
Cadmium Toxicity Is Regulated by Peroxisome Proliferator-Activated Receptor δ in Human Proximal Tubular Cells

Abstract
1. Introduction
2. Results
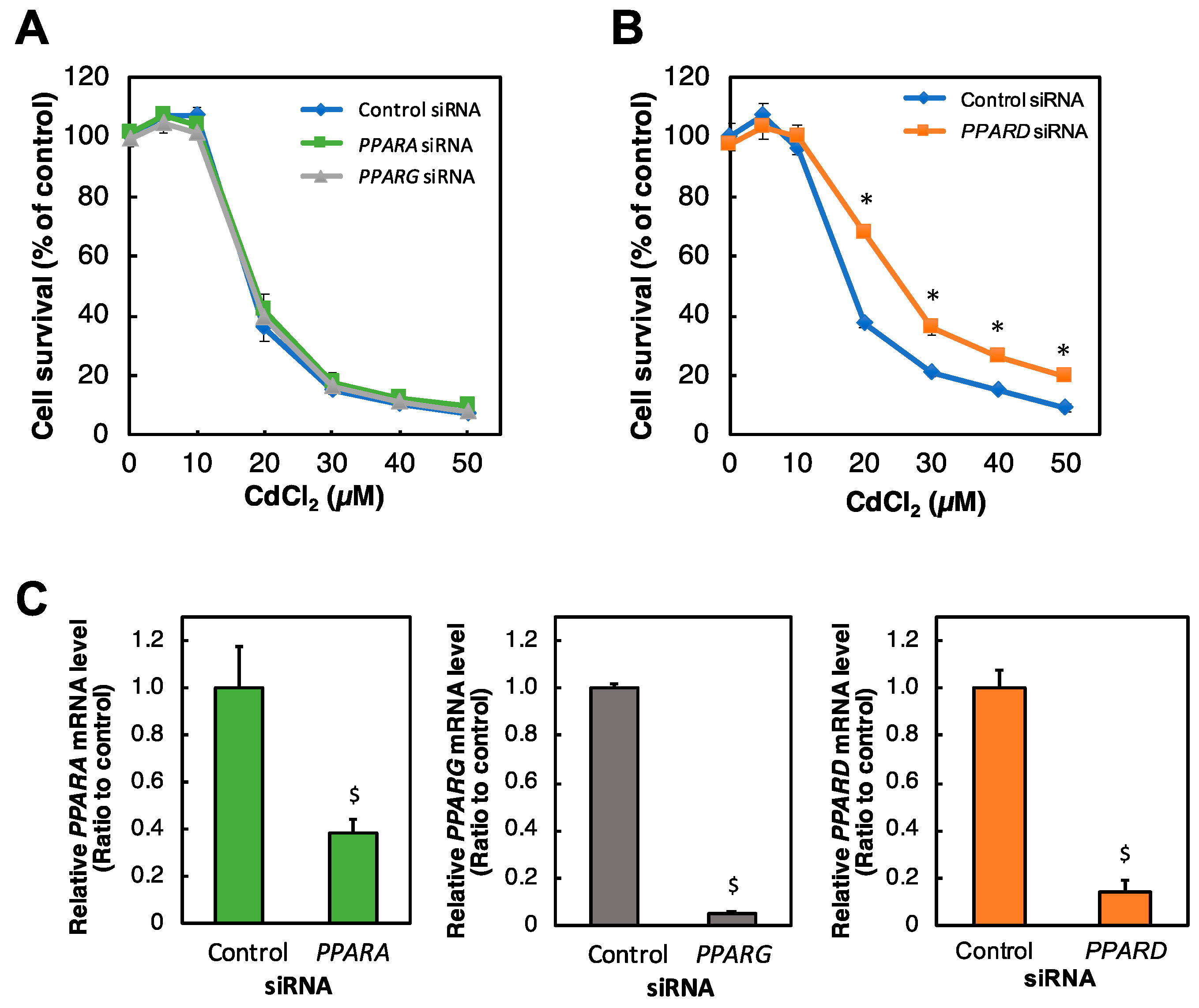
2.1. Identification of PPAR Affecting Cd Toxicity in HK-2 Cells
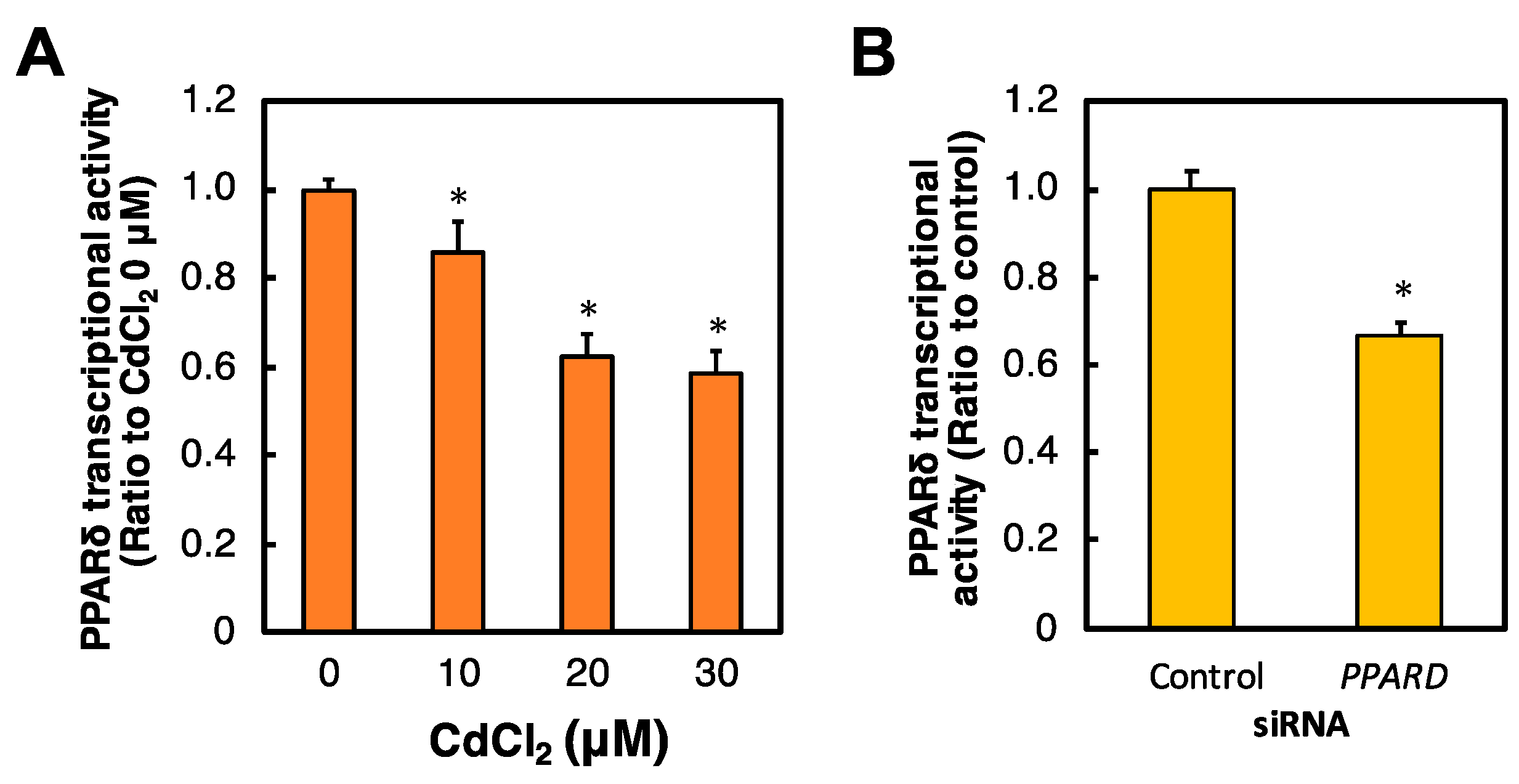
2.2. Effects of Cd and PPARD Knockdown on the PPARδ Transcriptional Activity in HK-2 Cells
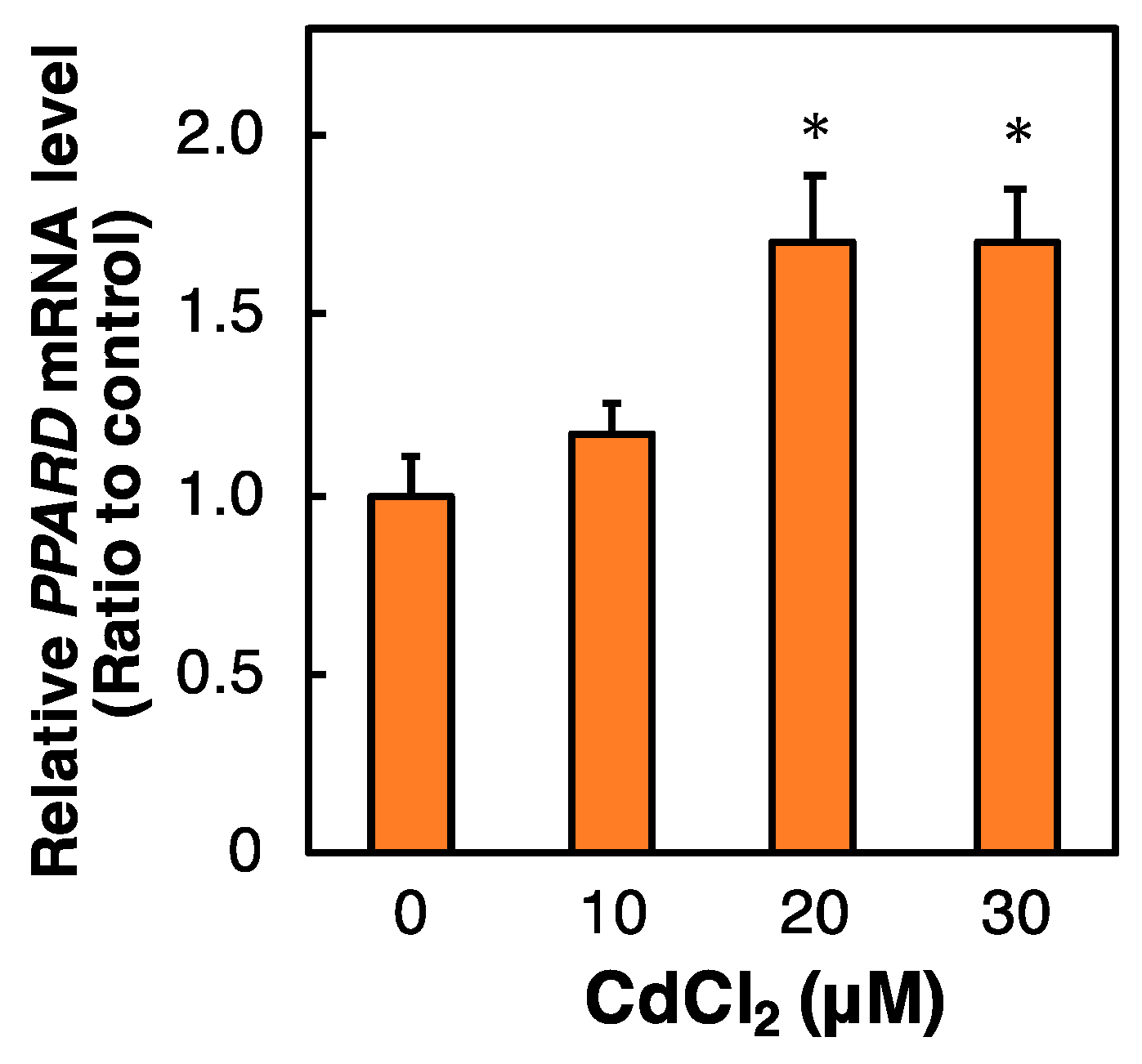
2.3. Effect of Cd on the PPARD mRNA Level in HK-2 Cells
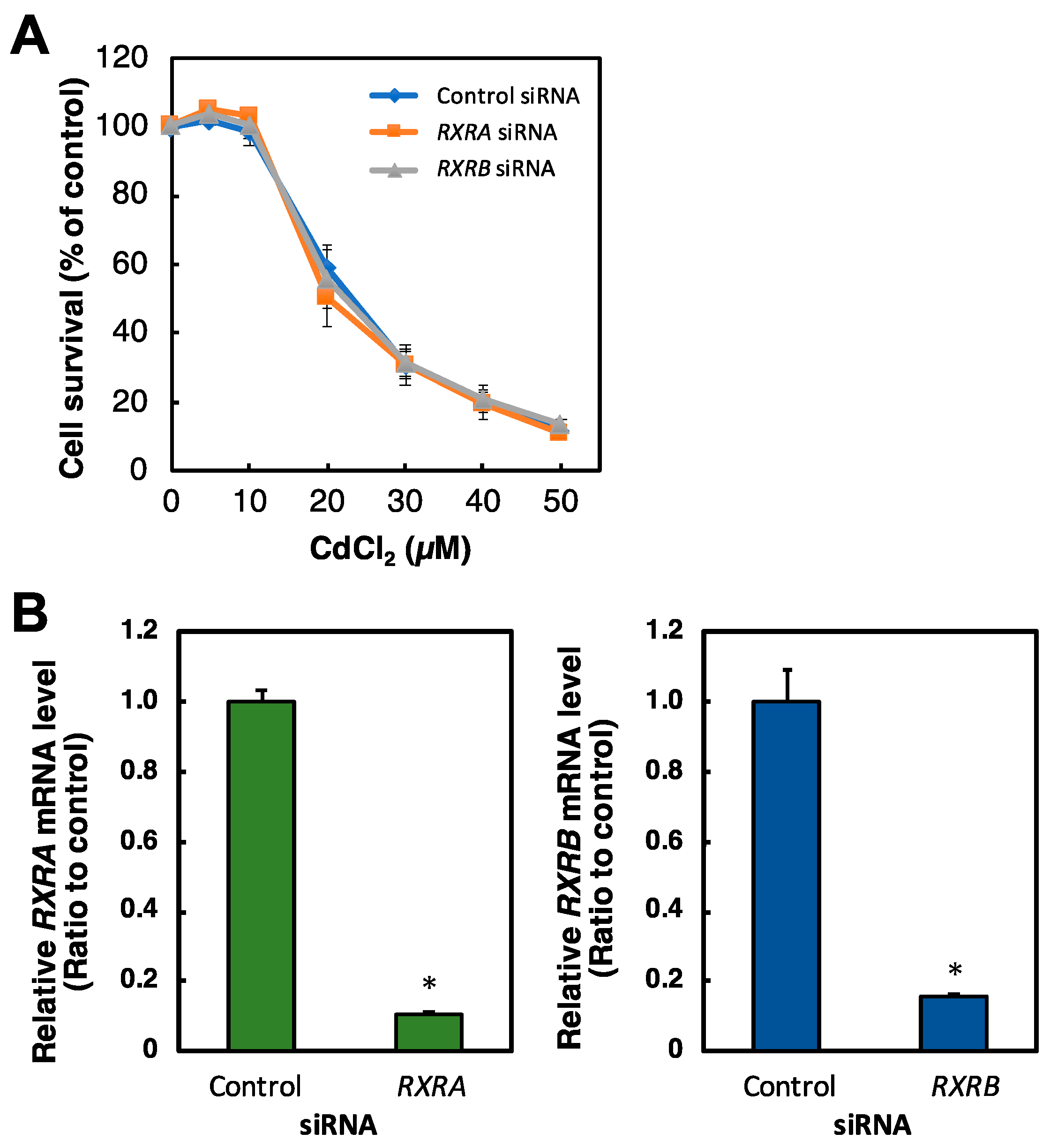
2.4. Effect of RXR Knockdown on the Viability of HK-2 Cells Treated with Cd
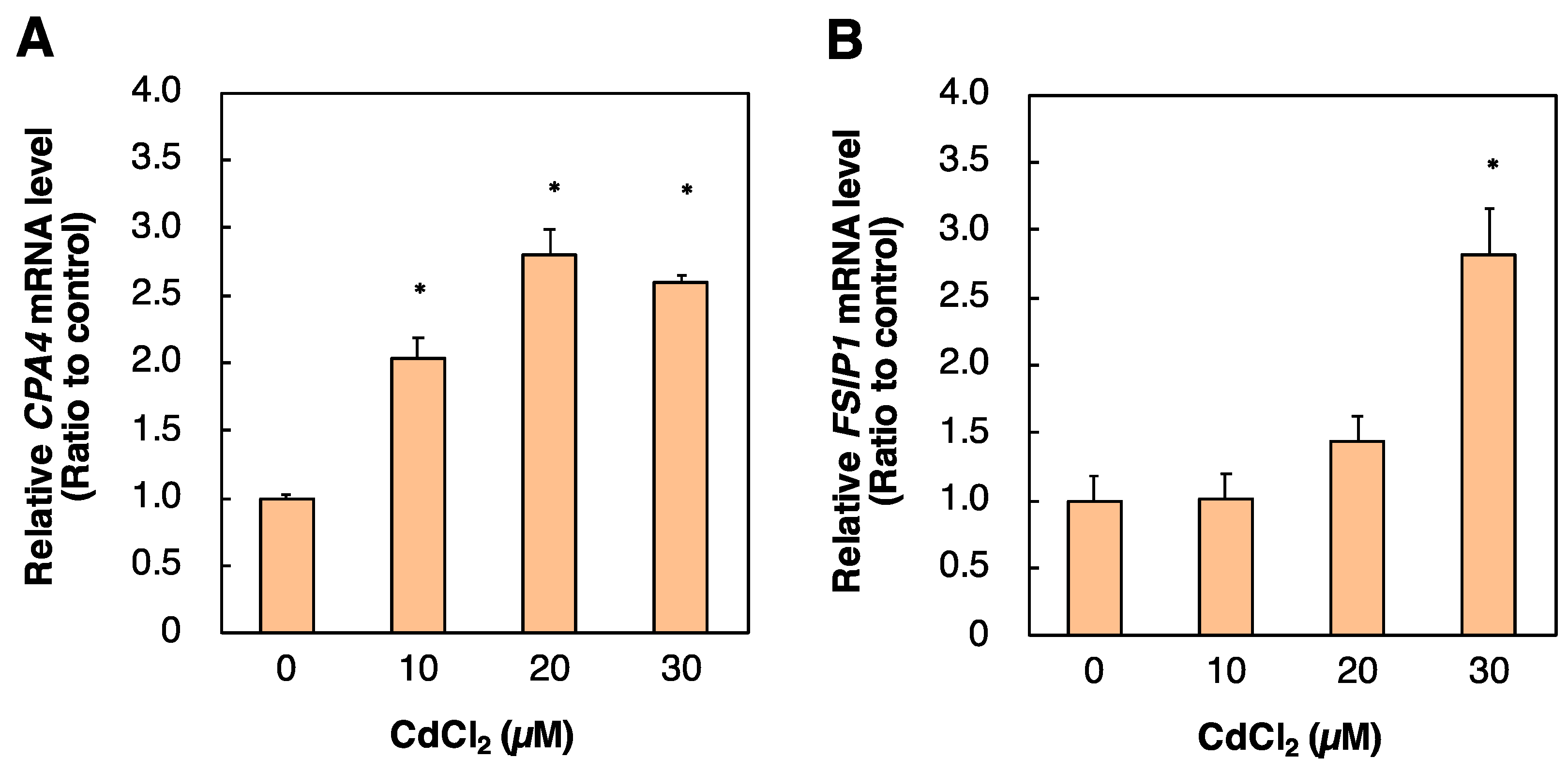
2.5. Identification of Genes Regulated by PPARD Knockdown in HK-2 Cells
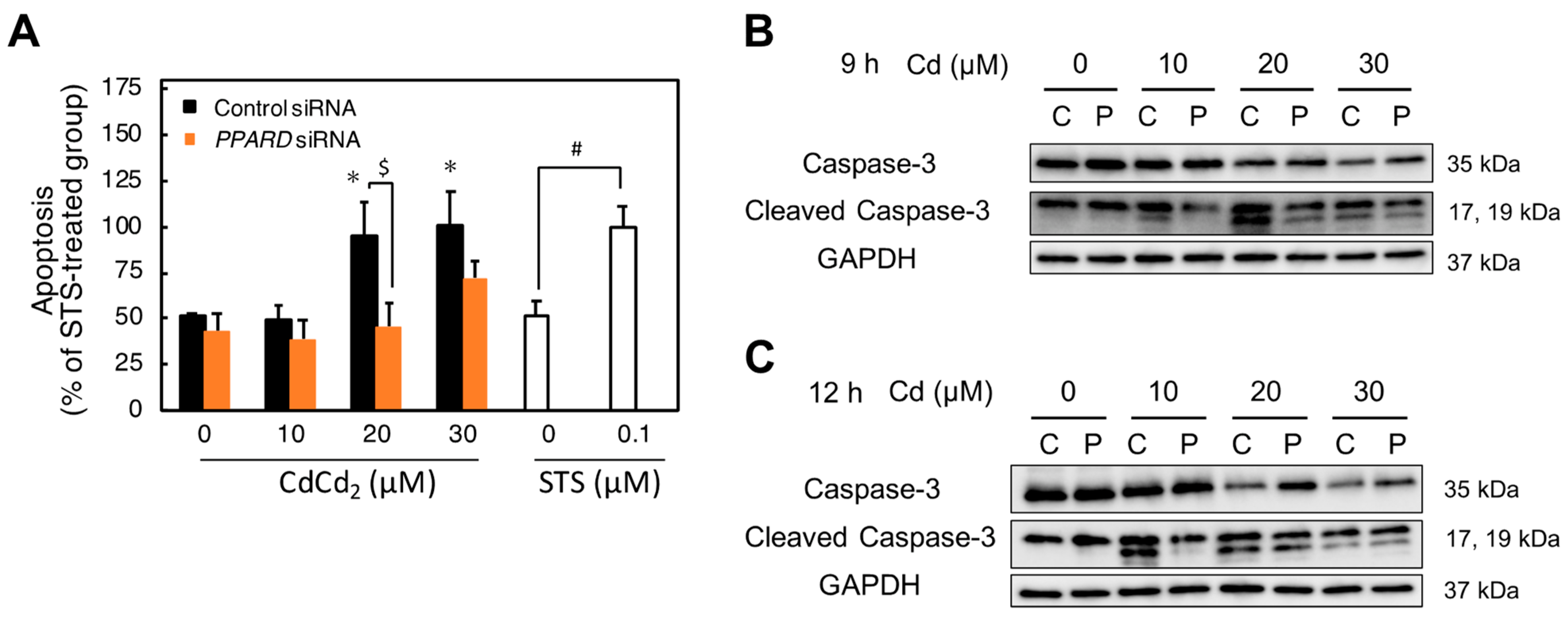
2.6. Involvement of PPARD Knockdown in Cd-Induced Apoptosis
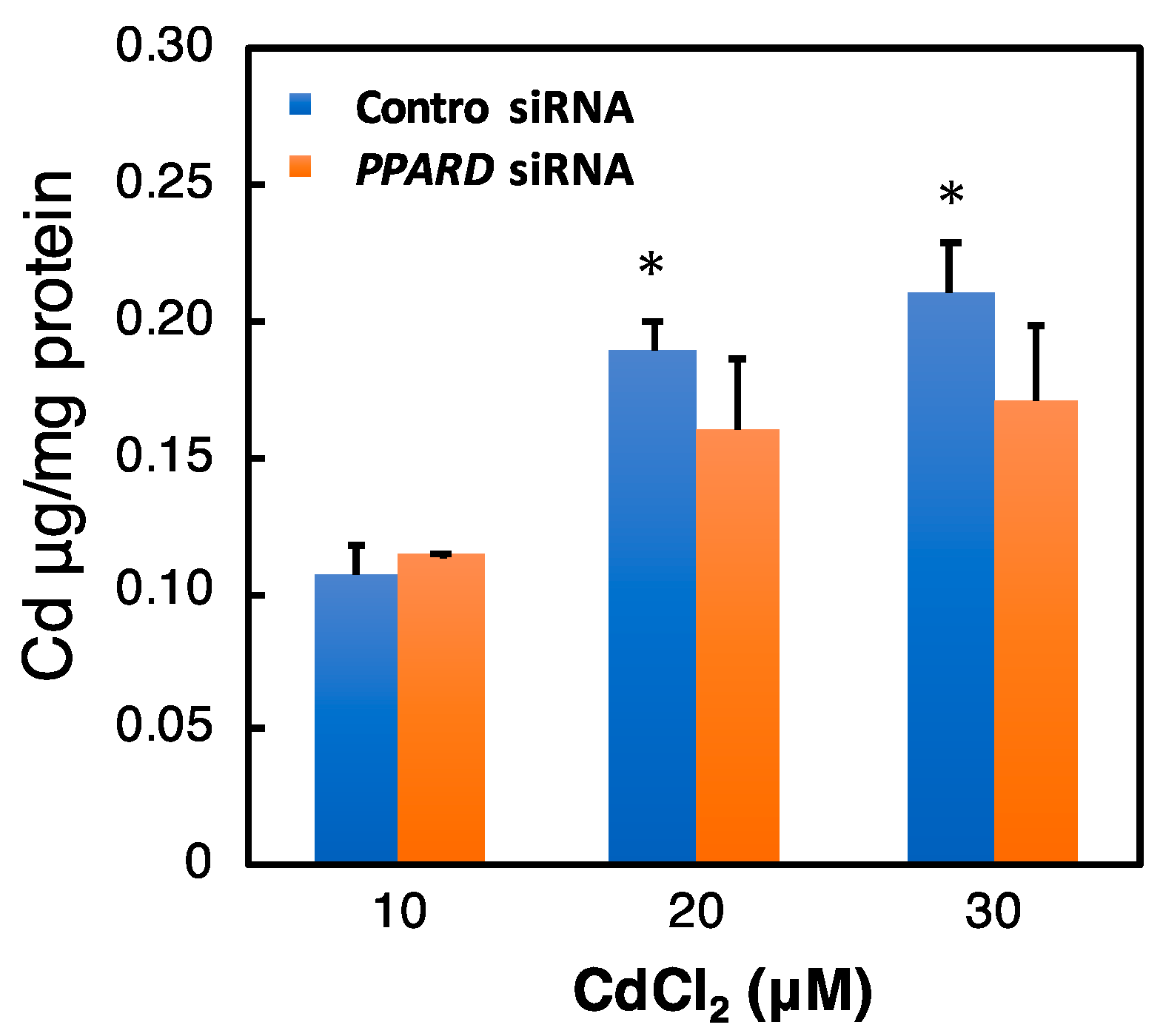
2.7. Effect of PPARD Knockdown on the Intracellular Cd Concentration
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Small Interfering RNA (siRNA) Transfection
4.3. Cell Survival Rate
4.4. RNA Extraction
4.5. DNA Microarray Analysis
4.6. Real-Time Reverse Transcription (RT)-PCR
4.7. Western Blot Analysis
4.8. Nuclear Protein Extraction
4.9. PPARδ Transcriptional Activity Assay
4.10. Apoptosis Assay
4.11. Determination of Cd Content
