
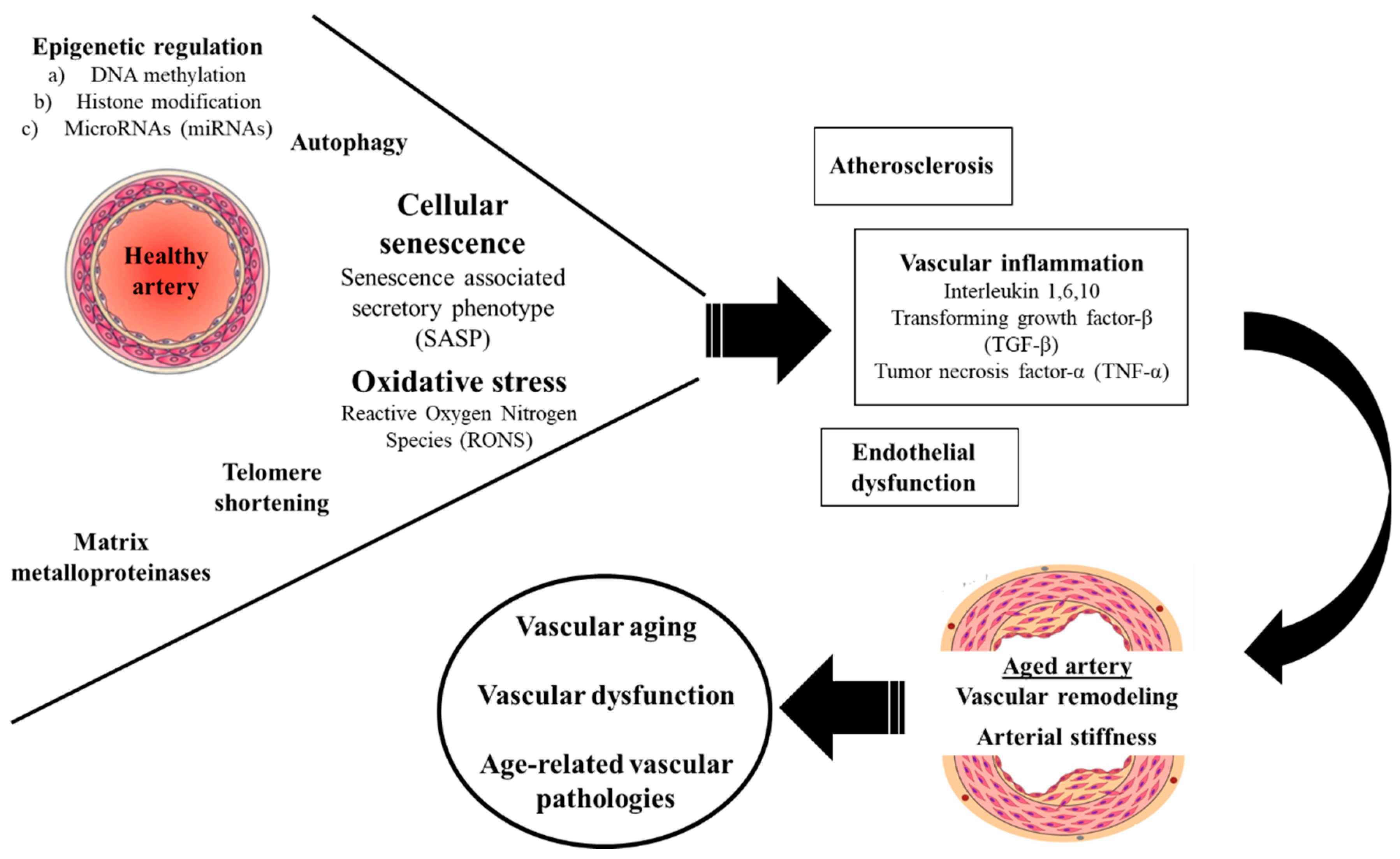
Theories and Molecular Basis of Vascular Aging: A Review of the Literature from VascAgeNet Group on Pathophysiological Mechanisms of Vascular Aging

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Methodology
2.1. Oxidative Stress
2.1.1. Role of Oxidative and Nitrosative Stress
2.1.2. Endogenous Sources of RONS
2.1.3. The Renin/Angiotensin Signaling Pathway
2.1.4. Exogenous Sources of RONS
2.2. Inflammation
2.2.1. Interleukin-1
2.2.2. Ιnterleukin-6
2.2.3. Ιnterleukin-10
2.2.4. Transforming Growth Factor Beta
2.2.5. Tumor Necrosis Factor-α
2.3. Extracellular Matrix Metalloproteinases
2.3.1. MMP-2
2.3.2. MMP-3
2.3.3. MMP-7
2.3.4. MMP-9
2.4. Epigenetic Regulation
2.4.1. DNA Methylation
2.4.2. Histone Modification
2.4.3. Non-Coding RNAs
2.5. Telomere Shortening
2.6. Cellular Senescence
2.7. Autophagy
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE | angiotensin converting enzyme |
| AMPK | AMP-activated protein kinase |
| AngII | angiotensin II |
| AP-1 | activator protein-1 |
| ApoE | apolipoprotein E |
| BCL6 | B-cell lymphoma 6 protein |
| BP | blood pressure |
| CAT | catalase |
| COX-2 | cyclooxygenase-2 |
| CRP | C-reactive protein |
| CTGF | connective tissue growth factor |
| CVD | cardiovascular disease |
| DDR | DNA damage response |
| DNMT | DNA methyltransferases |
| ECM | extracellular matrix |
| ECs | endothelial cells |
| EGFR | epidermal growth factor receptor |
| EPCs | endothelial progenitor cells |
| eNOS | endothelial nitric oxide synthase |
| FOXO | Forkhead box |
| GPX | glutathione-peroxidase |
| HATs | histone acetyltransferases |
| HB-EGF | heparin-binding epidermal growth factor |
| HDACs | histone deacetylases |
| HDMs | histone demethylases |
| HDL | high density lipoprotein |
| HIF-1α | hypoxia-inducible factor 1-alpha |
| HMTs | histone methyltransferases |
| Hpx | hemopexin |
| HUVECs | human umbilical endothelial cells |
| ICAM-1 | intercellular adhesion molecule-1 |
| IFN | interferon |
| IL | interleukin |
| IMT | intima-media thickness |
| KLF2 | Kruppel-like Factor 2 |
| LDL | low density lipoprotein |
| LKB1 | liver kinase B1 |
| LOX-1 | lectin-like oxidized low-density lipoprotein receptor-1 |
| LPOs | lipoxygenases |
| Lys | lysine |
| MAPKs | mitogen-activated protein kinases |
| MCP-1 | monocyte chemotactic protein-1 |
| MIP-1a | macrophage inflammatory protein-1 alpha |
| miRNAs | microRNAs |
| MMPs | matrix metalloproteinases |
| MPOs | myeloperoxidases |
| mTOR | mammalian target of rapamycin |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| ncRNAs | non-coding RNAs |
| NF-κB | nuclear factor-kb |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| oxLDL | oxidized low-density lipoprotein |
| PAI-1 | plasminogen activator inhibitor-type 1 |
| PAR-1 | protease activated receptor-1 |
| PARP | poly (ADP-ribose) polymerase |
| PDGF | platelet-derived growth factor |
| PCNA | proliferating cell nuclear antigen |
| PGC-1a | peroxisome proliferator-activated receptor-γ coactivator-1α |
| RAS | renin/angiotensin system |
| RONS | reactive oxygen and nitrogen species |
| ROS | reactive oxygen species |
| RSPO3 | pro-permeability factor R-spondin 3 |
| SASP | senescence-associated secretory phenotype |
| sGCβ1 | soluble guanylyl cyclase β1 |
| Sirt | sirtuin |
| SOD | superoxide dismutase |
| TGF-β | transforming growth factor-β |
| TIMPs | tissue inhibitors of matrix metalloproteinases |
| TLR | toll-like receptor |
| TNF | tumor necrosis factor |
| TNFS4 | tumor necrosis factor superfamily member 4 |
| TRF-2 | telomeric repeat-binding factor 2 |
| VCAM-1 | vascular cell adhesion molecule-1 |
| VEGF | vascular endothelial growth factor |
| VSMC | vascular smooth muscle cell |
References
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Freedman, V.A.; Martin, L.G.; Schoeni, R.F. Recent Trends in Disability and Functioning among Older Adults in the United States: A Systematic Review. JAMA 2002, 288, 3137–3146. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises: Part I: Aging Arteries: A “Set up” for Vascular Disease. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef]
- Paneni, F.; Diaz Cañestro, C.; Libby, P.; Lüscher, T.F.; Camici, G.G. The Aging Cardiovascular System: Understanding It at the Cellular and Clinical Levels. J. Am. Coll. Cardiol. 2017, 69, 1952–1967. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Tauzin, L. Alterations in Viscoelastic Properties Following Premature Birth May Lead to Hypertension and Cardiovascular Disease Development in Later Life. Acta Paediatr. Int. J. Paediatr. 2015, 104, 19–26. [Google Scholar] [CrossRef]
- Martyn, C.N.; Greenwald, S.E. Impaired Synthesis of Elastin in Walls of Aorta and Large Conduit Arteries during Early Development as an Initiating Event in Pathogenesis of Systemic Hypertension. Lancet 1997, 350, 953–955. [Google Scholar] [CrossRef]
- Martin, H.; Hu, J.; Gennser, G.; Norman, M. Impaired Endothelial Function and Increased Carotid Stiffness in 9-Year-Old Children with Low Birthweight. Circulation 2000, 102, 2739–2744. [Google Scholar] [CrossRef]
- Brodszki, J.; Länne, T.; Maršál, K.; Ley, D. Impaired Vascular Growth in Late Adolescence after Intrauterine Growth Restriction. Circulation 2005, 111, 2623–2628. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, X.; Wang, B.; Ren, C.; Hu, J.; Greenberg, D.A.; Chen, T.; Xie, L.; Jin, K. Age-Related Impairment of Vascular Structure and Functions. Aging Dis. 2017, 8, 590–610. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Machin, D.R.; Lesniewski, L.A. Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease. Circ. Res. 2018, 123, 825–848. [Google Scholar] [CrossRef] [PubMed]
- Laina, A.; Stellos, K.; Stamatelopoulos, K. Vascular Ageing: Underlying Mechanisms and Clinical Implications. Exp. Gerontol. 2018, 109, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circ. Res. 2018, 123, 849–867. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-morte, D.; Testa, G.; Cacciatore, F.; Bonaduce, D.; Abete, P. Oxidative Stress and Diseases. Oxidative Stress Dis. 2018, 47, 770–773. [Google Scholar] [CrossRef]
- Martínez-Revelles, S.; García-Redondo, A.B.; Avendaño, M.S.; Varona, S.; Palao, T.; Orriols, M.; Roque, F.R.; Fortuño, A.; Touyz, R.M.; Martínez-González, J.; et al. Lysyl Oxidase Induces Vascular Oxidative Stress and Contributes to Arterial Stiffness and Abnormal Elastin Structure in Hypertension: Role of P38MAPK. Antioxid. Redox Signal. 2017, 27, 379–397. [Google Scholar] [CrossRef]
- Sena, C.M.; Leandro, A.; Azul, L.; Seiça, R.; Perry, G. Vascular Oxidative Stress: Impact and Therapeutic Approaches. Front. Physiol. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Durackova, Z. Systems Biology of Free Radicals and Antioxidants; Springer: Berlin/Heidelberg, Germany, 2014; Volume 9783642300. [Google Scholar] [CrossRef]
- Salisbury, D.; Bronas, U. Reactive Oxygen and Nitrogen Species: Impact on Endothelial Dysfunction. Nurs. Res. 2015, 64, 53–66. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-morte, D.; Testa, G.; Cacciatore, F.; Bonaduce, D.; Abete, P. Oxidative Stress and Diseases; IntechOpen: London, UK, 2012; pp. 757–772. [Google Scholar] [CrossRef]
- Genestra, M. Oxyl Radicals, Redox-Sensitive Signalling Cascades and Antioxidants. Cell. Signal. 2007, 19, 1807–1819. [Google Scholar] [CrossRef]
