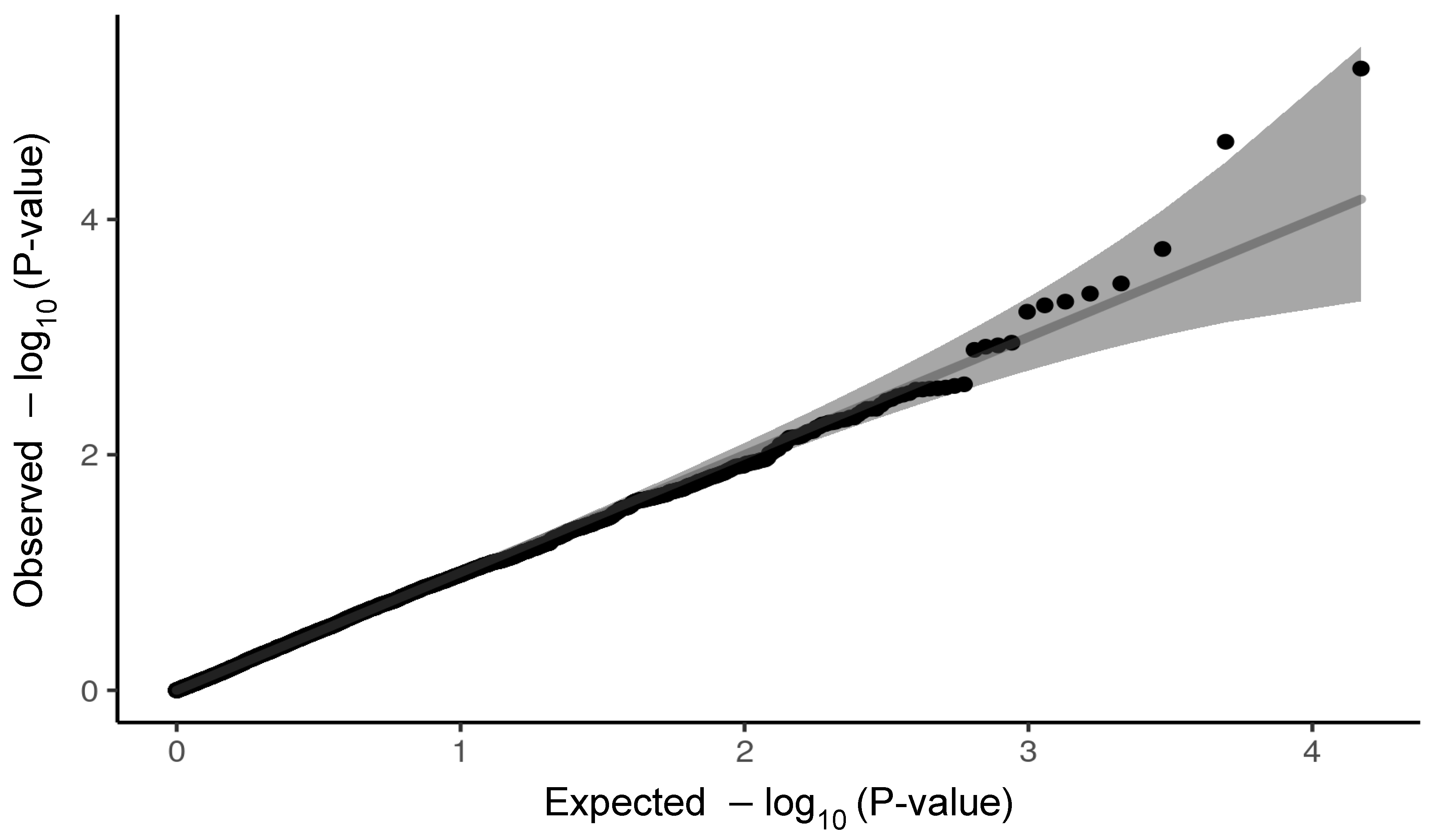
2.1. Variants Related to the Cardiovascular Panel and the Severe Outcome of COVID-19
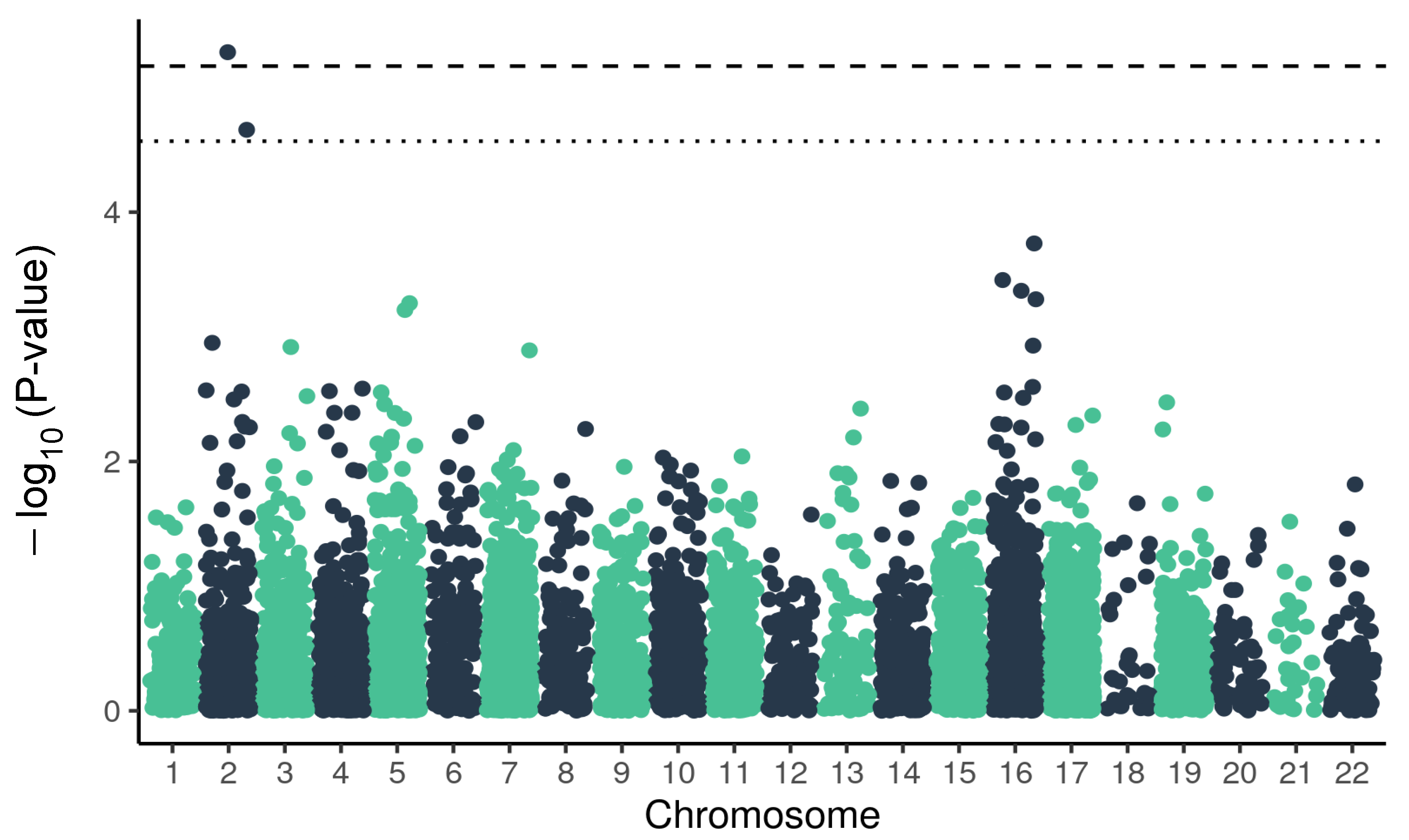
2.2. Variants Related to Pulmonary Panel and Severe Outcome of COVID-19
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Ethical Policy
4.3. Total Quality Management
4.4. Whole Genome Sequencing
4.5. Panels of Genes
4.6. Tag Variants
4.7. Genome-Wide Association Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Global excess deaths associated with COVID-19. January 2020–December 2021. 2022. Available online: https://www.who.int/data/stories/global-excess-deaths-associated-with-covid-19-january-2020-december-2021 (accessed on 7 June 2022).