4.12. Statistical Analysis
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Åkesson, A.; Barregard, L.; Bergdahl, I.A.; Nordberg, G.F.; Nordberg, M.; Skerfving, S. Non-Renal Effects and the Risk Assessment of Environmental Cadmium Exposure. Environ. Health Perspect. 2014, 122, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Järup, L.; Åkesson, A. Current status of cadmium as an environmental health problem. Toxicol. Appl. Pharmacol. 2009, 238, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Aoshima, K. Itai-itai disease: Lessons from the investigations of environmental epidemiology conducted in the 1970’s, with special reference to the studies of the Toyama Institute of Health. Nihon Eiseigaku Zasshi 2017, 72, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Lee, J.-Y.; Tokumoto, M.; Satoh, M. Cadmium Renal Toxicity via Apoptotic Pathways. Biol. Pharm. Bull. 2012, 35, 1892–1897. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Tokumoto, M.; Satoh, M. Novel Mechanisms of Cadmium-Induced Toxicity in Renal Cells. In Cadmium Toxicity: New Aspects in Human Disease, Rice Contamination, and Cytotoxicity; Himeno, S., Aoshima, K., Eds.; Springer: Singapore, 2019; pp. 153–162. [Google Scholar]
- Tokumoto, M.; Lee, J.-Y.; Satoh, M. Transcription Factors and Downstream Genes in Cadmium Toxicity. Biol. Pharm. Bull. 2019, 42, 1083–1088. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Tokumoto, M.; Fujiwara, Y.; Hasegawa, T.; Seko, Y.; Shimada, A.; Satoh, M. Accumulation of p53 via down-regulation of UBE2D family genes is a critical pathway for cadmium-induced renal toxicity. Sci. Rep. 2016, 6, 21968. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Tokumoto, M.; Hwang, G.-W.; Lee, M.-Y.; Satoh, M. Identification of ARNT-regulated BIRC3 as the target factor in cadmium renal toxicity. Sci. Rep. 2017, 7, 17287. [Google Scholar] [CrossRef]
- Lee, J.; Tokumoto, M.; Satoh, M. Cadmium toxicity mediated by the inhibition of SLC2A4 expression in human proximal Tubule cells. FASEB J. 2020, 35, e21236. [Google Scholar] [CrossRef]
- Tokumoto, M.; Lee, J.-Y.; Fujiwara, Y.; Satoh, M. Alteration of DNA binding activity of transcription factors in NRK-52E rat proximal tubular cells treated with cadmium. J. Toxicol. Sci. 2014, 39, 735–738. [Google Scholar] [CrossRef][Green Version]
- Burns, K.A.; Heuvel, J.P.V. Modulation of PPAR activity via phosphorylation. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2007, 1771, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Desvergne, B.; Wahli, W. Peroxisome Proliferator-Activated Receptors: Nuclear Control of Metabolism. Endocr. Rev. 1999, 20, 649–688. [Google Scholar] [CrossRef]