- Kubala, L.; Schmelzer, K.R.; Klinke, A.; Kolarova, H.; Baldus, S.; Hammock, B.D.; Eiserich, J.P. Modulation of Arachidonic and Linoleic Acid Metabolites in Myeloperoxidase-Deficient Mice during Acute Inflammation. Free Radic. Biol. Med. 2010, 48, 1311–1320. [Google Scholar] [CrossRef]
- Daugherty, A.; Dunn, J.L.; Rateri, D.L.; Heinecke, J.W. Myeloperoxidase, a Catalyst for Lipoprotein Oxidation, Is Expressed in Human Atherosclerotic Lesions. J. Clin. Investig. 1994, 94, 437–444. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Hazen, S.L. Myeloperoxidase, Modified Lipoproteins, and Atherogenesis. J. Lipid Res. 2009, 50, S346–S351. [Google Scholar] [CrossRef] [PubMed]
- Kettle, A.J.; Albrett, A.M.; Chapman, A.L.; Dickerhof, N.; Forbes, L.V.; Khalilova, I.; Turner, R. Measuring Chlorine Bleach in Biology and Medicine. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Rådmark, O.; Werz, O.; Steinhilber, D.; Samuelsson, B. 5-Lipoxygenase, a Key Enzyme for Leukotriene Biosynthesis in Health and Disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2015, 1851, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.E.; Kim, E.N.; Kim, M.Y.; Lim, J.H.; Jang, I.A.; Ban, T.H.; Shin, S.J.; Park, C.W.; Chang, Y.S.; Choi, B.S. Age-Associated Changes in the Vascular Renin-Angiotensin System in Mice. Oxid. Med. Cell. Longev. 2016, 2016, 6731093. [Google Scholar] [CrossRef]
- Touyz, R.M. Reactive Oxygen Species and Angiotensin II Signaling in Vascular Cells—Implications in Cardiovascular Disease. Brazilian J. Med. Biol. Res. 2004, 37, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Wolf, G. Free Radical Production and Angiotensin. Curr. Hypertens. Rep. 2000, 2, 167–173. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Harrison, D.G. Methods for Detection of Mitochondrial and Cellular Reactive Oxygen Species. Antioxid. Redox Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef] [PubMed]
- de Queiroz, T.M.; Monteiro, M.M.O.; Braga, V.A. Angiotensin-II-Derived Reactive Oxygen Species on Baroreflex Sensitivity during Hypertension: New Perspectives. Front. Physiol. 2013, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The Role of Antioxidants in the Chemistry of Oxidative Stress: A Review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Alberts-Grill, N.; Denning, T.L.; Rezvan, A.; Jo, H. The Role of the Vascular Dendritic Cell Network in Atherosclerosis. Am. J. Physiol. Cell Physiol. 2013, 305, C1–C21. [Google Scholar] [CrossRef] [PubMed]
- Zuliani, G.; Morieri, M.L.; Volpato, S.; Maggio, M.; Cherubini, A.; Francesconi, D.; Bandinelli, S.; Paolisso, G.; Guralnik, J.M.; Ferrucci, L. Insulin Resistance and Systemic Inflammation, but Not Metabolic Syndrome Phenotype, Predict 9 Years Mortality in Older Adults. Atherosclerosis 2014, 235, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Harris, T.B.; Guralnik, J.M.; Tracy, R.P.; Corti, M.-C.; Cohen, H.J.; Penninx, B.; Pahor, M.; Wallace, R.; Havlik, R.J. Serum IL-6 Level and the Development of Disability in Older Persons. Am. Geriatr. Soc. 1999, 47, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, E.; An, Y.; Zoli, M.; Simonsick, E.M.; Guralnik, J.M.; Bandinelli, S.; Boyd, C.M.; Ferrucci, L. Aging and the Burden of Multimorbidity: Associations with Inflammatory and Anabolic Hormonal Biomarkers. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 63–70. [Google Scholar] [CrossRef]
- Soysal, P.; Stubbs, B.; Lucato, P.; Luchini, C.; Solmi, M.; Peluso, R.; Sergi, G.; Isik, A.T.; Manzato, E.; Maggi, S.; et al. Inflammation and Frailty in the Elderly: A Systematic Review and Meta-Analysis. Ageing Res. Rev. 2016, 31, 1–8. [Google Scholar] [CrossRef]
- Montero, I.; Orbe, J.; Varo, N.; Beloqui, O.; Monreal, J.I.; Rodréguez, J.A.; Díez, J.; Libby, P.; Páramo, J.A. C-Reactive Protein Induces Matrix Metalloproteinase-1 and -10 in Human Endothelial Cells: Implications for Clinical and Subclinical Atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, 1369–1378. [Google Scholar] [CrossRef]
- Scicali, R.; Di Pino, A.; Urbano, F.; Ferrara, V.; Marchisello, S.; Di Mauro, S.; Scamporrino, A.; Filippello, A.; Piro, S.; Rabuazzo, A.M.; et al. Analysis of S100A12 Plasma Levels in Hyperlipidemic Subjects with or without Familial Hypercholesterolemia. Acta Diabetol. 2019, 56, 899–906. [Google Scholar] [CrossRef]
- Wang, W.; Deng, Z.; Li, L.; Li, J.; Jin, X. Association of Hyper-Sensitive C-Reactive Protein with Arterial Stiffness and Endothelial Function in Patients with Hyperlipidemia. Int. J. Clin. Exp. Med. 2016, 9, 23416–23424. [Google Scholar]
- Vicenová, B.; Vopálenský, V.; Burýšek, L.; Pospíšek, M. Emerging Role of Interleukin-1 in Cardiovascular Diseases. Physiol. Res. 2009, 58, 481–498. [Google Scholar] [CrossRef]
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The Origins of Age-Related Proinflammatory State. Blood 2005, 105, 2294–2299. [Google Scholar] [CrossRef]
- Kirii, H.; Niwa, T.; Yamada, Y.; Wada, H.; Saito, K.; Iwakura, Y.; Asano, M.; Moriwaki, H.; Seishima, M. Lack of Interleukin-1ß Decreases the Severity of Atherosclerosis in ApoE-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 656–660. [Google Scholar] [CrossRef]
- Merhi-Soussi, F.; Kwak, B.R.; Magne, D.; Chadjichristos, C.; Berti, M.; Pelli, G.; James, R.W.; Mach, F.; Gabay, C. Interleukin-1 Plays a Major Role in Vascular Inflammation and Atherosclerosis in Male Apolipoprotein E-Knockout Mice. Cardiovasc. Res. 2005, 66, 583–593. [Google Scholar] [CrossRef]
- Nicklin, M.J.H.; Hughes, D.E.; Barton, J.L.; Ure, J.M.; Duff, G.W. Arterial Inflammation in Mice Lacking the Interleukin 1 Receptor Antagonist Gene. J. Exp. Med. 2000, 191, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Fahey, E.; Doyle, S.L. IL-1 Family Cytokine Regulation of Vascular Permeability and Angiogenesis. Front. Immunol. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Libby, P.; Everett, B.M. Novel Antiatherosclerotic Therapies. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 538–545. [Google Scholar] [CrossRef]
- Tyrrell, D.J.; Goldstein, D.R. Ageing and Atherosclerosis: Vascular Intrinsic and Extrinsic Factors and Potential Role of IL-6. Nat. Rev. Cardiol. 2021, 18, 58–68. [Google Scholar] [CrossRef]
- Song, Y.; Shen, H.; Schenten, D.; Shan, P.; Lee, P.J.; Goldstein, D.R. Aging Enhances the Basal Production of IL-6 and CCL2 in Vascular Smooth Muscle Cells. Arter. Thromb Vasc Biol. 2013, 32, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P. Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature. Int. J. Mol. Sci. 2017, 18, 2563. [Google Scholar] [CrossRef]
- Hung, M.J.; Cherng, W.J.; Hung, M.Y.; Wu, H.T.; Pang, J.H.S. Interleukin-6 Inhibits Endothelial Nitric Oxide Synthase Activation and Increases Endothelial Nitric Oxide Synthase Binding to Stabilized Caveolin-1 in Human Vascular Endothelial Cells. J. Hypertens. 2010, 28, 940–951. [Google Scholar] [CrossRef]
- Schrader, L.I.; Kinzenbaw, D.A.; Johnson, A.W.; Faraci, F.M.; Didion, S.P. IL-6 Deficiency Protects against Angiotensin II-Induced Endothelial Dysfunction and Hypertrophy. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2576–2581. [Google Scholar] [CrossRef]