- Sousa, F.M.; Roelens, M.; Fricker, B.; Thiabaud, A.; Iten, A.; Cusini, A.; Flury, D.; Buettcher, M.; Zukol, F.; Balmelli, C.; et al. Risk factors for severe outcomes for COVID-19 patients hospitalised in Switzerland during the first pandemic wave, February to August 2020: Prospective observational cohort study. Swiss Med. Wkly. 2021, 151, w20547. [Google Scholar] [CrossRef]
- Zerbo, O.; Lewis, N.; Fireman, B.; Goddard, K.; Skarbinski, J.; Sejvar, J.J.; Azziz-Baumgartner, E.; Klein, N.P. Population-based assessment of risks for severe COVID-19 disease outcomes. Influ. Other Respir. Viruses 2022, 16, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, M.; Nguyen, D.T.; Vahidy, F.S.; Graviss, E.A. Risk factors for severity of COVID-19 in hospital patients age 18–29 years. PLoS ONE 2021, 16, e0255544. [Google Scholar] [CrossRef]
- Lippi, G.; Sanchis-Gomar, F.; Henry, B.M. Coronavirus disease 2019 (COVID-19): The portrait of a perfect storm. Ann. Transl. Med. 2020, 8, 497. [Google Scholar] [CrossRef]
- Team, E. The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19)—China, 2020. China CDC Wkly. 2020, 2, 113. [Google Scholar] [CrossRef]
- Kong, K.A.; Jung, S.; Yu, M.; Park, J.; Kang, I.S. Association Between Cardiovascular Risk Factors and the Severity of Coronavirus Disease 2019: Nationwide Epidemiological Study in Korea. Front. Cardiovasc. Med. 2021, 8, 732518. [Google Scholar] [CrossRef]
- Xu, J.; Xiao, W.; Liang, X.; Shi, L.; Zhang, P.; Wang, Y.; Wang, Y.; Yang, H. A meta-analysis on the risk factors adjusted association between cardiovascular disease and COVID-19 severity. BMC Public Health 2021, 21, 1533. [Google Scholar] [CrossRef]
- Calmes, D.; Graff, S.; Maes, N.; Frix, A.-N.; Thys, M.; Bonhomme, O.; Berg, J.; Debruche, M.; Gester, F.; Henket, M.; et al. Asthma and COPD Are Not Risk Factors for ICU Stay and Death in Case of SARS-CoV2 Infection. J. Allergy Clin. Immunol. Pract. 2021, 9, 160–169. [Google Scholar] [CrossRef]
- Lapić, I.; Antolic, M.R.; Horvat, I.; Premužić, V.; Palić, J.; Rogić, D.; Zadro, R. Association of polymorphisms in genes encoding prothrombotic and cardiovascular risk factors with disease severity in COVID-19 patients: A pilot study. J. Med. Virol. 2022, 94, 3669–3675. [Google Scholar] [CrossRef]
- Aveyard, P.; Gao, M.; Lindson, N.; Hartmann-Boyce, J.; Watkinson, P.; Young, D.; Coupland, C.A.C.; Tan, P.S.; Clift, A.K.; Harrison, D.; et al. Association between pre-existing respiratory disease and its treatment, and severe COVID-19: A population cohort study. Lancet Respir. Med. 2021, 9, 909–923. [Google Scholar] [CrossRef]
- Acevedo, N.; Escamilla-Gil, J.M.; Espinoza, H.; Regino, R.; Ramírez, J.; de Arco, L.F.; Dennis, R.; Torres-Duque, C.A.; Caraballo, L. Chronic Obstructive Pulmonary Disease Patients Have Increased Levels of Plasma Inflammatory Mediators Reported Upregulated in Severe COVID-19. Front. Immunol. 2021, 12, 678661. [Google Scholar] [CrossRef]
- Marçalo, R.; Neto, S.; Pinheiro, M.; Rodrigues, A.J.; Sousa, N.; Santos, M.A.S.; Simão, P.; Valente, C.; Andrade, L.; Marques, A.; et al. Evaluation of the genetic risk for COVID-19 outcomes in COPD and differences among worldwide populations. PLoS ONE 2022, 17, e0264009. [Google Scholar] [CrossRef]
- Girardin, J.-L.; Seixas, A.; Cejudo, J.R.; Osorio, R.S.; Avirappattu, G.; Reid, M.; Parthasarathy, S. Contribution of pulmonary diseases to COVID-19 mortality in a diverse urban community of New York. Chronic Respir. Dis. 2021, 18, 147997312098680. [Google Scholar] [CrossRef]