- Juge-Aubry, C.; Pernin, A.; Favez, T.; Burger, A.G.; Wahli, W.; Meier, C.A.; Desvergne, B. DNA Binding Properties of Peroxisome Proliferator-activated Receptor Subtypes on Various Natural Peroxisome Proliferator Response Elements. Importance of the 5’-flanking region. J. Biol. Chem. 1997, 272, 25252–25259. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N. The PPAR regulatory system in cardiac physiology and disease. Cardiovasc. Res. 2007, 73, 269–277. [Google Scholar] [CrossRef]
- Liu, Y.; Colby, J.K.; Zuo, X.; Jaoude, J.; Wei, D.; Shureiqi, I. The Role of PPAR-δ in Metabolism, Inflammation, and Cancer: Many Characters of a Critical Transcription Factor. Int. J. Mol. Sci. 2018, 19, 3339. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Gonzalez, F.J. Sorting out the functional role(s) of peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta) in cell proliferation and cancer. Biochim. Biophys. Acta 2009, 1796, 230–241. [Google Scholar] [PubMed]
- Wang, Y.X.; Zhang, C.L.; Yu, R.T.; Cho, H.K.; Nelson, M.C.; Bayuga-Ocampo, C.R.; Ham, J.; Kang, H.; Evans, R.M. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004, 2, e294. [Google Scholar] [CrossRef]