- Wassmann, S.; Stumpf, M.; Strehlow, K.; Schmid, A.; Schieffer, B.; Böhm, M.; Nickenig, G. Interleukin-6 Induces Oxidative Stress and Endothehal Dysfunction by Overexpression of the Angiotensin II Type 1 Receptor. Circ. Res. 2004, 94, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.W.; Kinzenbaw, D.A.; Modrick, M.L.; Faraci, F.M. Small-Molecule Inhibitors of Signal Transducer and Activator of Transcription 3 Protect against Angiotensin Ii-Induced Vascular Dysfunction and Hypertension. Hypertension 2013, 61, 437–442. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, S.; Ciechomska, M.; Cant, R.; Van Laar, J.M. Interleukin-6 (IL-6) Trans Signaling Drives a STAT3-Dependent Pathway That Leads to Hyperactive Transforming Growth Factor-β (TGF-β) Signaling Promoting SMAD3 Activation and Fibrosis via Gremlin Protein. J. Biol. Chem. 2014, 289, 9952–9960. [Google Scholar] [CrossRef] [PubMed]
- Mingomataj, E.; Bakiri, A.H. Regulator versus Effector Paradigm: Interleukin-10 as Indicator of the Switching Response. Clin. Rev. Allergy Immunol. 2016, 50, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Rutz, S.; Crellin, N.K.; Valdez, P.A.; Hymowitz, S.G. Regulation and Functions of the IL-10 Family of Cytokines in Inflammation and Disease. Annu. Rev. Immunol. 2011, 29, 71–109. [Google Scholar] [CrossRef] [PubMed]
- Kinzenbaw, D.A.; Chu, Y.; Peña Silva, R.A.; Didion, S.P.; Faraci, F.M. Interleukin-10 Protects against Aging-Induced Endothelial Dysfunction. Physiol. Rep. 2013, 1, 1–8. [Google Scholar] [CrossRef]
- Lima, V.V.; Zemse, S.M.; Chiao, C.W.; Bomfim, G.F.; Tostes, R.C.; Clinton Webb, R.; Giachini, F.R. Interleukin-10 Limits Increased Blood Pressure and Vascular RhoA/Rho-Kinase Signaling in Angiotensin II-Infused Mice. Life Sci. 2016, 145, 137–143. [Google Scholar] [CrossRef]
- Sikka, G.; Miller, K.L.; Steppan, J.; Pandey, D.; Jung, S.M.; Fraser, C.D.; Ellis, C.; Ross, D.; Vandegaer, K.; Bedja, D.; et al. Interleukin 10 Knockout Frail Mice Develop Cardiac and Vascular Dysfunction with Increased Age. Exp. Gerontol. 2013, 48, 128–135. [Google Scholar] [CrossRef]
- Strom, A.C.; Cross, A.J.; Cole, J.E.; Blair, P.A.; Leib, C.; Goddard, M.E.; Rosser, E.C.; Park, I.; Nilsson, A.H.; Nilsson, J.; et al. B Regulatory Cells Are Increased in Hypercholesterolemic Mice and Pro- Tect from Lesion Development via IL-10. Thromb Haemost. 2015, 114, 835–847. [Google Scholar]
- Arjuman, A.; Chandra, N.C. Effect of IL-10 on LOX-1 Expression, Signalling and Functional Activity: An Atheroprotective Response. Diabetes Vasc. Dis. Res. 2013, 10, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Rubic, T.; Lorenz, R.L. Downregulated CD36 and OxLDL Uptake and Stimulated ABCA1/G1 and Cholesterol Efflux as Anti-Atherosclerotic Mechanisms of Interleukin-10. Cardiovasc. Res. 2006, 69, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Boisvert, W.A. Interleukin-10 Protects against Atherosclerosis by Modulating Multiple Atherogenic Macrophage Function. Thromb. Haemost. 2015, 113, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Lustig, A.; Liu, H.B.; Metter, E.J.; An, Y.; Swaby, M.A.; Elango, P.; Ferrucci, L.; Hodes, R.J.; Weng, N.P. Telomere Shortening, Inflammatory Cytokines, and Anti-Cytomegalovirus Antibody Follow Distinct Ageassociated Trajectories in Humans. Front. Immunol. 2017, 8, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, D.B.; Firth, C.M.; Phillips, A.C.; Moss, P.; Baylis, D.; Syddall, H.; Sayer, A.A.; Cooper, C.; Lord, J.M. The Age-Related Increase in Low-Grade Systemic Inflammation (Inflammaging) Is Not Driven by Cytomegalovirus Infection. Aging Cell 2012, 11, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Rodríguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. TGF-β Signaling in Vascular Fibrosis. Cardiovasc. Res. 2007, 74, 196–206. [Google Scholar] [CrossRef]
- Samarakoon, R.; Higgins, S.P.; Higgins, C.E.; Higgins, P.J. TGF-Β1-Induced Plasminogen Activator Inhibitor-1 Expression in Vascular Smooth Muscle Cells Requires Pp60c-Src /EGFRY845 and Rho/ ROCK Signaling. J. Mol. Cell. Cardiol. 2008, 44, 527–538. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, D.; Spinetti, G.; Zhang, J.; Jiang, L.Q.; Pintus, G.; Monticone, R.; Lakatta, E.G. Matrix Metalloproteinase 2 Activation of Transforming Growth Factor-Β1 (TGF-Β1) and TGF-Β1-Type II Receptor Signaling within the Aged Arterial Wall. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1503–1509. [Google Scholar] [CrossRef]
- Wang, W.; Huang, X.R.; Canlas, E.; Oka, K.; Truong, L.D.; Bhowmick, N.A.; Ju, W.; Bottinger, E.P.; Lan, H.Y. Essential Role of Smad3 in Angiotensin II–Induced Vascular Fibrosis. Circ Res. 2006, 98, 1032–1039. [Google Scholar] [CrossRef]
- Toma, I.; McCaffrey, T.A. Transforming Growth Factor-β and Atherosclerosis: Interwoven Atherogenic and Atheroprotective Aspects. Cell Tissue Res. 2012, 347, 155–175. [Google Scholar] [CrossRef]
- Zhang, H.; Park, Y.; Wu, J.; Chen, X.P.; Lee, S.; Yang, J.; Dellsperger, K.C.; Zhang, C. Role of TNF-α in Vascular Dysfunction. Clin. Sci. 2009, 116, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.L.; Pendleton, L.C.; Levy, M.M.; Solomonson, L.P.; Eichler, D.C. Tumor Necrosis Factor-α Reduces Argininosuccinate Synthase Expression and Nitric Oxide Production in Aortic Endothelial Cells. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Labinskyy, N.; Smith, K.; Rivera, A.; Orosz, Z.; Ungvari, Z. Vasculoprotective Effects of Anti-Tumor Necrosis Factor-α Treatment in Aging. Am. J. Pathol. 2007, 170, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Rodríguez, L.; López-Hoyos, M.; Muñoz-Cacho, P.; Martínez-Taboada, V.M. Aging Is Associated with Circulating Cytokine Dysregulation. Cell. Immunol. 2012, 273, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Csiszar, A.; Ungvari, Z.; Koller, A.; Edwards, J.G.; Kaley, G. Proinflammatory Phenotype of Coronary Arteries Promotes Endothelial Apoptosis in Aging. Physiol. Genomics 2004, 17, 21–30. [Google Scholar] [CrossRef]
- Lacolley, P.; Regnault, V.; Avolio, A.P. Smooth Muscle Cell and Arterial Aging: Basic and Clinical Aspects. Cardiovasc. Res. 2018, 114, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Foote, K.; Bennett, M.R. Molecular Insights into Vascular Aging. Aging 2018, 10, 3647–3649. [Google Scholar] [CrossRef]
- Nagase, H.; Visse, R.; Murphy, G. Structure and Function of Matrix Metalloproteinases and TIMPs. Cardiovasc. Res. 2006, 69, 562–573. [Google Scholar] [CrossRef]
- Helena Laronha, J.C. Structure and Function Function of of Human. Cells 2020, 9, 1076. [Google Scholar] [CrossRef]
- Harvey, A.; Montezano, A.C.; Lopes, R.A.; Rios, F.; Touyz, R.M. Vascular Fibrosis in Aging and Hypertension: Molecular Mechanisms and Clinical Implications. Can. J. Cardiol. 2016, 32, 659–668. [Google Scholar] [CrossRef]
- Nagareddy, P.R.; Rajput, P.S.; Vasudevan, H.; McClure, B.; Kumar, U.; MacLeod, K.M.; McNeill, J. Inhibition of Matrix Metalloproteinase-2 Improves Endothelial Function and Prevents Hypertension in Insulin-Resistant Rats. Br. J. Pharmacol. 2012, 165, 705–715. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, J.; Telljohann, R.; Jiang, L.; Wu, J.; Monticone, R.E.; Kapoor, K.; Talan, M.; Lakatta, E.G. Chronic Matrix Metalloproteinase Inhibition Retards Age-Associated Arterial Proinflammation and Increase in Blood Pressure. Hypertension 2012, 60, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Puspitasari, Y.M.; Diaz-Canestro, C.; Liberale, L.; Guzik, T.J.; Flammer, A.J.; Bonetti, N.R.; Wüst, P.; Constantino, S.; Paneni, F.; Akhmedov, A.; et al. Therapeutic MMP-2 Knockdown Blunts Age-Dependent Carotid Stiffness by Decreasing Elastin Degradation and Augmenting Enos Activation. Atherosclerosis 2021, 331, e29–e30. [Google Scholar] [CrossRef]