- Diebold, I.; Schön, U.; Horvath, R.; Schwartz, O.; Holinski-Feder, E.; Kölbel, H.; Abicht, A. HADHA and HADHB gene associated phenotypes—Identification of rare variants in a patient cohort by Next Generation Sequencing. Mol. Cell. Probes 2019, 44, 14–20. [Google Scholar] [CrossRef]
- Miklas, J.W.; Clark, E.; Levy, S.; Detraux, D.; Leonard, A.; Beussman, K.; Showalter, M.R.; Smith, A.T.; Hofsteen, P.; Yang, X.; et al. TFPa/HADHA is required for fatty acid beta-oxidation and cardiolipin re-modeling in human cardiomyocytes. Nat. Commun. 2019, 10, 4671. [Google Scholar] [CrossRef] [Green Version]
- Dessein, A.-F.; Hebbar, E.; Vamecq, J.; Lebredonchel, E.; Devos, A.; Ghoumid, J.; Mention, K.; Dobbelaere, D.; Chevalier-Curt, M.J.; Fontaine, M.; et al. A novel HADHA variant associated with an atypical moderate and late-onset LCHAD deficiency. Mol. Genet. Metab. Rep. 2022, 31, 100860. [Google Scholar] [CrossRef]
- Bennett, M.J.; Spotswood, S.D.; Ross, K.F.; Comfort, S.; Koonce, R.; Boriack, R.L.; Ijlst, L.; Wanders, R.J. Fatal Hepatic Short-Chain l-3-Hydroxyacyl-Coenzyme a Dehydrogenase Deficiency: Clinical, Biochemical, and Pathological Studies on Three Subjects with This Recently Identified Disorder of Mitochondrial β-Oxidation. Pediatr. Dev. Pathol. 1999, 2, 337–345. [Google Scholar] [CrossRef]
- Wongkittichote, P.; Watson, J.R.; Leonard, J.M.; Toolan, E.R.; Dickson, P.I.; Grange, D.K. FatalCOVID-19 infection in a patient with long-chain3-hydroxyacyl-CoAdehydrogenase deficiency: A case report. JIMD Rep. 2020, 56, 40–45. [Google Scholar] [CrossRef]
- Takeuchi, K.; Xu, Y.; Kitano, M.; Chiyonobu, K.; Abo, M.; Ikegami, K.; Ogawa, S.; Ikejiri, M.; Kondo, M.; Gotoh, S.; et al. Copy number variation in DRC1 is the major cause of primary ciliary dyskinesia in the Japanese population. Mol. Genet. Genom. Med. 2020, 8, e1137. [Google Scholar] [CrossRef] [Green Version]
- Morimoto, K.; Hijikata, M.; Zariwala, M.A.; Nykamp, K.; Inaba, A.; Guo, T.; Yamada, H.; Truty, R.; Sasaki, Y.; Ohta, K.; et al. Recurring large deletion in DRC1 (CCDC164) identified as causing primary ciliary dyskinesia in two Asian patients. Mol. Genet. Genom. Med. 2019, 7, e838. [Google Scholar] [CrossRef] [Green Version]
- Keicho, N.; Hijikata, M.; Morimoto, K.; Homma, S.; Taguchi, Y.; Azuma, A.; Kudoh, S. Primary ciliary dyskinesia caused by a large homozygous deletion including exons 1–4 of DRC1 in Japanese patients with recurrent sinopulmonary infection. Mol. Genet. Genom. Med. 2020, 8, e1033. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, A.T.; Ganesh, R.; Chinnappa, J.; Kinimi, I.; Lucas, J. Primary ciliary dyskinesia due to DRC1/CCDC164 gene mutation. Lung India 2020, 37, 179. [Google Scholar] [CrossRef]
- Gazon, H.; Juszczak, D.; Fadeur, M.; Camby, S.; Meuris, C.; Thys, M.; Jacques, J.; Henket, M.; Léonard, P.; Frippiat, F.; et al. Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477. [Google Scholar]
- Kaja, E.; Lejman, A.; Sielski, D.; Sypniewski, M.; Gambin, T.; Dawidziuk, M.; Suchocki, T.; Golik, P.; Wojtaszewska, M.; Mroczek, M.; et al. The Thousand Polish Genomes—A Database of Polish Variant Allele Frequencies. Int. J. Mol. Sci. 2022, 23, 4532. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Panels. 2022. Available online: https://blueprintgenetics.com/tests/panels/ (accessed on 7 June 2022).