- Berchtold, L.A.; Miani, M.; Diep, T.A.; Madsen, A.N.; Cigliola, V.; Colli, M.; Krivokapic, J.M.; Pociot, F.; Eizirik, D.L.; Meda, P.; et al. Pannexin-2-deficiency sensitizes pancreatic β-cells to cytokine-induced apoptosis in vitro and impairs glucose tolerance in vivo. Mol. Cell. Endocrinol. 2017, 448, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Egaña, I.; Kaito, H.; Nitzsche, A.; Becker, L.; Ballester-Lopez, C.; Niaudet, C.; Petkova, M.; Liu, W.; Vanlandewijck, M.; Vernaleken, A.; et al. Female mice lacking Pald1 exhibit endothelial cell apoptosis and emphysema. Sci. Rep. 2017, 7, 15453. [Google Scholar] [CrossRef]
- Fu, Y.; Su, L.; Cai, M.; Yao, B.; Xiao, S.; He, Q.; Xu, L.; Yang, L.; Zhao, C.; Wan, T.; et al. Downregulation of CPA4 inhibits non small–cell lung cancer growth by suppressing the AKT/c-MYC pathway. Mol. Carcinog. 2019, 58, 2026–2039. [Google Scholar] [CrossRef]
- Johnson, J.D.; Han, Z.; Otani, K.; Ye, H.; Zhang, Y.; Wu, H.; Horikawa, Y.; Misler, S.; Bell, G.I.; Polonsky, K.S. RyR2 and Calpain-10 Delineate a Novel Apoptosis Pathway in Pancreatic Islets. J. Biol. Chem. 2004, 279, 24794–24802. [Google Scholar] [CrossRef]