- Lee, H.Y.; You, H.J.; Won, J.Y.; Youn, S.W.; Cho, H.J.; Park, K.W.; Park, W.Y.; Seo, J.S.; Park, Y.B.; Walsh, K.; et al. Forkhead Factor, FOXO3a, Induces Apoptosis of Endothelial Cells through Activation of Matrix Metalloproteinases. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 302–308. [Google Scholar] [CrossRef]
- Silence, J.; Lupu, F.; Collen, D.; Lijnen, H.R. Persistence of Atherosclerotic Plaque but Reduced Aneurysm Formation in Mice with Stromelysin-1 (MMP-3) Gene Inactivation. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; George, S.J.; Newby, A.C.; Jackson, C.L. Divergent Effects of Matrix Metalloproteinases 3, 7, 9, and 12 on Atherosclerotic Plaque Stability in Mouse Brachiocephalic Arteries. Proc. Natl. Acad. Sci. USA 2005, 102, 15575–15580. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.; Aukrust, P.; Russell, D.; Krohg-Sørensen, K.; Almås, T.; Bundgaard, D.; Bjerkeli, V.; Sagen, E.L.; Michelsen, A.E.; Dahl, T.B.; et al. Matrix Metalloproteinase 7 Is Associated with Symptomatic Lesions and Adverse Events in Patients with Carotid Atherosclerosis. PLoS ONE 2014, 9, e84935. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Park, J.H.; Jang, W.; Hwang, I.K.; Kim, I.J.; Kim, H.J.; Cho, K.H.; Lee, S.T. Apolipoprotein A-IV Is a Novel Substrate for Matrix Metalloproteinases. J. Biochem. 2012, 151, 291–298. [Google Scholar] [CrossRef]
- Williams, H.; Johnson, J.L.; Jackson, C.L.; White, S.J.; George, S.J. MMP-7 Mediates Cleavage of N-Cadherin and Promotes Smooth Muscle Cell Apoptosis. Cardiovasc. Res. 2010, 87, 137–146. [Google Scholar] [CrossRef]
- Hao, L.; Du, M.; Lopez-Campistrous, A.; Fernandez-Patron, C. Agonist-Induced Activation of Matrix Metalloproteinase-7 Promotes Vasoconstriction through the Epidermal Growth Factor-Receptor Pathway. Circ. Res. 2004, 94, 68–76. [Google Scholar] [CrossRef]
- Florence, J.M.; Krupa, A.; Booshehri, L.M.; Allen, T.C.; Kurdowska, A.K. Metalloproteinase-9 Contributes to Endothelial Dysfunction in Atherosclerosis via Protease Activated Receptor-1. PLoS ONE 2017, 12, e0171427. [Google Scholar] [CrossRef] [PubMed]
- Tziakas, D.N.; Lazarides, M.K.; Tentes, I.K.; Georgiadis, G.S.; Eleftheriadou, E.; Chalikias, G.K.; Kortsaris, A.; Hatseras, D.I. Gelatinases [Matrix Metalloproteinase-2 (MMP-2) and MMP-9] Induce Carotid Plaque Instability but Their Systemic Levels Are Not Predictive of Local Events. Ann. Vasc. Surg. 2005, 19, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Luttun, A.; Lutgens, E.; Manderveld, A.; Maris, K.; Collen, D.; Carmeliet, P.; Moons, L. Loss of Matrix Metalloproteinase-9 or Matrix Metalloproteinase-12 Protects Apolipoprotein E-Deficient Mice against Atherosclerotic Media Destruction but Differentially Affects Plaque Growth. Circulation 2004, 109, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.T.; Collins, E.T.; Marine, L.A.; Uberti, M.G.; Uchida, H.; Leidenfrost, J.E.; Khan, F.F.; Boc, K.P.; Abendschein, D.R.; Parks, W.C. Matrix Metalloproteinase-9 Modulation by Resident Arterial Cells Is Responsible for Injury-Induced Accelerated Atherosclerotic Plaque Development in Apolipoprotein E-Deficient Mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1020–1025. [Google Scholar] [CrossRef]
- Galis, Z.S.; Johnson, C.; Godin, D.; Magid, R.; Shipley, J.M.; Senior, R.M.; Ivan, E. Targeted Disruption of the Matrix Metalloproteinase-9 Gene Impairs Smooth Muscle Cell Migration and Geometrical Arterial Remodeling. Circ. Res. 2002, 91, 852–859. [Google Scholar] [CrossRef]
- Pyo, R.; Lee, J.K.; Shipley, J.M.; Curci, J.A.; Mao, D.; Ziporin, S.J.; Ennis, T.L.; Shapiro, S.D.; Senior, R.M.; Thompson, R.W. Targeted Gene Disruption of Matrix Metalloproteinase-9 (Gelatinase B) Suppresses Development of Experimental Abdominal Aortic Aneurysms. J. Clin. Investig. 2000, 105, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Ramirez Correa, G.A.; Zacchigna, S.; Arsic, N.; Zentilin, L.; Salvi, A.; Sinagra, G.; Giacca, M. Potent Inhibition of Arterial Intimal Hyperplasia by TIMP1 Gene Transfer Using AAV Vectors. Mol. Ther. 2004, 9, 876–884. [Google Scholar] [CrossRef]
- Allaire, E.; Forough, R.; Clowes, M.; Starcher, B.; Clowes, A.W. Local Overexpression of TIMP-1 Prevents Aortic Aneurysm Degeneration and Rupture in a Rat Model. J. Clin. Investig. 1998, 102, 1413–1420. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Baker, A.H.; Oka, K.; Chan, L.; Newby, A.C.; Jackson, C.L.; George, S.J. Suppression of Atherosclerotic Plaque Progression and Instability by Tissue Inhibitor of Metalloproteinase-2: Involvement of Macrophage Migration and Apoptosis. Circulation 2006, 113, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Kassiri, Z. Biology of Tissue Inhibitor of Metalloproteinase 3 (TIMP3), and Its Therapeutic Implications in Cardiovascular Pathology. Front. Physiol. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Son, D.J.; Jung, Y.Y.; Seo, Y.S.; Park, H.; Lee, D.H.; Kim, S.; Roh, Y.S.; Han, S.B.; Yoon, D.Y.; Hong, J.T. Interleukin-32α Inhibits Endothelial Inflammation, Vascular Smooth Muscle Cell Activation, and Atherosclerosis by Upregulating Timp3 and Reck through Suppressing MicroRNA-205 Biogenesis. Theranostics 2017, 7, 2186–2203. [Google Scholar] [CrossRef] [PubMed]
- Stöhr, R.; Cavalera, M.; Menini, S.; Mavilio, M.; Casagrande, V.; Rossi, C.; Urbani, A.; Cardellini, M.; Pugliese, G.; Menghini, R.; et al. Loss of TIMP3 Exacerbates Atherosclerosis in ApoE Null Mice. Atherosclerosis 2014, 235, 438–443. [Google Scholar] [CrossRef]
- Basu, R.; Lee, J.; Morton, J.S.; Takawale, A.; Fan, D.; Kandalam, V.; Wang, X.; Davidge, S.T.; Kassiri, Z. TIMP3 Is the Primary TIMP to Regulate Agonist-Induced Vascular Remodelling and Hypertension. Cardiovasc. Res. 2013, 98, 360–371. [Google Scholar] [CrossRef] [PubMed]
- McNulty, M.; Spiers, P.; McGovern, E.; Feely, J. Aging Is Associated with Increased Matrix Metalloproteinase-2 Activity in the Human Aorta. Am. J. Hypertens. 2005, 18, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharmacol. 2018, 81, 241–330. [Google Scholar] [CrossRef] [PubMed]
- Beaudeux, J.L.; Giral, P.; Bruckert, E.; Bernard, M.; Foglietti, M.J.; Chapman, M.J. Serum Matrix Metalloproteinase-3 and Tissue Inhibitor of Metalloproteinases-1 as Potential Markers of Carotid Atherosclerosis in Infraclinical Hyperlipidemia. Atherosclerosis 2003, 169, 139–146. [Google Scholar] [CrossRef]
- McCawley, L.J.; Matrisian, L.M. Matrix Metalloproteinases: They’re Not Just for Matrix Anymore! Curr. Opin. Cell Biol. 2001, 13, 534–540. [Google Scholar] [CrossRef]
- Yabluchanskiy, A.; Ma, Y.; Iyer, R.P.; Hall, M.E.; Lindsey, M.L. Matrix Metalloproteinase-9: Many Shades of Function in Cardiovascular Disease. Physiology 2013, 28, 391–403. [Google Scholar] [CrossRef]
- Loftus, I.M.; Naylor, A.R.; Goodall, S.; Crowther, M.; Jones, L.; Bell, P.R.F.; Thompson, M.M. Increased Matrix Metalloproteinase-9 Activity in Unstable Carotid Plaques. Stroke 2000, 31, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, X.; Feng, Y.; Dong, G.; Wang, Y.; Yang, J. The Role of Matrix Metalloproteinase-9 in Atherosclerotic Plaque Instability. Mediators Inflamm. 2020, 2020, 3872367. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Pushpakumar, S.; Muradashvili, N.; Kundu, S.; Tyagi, S.C.; Sen, U. Regulation and Involvement of Matrix Metalloproteinases in Vascular Diseases. Front. Biosci. 2016, 21, 89–118. [Google Scholar]