- Hill, W.G.; Robertson, A. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 1968, 38, 226–231. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Genomic Position (GRCh38) | Variant | Raw p-Value | BF | FDR | OR | SE | MAF |
|---|---|---|---|---|---|---|---|---|
| HADHA | chr2:26242159 | rs56218721 G > C | 0.000014 | 0.28 | 0.28 | 0.59 | 0.75 | 0.47 |
| Gene | Genomic Position (GRCh38) | Variant | Raw p-Value | BF | FDR | OR | SE | MAF |
|---|---|---|---|---|---|---|---|---|
| DRC1 | chr2:26447115 | rs10193369 A > T | 0.0000052 | 0.038 | 0.039 | 0.56 | 0.13 | 0.48 |
| DRC1 | chr2:26414115 | rs3067393 C > CATT | 0.000022 | 0.16 | 0.08 | 1.71 | 0.13 | 0.43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sypniewski, M.; Król, Z.J.; Szyda, J.; Kaja, E.; Mroczek, M.; Suchocki, T.; Lejman, A.; Stępień, M.; Topolski, P.; Dąbrowski, M.; et al. Gene Variants Related to Cardiovascular and Pulmonary Diseases May Correlate with Severe Outcome of COVID-19. Int. J. Mol. Sci. 2022, 23, 8696. https://doi.org/10.3390/ijms23158696
Sypniewski M, Król ZJ, Szyda J, Kaja E, Mroczek M, Suchocki T, Lejman A, Stępień M, Topolski P, Dąbrowski M, et al. Gene Variants Related to Cardiovascular and Pulmonary Diseases May Correlate with Severe Outcome of COVID-19. International Journal of Molecular Sciences. 2022; 23(15):8696. https://doi.org/10.3390/ijms23158696
Chicago/Turabian StyleSypniewski, Mateusz, Zbigniew J. Król, Joanna Szyda, Elżbieta Kaja, Magdalena Mroczek, Tomasz Suchocki, Adrian Lejman, Maria Stępień, Piotr Topolski, Maciej Dąbrowski, and et al. 2022. "Gene Variants Related to Cardiovascular and Pulmonary Diseases May Correlate with Severe Outcome of COVID-19" International Journal of Molecular Sciences 23, no. 15: 8696. https://doi.org/10.3390/ijms23158696
APA StyleSypniewski, M., Król, Z. J., Szyda, J., Kaja, E., Mroczek, M., Suchocki, T., Lejman, A., Stępień, M., Topolski, P., Dąbrowski, M., Kotlarz, K., Aplas, A., Wasiak, M., Wojtaszewska, M., Zawadzki, P., Pawlak, A., Gil, R., Dobosz, P., & Stojak, J. (2022). Gene Variants Related to Cardiovascular and Pulmonary Diseases May Correlate with Severe Outcome of COVID-19. International Journal of Molecular Sciences, 23(15), 8696. https://doi.org/10.3390/ijms23158696