- Kim, N.; Yu, L.; Dawe, R.; Petyuk, V.A.; Gaiteri, C.; De Jager, P.L.; Schneider, J.A.; Arfanakis, K.; Bennett, D.A. Microstructural changes in the brain mediate the association of AK4, IGFBP5, HSPB2, and ITPK1 with cognitive decline. Neurobiol. Aging 2019, 84, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, H.; Sun, L.; Zhao, D.; Liu, P.; Yan, M.; Zaidi, N.; Izadmehr, S.; Gupta, A.; Abu-Amer, W.; et al. FSIP1 binds HER2 directly to regulate breast cancer growth and invasiveness. Proc. Natl. Acad. Sci. USA 2017, 114, 7683–7688. [Google Scholar] [CrossRef] [PubMed]
- Poungvarin, N.; Lee, J.K.; Yechoor, V.K.; Li, M.V.; Assavapokee, T.; Suksaranjit, P.; Thepsongwajja, J.J.; Saha, P.K.; Oka, K.; Chan, L. Carbohydrate response element-binding protein (ChREBP) plays a pivotal role in beta cell glucotoxicity. Diabetologia 2012, 55, 1783–1796. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Kakinuma, S.; Nishimura, M.; Kobayashi, Y.; Nagata, K.; Shimada, Y. Interleukin-9 receptor gene is transcriptionally regulated by nucleolin in T-Cell lymphoma cells. Mol. Carcinog. 2011, 51, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.P.; Chi, A.L.; Ai, W.; Takaishi, S.; Dubeykovskaya, Z.; Quante, M.; Fox, J.G.; Wang, T.C. p53 inhibition of AP1-dependent TFF2 expression induces apoptosis and inhibits cell migration in gastric cancer cells. Am. J. Physiol. Liver Physiol. 2009, 297, G385–G396. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Cao, Y.; Zhang, X.; Zhao, H. Increased CCL19 expression is associated with progression in cervical cancer. Oncotarget 2017, 8, 73817–73825. [Google Scholar] [CrossRef] [PubMed]