- Raffetto, J.D.; Khalil, R.A. Matrix Metalloproteinases and Their Inhibitors in Vascular Remodeling and Vascular Disease. Biochem. Pharmacol. 2008, 75, 346–359. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Tabaei, S.; Tabaee, S.S. DNA Methylation Abnormalities in Atherosclerosis. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Li, S.; Liu, Y.S. Roles and Mechanisms of DNA Methylation in Vascular Aging and Related Diseases. Front. Cell Dev. Biol. 2021, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Kumar, S.; Vikram, A.; Hoffman, T.A.; Naqvi, A.; Lewarchik, C.M.; Kim, Y.R.; Irani, K. Histone and DNA Methylation-Mediated Epigenetic Downregulation of Endothelial Kruppel-like Factor 2 by Low-Density Lipoprotein Cholesterol. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1936–1942. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Huang, Y.T.; Huang, T.S.; Pang, W.; Zhu, J.J.; Liu, Y.F.; Tang, R.Z.; Zhao, C.R.; Yao, W.J.; Li, Y.S.; et al. The Mammalian Target of Rapamycin and DNA Methyltransferase 1 Axis Mediates Vascular Endothelial Dysfunction in Response to Disturbed Flow. Sci. Rep. 2017, 7, 14996. [Google Scholar] [CrossRef]
- Xu, L.; Hao, H.; Hao, Y.; Wei, G.; Li, G.; Ma, P.; Xu, L.; Ding, N.; Ma, S.; Chen, A.F.; et al. Aberrant MFN2 Transcription Facilitates Homocysteine-Induced VSMCs Proliferation via the Increased Binding of c-Myc to DNMT1 in Atherosclerosis. J. Cell. Mol. Med. 2019, 23, 4611–4626. [Google Scholar] [CrossRef]
- Zhu, L.; Jia, L.; Liu, Z.; Zhang, Y.; Wang, J.; Yuan, Z.; Hui, R. Elevated Methylation of FOXP3 (Forkhead Box P3)-TSDR (Regulatory T-Cell-Specific Demethylated Region) Is Associated with Increased Risk for Adverse Outcomes in Patients with Acute Coronary Syndrome. Hypertension 2019, 74, 581–589. [Google Scholar] [CrossRef]
- Kim, J.Y.; Choi, B.G.; Jelinek, J.; Kim, D.H.; Lee, S.H.; Cho, K.; Rha, S.H.; Lee, Y.H.; Jin, H.S.; Choi, D.K.; et al. Promoter Methylation Changes in ALOX12 and AIRE1: Novel Epigenetic Markers for Atherosclerosis. Clin. Epigenetics 2020, 12, 1–13. [Google Scholar] [CrossRef]
- Aavik, E.; Lumivuori, H.; Leppänen, O.; Wirth, T.; Häkkinen, S.K.; Bräsen, J.H.; Beschorner, U.; Zeller, T.; Braspenning, M.; Van Criekinge, W.; et al. Global DNA Methylation Analysis of Human Atherosclerotic Plaques Reveals Extensive Genomic Hypomethylation and Reactivation at Imprinted Locus 14q32 Involving Induction of a MiRNA Cluster. Eur. Heart J. 2015, 36, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, X.; Yang, M.; Yang, H.; Xu, N.; Fan, X.; Liu, G.; Jiang, X.; Fan, J.; Zhang, L.; et al. DNA Methylome Profiling Reveals Epigenetic Regulation of Lipoprotein-Associated Phospholipase A2 in Human Vulnerable Atherosclerotic Plaque. Clin. Epigenetics 2021, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Istas, G.; Declerck, K.; Pudenz, M.; Szic, K.S.V.; Lendinez-Tortajada, V.; Leon-Latre, M.; Heyninck, K.; Haegeman, G.; Casasnovas, J.A.; Tellez-Plaza, M.; et al. Identification of Differentially Methylated BRCA1 and CRISP2 DNA Regions as Blood Surrogate Markers for Cardiovascular Disease. Sci. Rep. 2017, 7, 5120. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The Language of Covalent Histone Modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.H.; Di Guo, D.; Guan, Y.; Chi, C.Y.; Liu, B.D. Changes of DNA Methylation of Isoetes Sinensis under Pb and Cd Stress. Environ. Sci. Pollut. Res. 2019, 26, 3428–3435. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The Many Roles of Histone Deacetylases in Development and Physiology: Implications for Disease and Therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.M.; Wagner, R.; Rzucidlo, E.M. Age-Related Loss of SirT1 Expression Results in Dysregulated Human Vascular Smooth Muscle Cell Function. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, 533–541. [Google Scholar] [CrossRef]
- Halasa, M.; Adamczuk, K.; Adamczuk, G.; Afshan, S.; Stepulak, A.; Cybulski, M.; Wawruszak, A. Deacetylation of Transcription Factors in Carcinogenesis. Int. J. Mol. Sci. 2021, 22, 1810. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Koya, D. The Protective Role of Sirt1 in Vascular Tissue: Its Relationship to Vascular Aging and Atherosclerosis. Aging 2016, 8, 2290–2307. [Google Scholar] [CrossRef]
- Mattagajasingh, I.; Kim, C.S.; Naqvi, A.; Yamamori, T.; Hoffman, T.A.; Jung, S.B.; DeRicco, J.; Kasuno, K.; Irani, K. SIRT1 Promotes Endothelium-Dependent Vascular Relaxation by Activating Endothelial Nitric Oxide Synthase. Proc. Natl. Acad. Sci. USA 2007, 104, 14855–14860. [Google Scholar] [CrossRef]
- Donato, A.J.; Magerko, K.A.; Lawson, B.R.; Durrant, J.R.; Lesniewski, L.A.; Seals, D.R. SIRT-1 and Vascular Endothelial Dysfunction with Ageing in Mice and Humans. J. Physiol. 2011, 589, 4545–4554. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Akishita, M.; Eto, M.; Iijima, K.; Kaneki, M.; Ouchi, Y. Sirt1 Modulates Premature Senescence-like Phenotype in Human Endothelial Cells. J. Mol. Cell. Cardiol. 2007, 43, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Man, A.W.C.; Yang, K.; Guo, Y.; Xu, C.; Tse, H.F.; Han, W.; Bloksgaard, M.; De Mey, J.G.R.; Vanhoutte, P.M.; et al. Endothelial SIRT1 Prevents Adverse Arterial Remodeling by Facilitating HERC2-Mediated Degradation of Acetylated LKB1. Oncotarget 2016, 7, 39065–39081. [Google Scholar] [CrossRef]
- Olmos, Y.; Sánchez-Gómez, F.J.; Wild, B.; García-Quintans, N.; Cabezudo, S.; Lamas, S.; Monsalve, M. SirT1 Regulation of Antioxidant Genes Is Dependent on the Formation of a FoxO3a/PGC-1α Complex. Antioxid. Redox Signal. 2013, 19, 1507–1521. [Google Scholar] [CrossRef] [PubMed]
- D’Onofrio, N.; Servillo, L.; Balestrieri, M.L. SIRT1 and SIRT6 Signaling Pathways in Cardiovascular Disease Protection. Antioxid. Redox Signal. 2018, 28, 711–732. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.X.; Van Tits, L.J.; Lohmann, C.; Arsiwala, T.; Winnik, S.; Tailleux, A.; Stein, S.; Gomes, A.P.; Suri, V.; Ellis, J.L.; et al. The Sirt1 Activator SRT3025 Provides Atheroprotection in Apoe−/− Mice by Reducing Hepatic Pcsk9 Secretion and Enhancing Ldlr Expression. Eur. Heart J. 2015, 36, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef]
- Gillum, M.P.; Erion, D.M.; Shulman, G.I. Sirtuin-1 Regulation of Mammalian Metabolism. Trends Mol. Med. 2011, 17, 8–13. [Google Scholar] [CrossRef]
- Wan, Y.Z.; Gao, P.; Zhou, S.; Zhang, Z.Q.; Hao, D.L.; Lian, L.S.; Li, Y.J.; Chen, H.Z.; Liu, D.P. SIRT1-Mediated Epigenetic Downregulation of Plasminogen Activator Inhibitor-1 Prevents Vascular Endothelial Replicative Senescence. Aging Cell 2014, 13, 890–899. [Google Scholar] [CrossRef]
- Zhou, S.; Chen, H.Z.; Wan, Y.Z.; Zhang, Q.J.; Wei, Y.S.; Huang, S.; Liu, J.J.; Lu, Y.B.; Zhang, Z.Q.; Yang, R.F.; et al. Repression of P66Shc Expression by SIRT1 Contributes to the Prevention of Hyperglycemia-Induced Endothelial Dysfunction. Circ. Res. 2011, 109, 639–648. [Google Scholar] [CrossRef]
- Cutolo, M.; Soldano, S.; Contini, P.; Sulli, A.; Seriolo, B.; Montagna, P.; Brizzolara, R. Intracellular NF-KκB Decrease and IKBaα Increase in Human Macrophages Following CTLA4-Ig Treatment. Clin. Exp. Rheumatol. 2013, 31, 943–946. [Google Scholar] [PubMed]
- Zhang, R.; Chen, H.Z.; Liu, J.J.; Jia, Y.Y.; Zhang, Z.Q.; Yang, R.F.; Zhang, Y.; Xu, J.; Wei, Y.S.; Liu, D.P.; et al. SIRT1 Suppresses Activator Protein-1 Transcriptional Activity and Cyclooxygenase-2 Expression in Macrophages. J. Biol. Chem. 2010, 285, 7097–7110. [Google Scholar] [CrossRef] [PubMed]
- Fry, J.L.; Al Sayah, L.; Weisbrod, R.M.; Van Roy, I.; Weng, X.; Cohen, R.A.; Bachschmid, M.M.; Seta, F. Vascular Smooth Muscle Sirtuin-1 Protects against Diet-Induced Aortic Stiffness. Hypertension 2016, 68, 775–784. [Google Scholar] [CrossRef]
- Pan, P.W.; Feldman, J.L.; Devries, M.K.; Dong, A.; Edwards, A.M.; Denu, J.M. Structure and Biochemical Functions of SIRT6. J. Biol. Chem. 2011, 286, 14575–14587. [Google Scholar] [CrossRef] [PubMed]
- Cardus, A.; Uryga, A.K.; Walters, G.; Erusalimsky, J.D. SIRT6 Protects Human Endothelial Cells from DNA Damage, Telomere Dysfunction, and Senescence. Cardiovasc. Res. 2013, 97, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.; Woo, Y.M.; Moon, S.; Lee, J.; Park, H.; Jang, H.; Bae, S.; Park, K.; Heo, J.H.; Choi, Y. Sirtuin 6 Deficiency Induces Endothelial Cell Senescence via Downregulation of Forkhead Box M1 Expression. Aging 2020, 12, 20946–20967. [Google Scholar] [CrossRef]
- Xu, S.; Yin, M.; Koroleva, M.; Mastrangelo, M.A.; Zhang, W.; Bai, P.; Little, P.J.; Jin, Z.G. SIRT6 Protects against Endothelial Dysfunction and Atherosclerosis in Mice. Aging 2016, 8, 1064–1082. [Google Scholar] [CrossRef]
- Wang, T.; Sun, C.; Hu, L.; Gao, E.; Li, C.; Wang, H.; Sun, D. Sirt6 Stabilizes Atherosclerosis Plaques by Promoting Macrophage Autophagy and Reducing Contact with Endothelial Cells. Biochem. Cell Biol. 2020, 98, 120–129. [Google Scholar] [CrossRef]
- Liu, H.; Chen, T.; Li, N.; Wang, S.; Bu, P. Role of SIRT3 in Angiotensin II-Induced Human Umbilical Vein Endothelial Cells Dysfunction. BMC Cardiovasc. Disord. 2015, 15, 1–7. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Pandey, A.; Xiao, L.; Arslanbaeva, L.; Sidorova, T.; Lopez, M.G.; Billings, F.T.; Verdin, E.; Auwerx, J.; Harrison, D.G.; et al. Mitochondrial Deacetylase SIRT3 Reduces Vascular Dysfunction and Hypertension While SIRT3 Depletion in Essential Hypertension Is Linked to Vascular Inflammation and Oxidative Stress. Circ. Res. 2020, 126, 439–452. [Google Scholar] [CrossRef]
- Liu, J.; Wu, X.; Wang, X.; Zhang, Y.; Bu, P.; Zhang, Q.; Jiang, F. Global Gene Expression Profiling Reveals Functional Importance of Sirt2 in Endothelial Cells under Oxidative Stress. Int. J. Mol. Sci. 2013, 14, 5633–5649. [Google Scholar] [CrossRef]
- Araki, S.; Izumiya, Y.; Rokutanda, T.; Ianni, A.; Hanatani, S.; Kimura, Y.; Onoue, Y.; Senokuchi, T.; Yoshizawa, T.; Yasuda, O.; et al. Sirt7 Contributes to Myocardial Tissue Repair by Maintaining Transforming Growth Factor-β Signaling Pathway. Circulation 2015, 132, 1081–1093. [Google Scholar] [CrossRef] [PubMed]
- Beermann, J.; Piccoli, M.T.; Viereck, J.; Thum, T. Non-Coding Rnas in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef]
- Ding, Q.; Shao, C.; Rose, P.; Zhu, Y.Z. Epigenetics and Vascular Senescence–Potential New Therapeutic Targets? Front. Pharmacol. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Menghini, R.; Casagrande, V.; Cardellini, M.; Martelli, E.; Terrinoni, A.; Amati, F.; Vasa-Nicotera, M.; Ippoliti, A.; Novelli, G.; Melino, G.; et al. MicroRNA 217 Modulates Endothelial Cell Senescence via Silent Information Regulator 1. Circulation 2009, 120, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- De Yébenes, V.G.; Briones, A.M.; Martos-Folgado, I.; Mur, S.M.; Oller, J.; Bilal, F.; González-Amor, M.; Méndez-Barbero, N.; Silla-Castro, J.C.; Were, F.; et al. Aging-Associated MiR-217 Aggravates Atherosclerosis and Promotes Cardiovascular Dysfunction. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2408–2424. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Li, J.; Chen, A.F. MicroRNA-34a Induces Endothelial Progenitor Cell Senescence and Impedes Its Angiogenesis via Suppressing Silent Information Regulator 1. Am. J. Physiol. Endocrinol. Metab. 2010, 299, 110–116. [Google Scholar] [CrossRef]
- Zhu, S.; Deng, S.; Ma, Q.; Zhang, T.; Jia, C.; Zhuo, D.; Yang, F.; Wei, J.; Wang, L.; Dykxhoorn, D.M.; et al. MicroRNA-10A* and MicroRNA-21 Modulate Endothelial Progenitor Cell Senescence via Suppressing High-Mobility Group A2. Circ. Res. 2013, 112, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.G.; Zheng, B.; Han, M.; Fang, X.M.; Li, H.X.; Miao, S.B.; Su, M.; Han, Y.; Shi, H.J.; Wen, J.K. MiR-146a and Krüppel-like Factor 4 Form a Feedback Loop to Participate in Vascular Smooth Muscle Cell Proliferation. EMBO Rep. 2011, 12, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Cordes, K.R.; Sheehy, N.T.; White, M.; Berry, E.; Sarah, U.; Muth, A.N.; Lee, T.; Miano, J.M.; Ivey, K.N. MiR-145 and MiR-143 Regulate Smooth Muscle Cell Fate Decisions. Nature 2010, 460, 705–710. [Google Scholar] [CrossRef]
- Farina, F.M.; Hall, I.F.; Serio, S.; Zani, S.; Climent, M.; Salvarani, N.; Carullo, P.; Civilini, E.; Condorelli, G.; Elia, L.; et al. MiR-128-3p Is a Novel Regulator of Vascular Smooth Muscle Cell Phenotypic Switch and Vascular Diseases. Circ. Res. 2020, 126, e120–e135. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chen, Q.; He, S.; Yang, M.; Maguire, E.M.; An, W.; Afzal, T.A.; Luong, L.A.; Zhang, L.; Xiao, Q. MiR-22 Is a Novel Mediator of Vascular Smooth Muscle Cell Phenotypic Modulation and Neointima Formation. Circulation 2018, 137, 1824–1841. [Google Scholar] [CrossRef]
- Olivieri, F.; Rippo, M.R.; Monsurrò, V.; Salvioli, S.; Capri, M.; Procopio, A.D.; Franceschi, C. MicroRNAs Linking Inflamm-Aging, Cellular Senescence and Cancer. Ageing Res. Rev. 2013, 12, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Choi, S.; Kim, S.; Kim, J.; Lee, D.K.; Park, W.; Kim, T.; Jung, J.; Hwang, J.Y.; Won, M.H.; et al. NF-ΚB-Responsive MiR-155 Induces Functional Impairment of Vascular Smooth Muscle Cells by Downregulating Soluble Guanylyl Cyclase. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nazari-Jahantigh, M.; Wei, Y.; Noels, H.; Akhtar, S.; Zhou, Z.; Koenen, R.R.; Heyll, K.; Gremse, F.; Kiessling, F.; Grommes, J.; et al. MicroRNA-155 Promotes Atherosclerosis by Repressing Bcl6 in Macrophages. J. Clin. Investig. 2012, 122, 4190–4202. [Google Scholar] [CrossRef] [PubMed]
- Uryga, A.K.; Bennett, M.R. Ageing Induced Vascular Smooth Muscle Cell Senescence in Atherosclerosis. J. Physiol. 2016, 594, 2115–2124. [Google Scholar] [CrossRef]
- Fyhrquist, F.; Saijonmaa, O.; Strandberg, T. The Roles of Senescence and Telomere Shortening in Cardiovascular Disease. Nat. Rev. Cardiol. 2013, 10, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Samuel, I.; Bloom, A.J.D. The Role of Senescence, Telomere Dysfunction and Shelterin in Vascular Aging. Microcirculation 2019, 26, e12487. [Google Scholar] [CrossRef]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular Smooth Muscle Cells Undergo Telomere-Based Senescence in Human Atherosclerosis: Effects of Telomerase and Oxidative Stress. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef]
- Bhayadia, R.; Schmidt, B.M.W.; Melk, A.; Hömme, M. Senescence-Induced Oxidative Stress Causes Endothelial Dysfunction. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2016, 71, 161–169. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial Cell Senescence in Human Atherosclerosis: Role of Telomere in Endothelial Dysfunction. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Uryga, A.K.; Reinhold, J.; Figg, N.; Baker, L.; Finigan, A.; Gray, K.; Kumar, S.; Clarke, M.; Bennett, M. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation 2015, 132, 1909–1919. [Google Scholar] [CrossRef]