- Zong, D.; Jiang, N.; Xu, J.-H.; Wang, D.-J.; Zhu, H.-F.; Wu, L.-R.; Chen, C.; Yin, L.; He, X. ZNF488 is an independent prognostic indicator in nasopharyngeal carcinoma and promotes cell adhesion and proliferation via collagen IV/FAK/AKT/Cyclin D1 pathway. Cancer Manag. Res. 2019, ume 11, 5871–5882. [Google Scholar] [CrossRef]
- Chitranshi, N.; Dheer, Y.; Gupta, V.; Abbasi, M.; Mirzaei, M.; You, Y.; Chung, R.; Graham, S.L.; Gupta, V. PTPN11 induces endoplasmic stress and apoptosis in SH-SY5Y cells. Neuroscience 2017, 364, 175–189. [Google Scholar] [CrossRef]
- Ding, K.; Yu, Z.H.; Yu, C.; Jia, Y.Y.; He, L.; Liao, C.S.; Li, J.; Zhang, C.J.; Li, Y.J.; Wu, T.C.; et al. Effect of gga-miR-155 on cell proliferation, apoptosis and invasion of Marek’s disease virus (MDV) transformed cell line MSB1 by targeting RORA. BMC Vet. Res. 2020, 16, 23. [Google Scholar] [CrossRef] [PubMed]
- Geraldo, L.H.M.; de Sampaio Spohr, T.C.L.; do Amaral, R.F.; da Fonseca, A.C.C.; Garcia, C.; de Almeida Mendes, F.; Freitas, C.; Fabio dosSantos, M.; Lima, F.R.S. Role of lysophosphatidic acid and its receptors in health and disease: Novel therapeutic strategies. Signal Transduct. Target. Ther. 2021, 6, 45. [Google Scholar] [CrossRef]