- Ogami, M.; Ikura, Y.; Ohsawa, M.; Matsuo, T.; Kayo, S.; Yoshimi, N.; Hai, E.; Shirai, N.; Ehara, S.; Komatsu, R.; et al. Telomere Shortening in Human Coronary Artery Diseases. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 546–550. [Google Scholar] [CrossRef]
- Nzietchueng, R.; Elfarra, M.; Nloga, J.; Labat, C.; Carteaux, J.P.; Maureira, P.; Lacolley, P.; Villemot, J.P.; Benetos, A. Telomere Length in Vascular Tissues from Patients with Atherosclerotic Disease. J. Nutr. Health Aging 2011, 15, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Raymond, A.R.; Norton, G.R.; Woodiwiss, A.J.; Brooksbank, R.L. Impact of Gender and Menopausal Status on Relationships between Biological Aging, as Indexed by Telomere Length, and Aortic Stiffness. Am. J. Hypertens. 2015, 28, 623–630. [Google Scholar] [CrossRef]
- McDonnell, B.J.; Yasmin; Butcher, L.; Cockcroft, J.R.; Wilkinson, I.B.; Erusalimsky, J.D.; McEniery, C.M. The Age-Dependent Association between Aortic Pulse Wave Velocity and Telomere Length. J. Physiol. 2017, 595, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Jeanclos, E.; Schork, N.J.; Kyvik, K.O.; Kimura, M.; Skurnick, J.H.; Aviv, A. Telomere Length Inversely Correlates with Pulse Pressure and Is Highly Familial. Hypertension 2000, 36, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Chen, A.F.; Wang, H.Z.; Xie, L.Y.; Sui, K.X.; Zhang, Q.Y. Association of Shorter Mean Telomere Length with Large Artery Stiffness in Patients with Coronary Heart Disease. Aging Male 2011, 14, 27–32. [Google Scholar] [CrossRef] [PubMed]
- van der Harst, P.; van der Steege, G.; de Boer, R.A.; Voors, A.A.; Hall, A.S.; Mulder, M.J.; van Gilst, W.H.; van Veldhuisen, D.J. Telomere Length of Circulating Leukocytes Is Decreased in Patients With Chronic Heart Failure. J. Am. Coll. Cardiol. 2007, 49, 1459–1464. [Google Scholar] [CrossRef]
- Brouilette, S.; Singh, R.K.; Thompson, J.R.; Goodall, A.H.; Samani, N.J. White Cell Telomere Length and Risk of Premature Myocardial Infarction. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 842–846. [Google Scholar] [CrossRef] [PubMed]
- Benetos, A.; Gardner, J.P.; Zureik, M.; Labat, C.; Xiaobin, L.; Adamopoulos, C.; Temmar, M.; Bean, K.E.; Thomas, F.; Aviv, A. Short Telomeres Are Associated with Increased Carotid Atherosclerosis in Hypertensive Subjects. Hypertension 2004, 43, 182–185. [Google Scholar] [CrossRef]
- Haycock, P.C.; Heydon, E.E.; Kaptoge, S.; Butterworth, A.S.; Thompson, A.; Willeit, P. Leucocyte Telomere Length and Risk of Cardiovascular Disease: Systematic Review and Meta- Analysis. BMJ 2014, 349, g4227. [Google Scholar] [CrossRef]
- D’Mello, M.J.J.; Ross, S.A.; Briel, M.; Anand, S.S.; Gerstein, H.; Paré, G. Association between Shortened Leukocyte Telomere Length and Cardiometabolic Outcomes: Systematic Review and Meta-Analysis. Circ. Cardiovasc. Genet. 2015, 8, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Farzaneh-Far, R.; Cawthon, R.M.; Na, B.; Browner, W.S.; Schiller, N.B.; Whooley, M.A. Prognostic Value of Leukocyte Telomere Length in Patients with Stable Coronary Artery Disease: Data from the Heart and Soul Study (R1). Arter. Thromb. Vasc. Biol. 2008, 28, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular Senescence in Ageing: From Mechanisms to Therapeutic Opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and Aging: Causes, Consequences, and Therapeutic Avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Schmeer, C.; Kretz, A.; Wengerodt, D.; Stojiljkovic, M.; Witte, O.W. Dissecting Aging and Senescence-Current Concepts and Open Lessons. Cells 2019, 8, 1446. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Morton, J.P.; Athineos, D.; Kang, T.; Lasitschka, F.; Andrulis, M.; Pascual, G.; et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat. Cell Biol. 2014, 15, 978–990. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Machado-Oliveira, G.; Ramos, C.; Marques, A.R.; Vieira, O.V. Cell Senescence, Multiple Organelle Dysfunction and Atherosclerosis. Cells 2020, 9, 2146. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; Van Deursen, J.M. Senescent Intimal Foam Cells Are Deleterious at All Stages of Atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Gardner, S.E.; Humphry, M.; Bennett, M.R.; Clarke, M.C.H. Senescent Vascular Smooth Muscle Cells Drive Inflammation through an Interleukin-1α-Dependent Senescence-Associated Secretory Phenotype. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1963–1974. [Google Scholar] [CrossRef]
- Alique, M.; Ruíz-torres, M.P.; Bodega, G.; Noci, M.V.; Bohórquez, L.; Luna, C.; Luque, R.; Carmona, A.; Ramírez, R. Microvesicles from the Plasma of Elderly Subjects and from Senescent Endothelial Cells Promote Vascular Calcification. Aging 2017, 9, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; Zhang, X.; Kumar, A.; Atkinson, E.J.; Zhu, Y.; Jachim, S.; Mazula, D.L.; Brown, A.K.; Berning, M.; Aversa, Z.; et al. The Senescence-Associated Secretome as an Indicator of Age and Medical Risk. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ Heel of Senescent Cells: From Transcriptome to Senolytic Drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Lesniewski, L.A.; Seals, D.R.; Walker, A.E.; Henson, G.D.; Blimline, M.W.; Trott, D.W.; Bosshardt, G.C.; LaRocca, T.J.; Lawson, B.R.; Zigler, M.C.; et al. Dietary Rapamycin Supplementation Reverses Age-Related Vascular Dysfunction and Oxidative Stress, While Modulating Nutrient-Sensing, Cell Cycle, and Senescence Pathways. Aging Cell 2017, 16, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Mattison, J.A.; Wang, M.; Bernier, M.; Zhang, J.; Park, S.S.; Maudsley, S.; An, S.S.; Santhanam, L.; Martin, B.; Faulkner, S.; et al. Resveratrol Prevents High Fat/Sucrose Diet-Induced Central Arterial Wall Inflammation and Stiffening in Nonhuman Primates Julie. Cell Metab. 2014, 20, 183–190. [Google Scholar] [CrossRef]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and Regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef]
- Madeo, F.; Zimmermann, A.; Maiuri, M.C.; Kroemer, G. Essential Role for Autophagy in Life Span Extension. J. Clin. Investig. 2015, 125, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, M.; Sedej, S.; Carmona-Gutierrez, D.; Madeo, F.; Kroemer, G. Autophagy in Cardiovascular Aging. Circ. Res. 2018, 123, 803–824. [Google Scholar] [CrossRef]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a Promoter of Longevity: Insights from Model Organisms. Nat. Rev. Mol. Cell Biol. 2019, 19, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Nussenzweig, S.C.; Verma, S.; Finkel, T. The Role of Autophagy in Vascular Biology. Circ. Res. 2015, 116, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed Interplay between Autophagy and Inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy Impairment with Lysosomal and Mitochondrial Dysfunction Is an Important Characteristic of Oxidative Stress-Induced Senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef]
- Larocca, T.J.; Henson, G.D.; Thorburn, A.; Sindler, A.L.; Pierce, G.L.; Seals, D.R. Translational Evidence That Impaired Autophagy Contributes to Arterial Ageing. J. Physiol. 2012, 590, 3305–3316. [Google Scholar] [CrossRef]
- Bharath, L.P.; Cho, J.M.; Park, S.K.; Ruan, T.; Li, Y.; Mueller, R.; Bean, T.; Reese, V.; Richardson, R.S.; Cai, J.; et al. Endothelial Cell Autophagy Maintains Shear-Stress-Induced Nitric Oxide Generation via Glycolysis-Dependent Purinergic Signaling to ENOS. Arter. Thromb Vasc Biol. 2017, 37, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; da Costa Martins, P.A.; Bitsch, N.; Pintelon, I.; de Meyer, G.R.Y.; Martinet, W.; Schrijvers, D.M. Defective Autophagy in Vascular Smooth Muscle Cells Accelerates Senescence and Promotes Neointima Formation and Atherogenesis. Autophagy 2015, 11, 2014–2032. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Oba, M.; Suzuki, M.; Takahashi, A.; Yamamuro, T.; Fujiwara, M.; Ikenaka, K.; Minami, S.; Tabata, N.; Yamamoto, K.; et al. Suppression of Autophagic Activity by Rubicon Is a Signature of Aging. Nat. Commun. 2019, 10, 847. [Google Scholar] [CrossRef] [PubMed]
- Donato, A.J.; Walker, A.E.; Magerko, K.A.; Bramwell, R.C.; Black, A.D.; Henson, G.D.; Lawson, B.R.; Lesniewski, L.A.; Seals, D.R. Life-Long Caloric Restriction Reduces Oxidative Stress and Preserves Nitric Oxide Bioavailability and Function in Arteries of Old Mice. Aging Cell 2013, 12, 772–783. [Google Scholar] [CrossRef]
- Martens, C.R.; Seals, D.R. Practical Alternatives to Chronic Caloric Restriction for Optimizing Vascular Function with Ageing. J. Physiol. 2016, 594, 7177–7195. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| MMP and TIMP Class | Overexpression | Deficiency |
|---|---|---|
| MMP-2 | (a) Increased TGF-β1 and SMAD signaling leading to
(b) Endothelial dysfunction due to decreased NO production [83] (c) Increased intima-media thickening and vascular fibrosis [70] | (a) Reduced elastin fiber degeneration and collagen deposition [84] (b) Enhanced eNOS activation [85] |
| MMP-3 | Apoptosis of ECs [86] | Accelerated plaque growth rate with increased macrophage and decreased VSMC composition [87,88] |
| MMP-7 | (a) Atherosclerosis and plaque instability through collagen and matrix modulation and cleavage of apolipoprotein A-IV [89,90] (b) VSMC apoptosis through cleavage of n-cadherin [91] (c) Vasoconstriction through shedding of the HB-EGF and subsequent activation of EGFR [92] | Increased accumulation of VSMCs within the atherosclerotic plaques |
| MMP-9 | (a) Apoptosis in ECs through cleavage of PAR-1 [93] (b) Migration of VSMCs and contribution to atherosclerotic plaque instability and intraplaque hemorrhage [94] | (a) Reduction in size of atherosclerotic lesions and plaque burden [95,96] (b) Inhibition of VSMCs’ migration and restriction of vascular remodeling [97] (c) Prevention of formation of abdominal aortic aneurysms [98] |
| TIMP-1 | (a) Reduction in intimal formation through decreased collagen deposition and increased elastin accumulation [99] (b) Protection against aneurysm formation and rupture through prevention of elastin degradation [100] | |
| TIMP-2 | Suppression of atherosclerotic plaque progression through inhibition of migration and apoptosis of macrophages and foam cells [101] | |
| TIMP-3 | (a) Reduced intimal formation through apoptosis of VSMC [102] (b) Atherosclerosis through inhibition of EC inflammation and VSMC proliferation and migration [103] (c) Accumulation of inflammatory monocytes/macrophages within the vascular wall [104] | (a) Enhanced inflammation and atherosclerosis through increased (b) Adverse vascular remodeling and vascular aneurysm formation through loss of elastic lamellae and inflammation [105] |
| Recruitment of EC migration [139] |
| Delay of the aging and dysfunction of EPCs [140] |
| Inhibition of aging of ECs by binding the PAI-1 promoter and by deacetylation of histone H4K16 [141] |
| Promotion of endothelial KLF2 expression which enables transition of ECs to a “vaso-protective” state [141] |
| Mitigation of hyperglycaemia-induced endothelial dysfunction due to ROS production by inhibiting vascular p66Shc gene transcription [142] |
| Alleviation of oxidative stress and inflammation by the inhibition of NF-κB signaling pathway [143] |
| Activation of eNOS and promotion of NO production by the deacetylation of eNOS on Lys496 and Lys506 [132] |
| Reduction of COX-2 expression through downregulation of transcription factor AP-1 in macrophages [144] |
| Reduction of arterial remodeling and stiffness by alleviation of oxidative stress in VSMCs [145] |
| Deacetylation and activation of the FOXO 1, 3, and 4 transcription factors leading to the expression of several antioxidant genes [136] |
| miR-10A | Propagation of senescence of EPCs through suppression of the high-mobility group A2 molecule [160] |
| miR-21 | Propagation of senescence of EPCs through suppression of the high-mobility group A2 molecule [160] |
| miR-22 | Inhibition of VSMC proliferation and migration and neointima formation 164 |
| miR-34a | Suppression of EC proliferation and promotion of EC senescence in part through Sirt1 inhibition [159] Impairment of EPC-mediated angiogenesis through suppression of silent information regulator 1 [159] |
| miR-126 | Reduction of endothelial inflammation through inhibition of VCAM-1 expression [165] |
| miR-128 | Reduction of VSMC proliferation, migration, and contractility [163] |
| miR-143 | Inhibition of VSMC proliferation through targeting the transcription factor Elk-1 [162] |
| miR-145 | Inhibition of VSMC proliferation through targeting the transcription factor myocardin [162] |
| miR-146a | Promotion of VSMC proliferation and vascular neointimal hyperplasia through targeting KLF4 [161] |
| miR-155 | Promotion of atherosclerosis through repression of macrophage BCL6 expression [167] Endothelial dysfunction and vasoconstriction through downregulation of eNOS and sGCβ1 expression [166] |
| miR-217 | Acceleration of EC senescence, endothelial dysfunction and development of atherosclerosis through Sirt1 downregulation [157,158] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gkaliagkousi, E.; Lazaridis, A.; Dogan, S.; Fraenkel, E.; Tuna, B.G.; Mozos, I.; Vukicevic, M.; Yalcin, O.; Gopcevic, K. Theories and Molecular Basis of Vascular Aging: A Review of the Literature from VascAgeNet Group on Pathophysiological Mechanisms of Vascular Aging. Int. J. Mol. Sci. 2022, 23, 8672. https://doi.org/10.3390/ijms23158672
Gkaliagkousi E, Lazaridis A, Dogan S, Fraenkel E, Tuna BG, Mozos I, Vukicevic M, Yalcin O, Gopcevic K. Theories and Molecular Basis of Vascular Aging: A Review of the Literature from VascAgeNet Group on Pathophysiological Mechanisms of Vascular Aging. International Journal of Molecular Sciences. 2022; 23(15):8672. https://doi.org/10.3390/ijms23158672
Chicago/Turabian StyleGkaliagkousi, Eugenia, Antonios Lazaridis, Soner Dogan, Emil Fraenkel, Bilge Guvenc Tuna, Ioana Mozos, Milica Vukicevic, Ozlem Yalcin, and Kristina Gopcevic. 2022. "Theories and Molecular Basis of Vascular Aging: A Review of the Literature from VascAgeNet Group on Pathophysiological Mechanisms of Vascular Aging" International Journal of Molecular Sciences 23, no. 15: 8672. https://doi.org/10.3390/ijms23158672
APA StyleGkaliagkousi, E., Lazaridis, A., Dogan, S., Fraenkel, E., Tuna, B. G., Mozos, I., Vukicevic, M., Yalcin, O., & Gopcevic, K. (2022). Theories and Molecular Basis of Vascular Aging: A Review of the Literature from VascAgeNet Group on Pathophysiological Mechanisms of Vascular Aging. International Journal of Molecular Sciences, 23(15), 8672. https://doi.org/10.3390/ijms23158672