- Suchanski, J.; Grzegrzolka, J.; Owczarek, T.; Pasikowski, P.; Piotrowska, A.; Kocbach, B.; Nowak, A.; Dziegiel, P.; Wojnar, A.; Ugorski, M. Sulfatide decreases the resistance to stress-induced apoptosis and increases P-selectin-mediated adhesion: A two-edged sword in breast cancer progression. Breast Cancer Res. 2018, 20, 133. [Google Scholar] [CrossRef] [PubMed]
- Tokumoto, M.; Fujiwara, Y.; Shimada, A.; Hasegawa, T.; Seko, Y.; Nagase, H.; Satoh, M. Cadmium toxicity is caused by accumulation of p53 through the down-regulation of Ube2d family genes in vitro and in vivo. J. Toxicol. Sci. 2011, 36, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.S.; Eun, S.Y.; Ham, S.A.; Yoo, T.; Lee, W.J.; Paek, K.S.; Do, J.T.; Lim, D.S.; Seo, H.G. PPARδ modulates oxLDL-induced apoptosis of vascular smooth muscle cells through a TGF-β/FAK signaling axis. Int. J. Biochem. Cell Biol. 2015, 62, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.K.; Liou, J.Y. Cyclooxygenase inhibitors induce colon cancer cell apoptosis Via PPARdelta --> 14-3-3epsilon pathway. Methods Mol. Biol. 2009, 512, 295–307. [Google Scholar] [PubMed]
- Jordan, S.D.; Kriebs, A.; Vaughan, M.; Duglan, D.; Fan, W.; Henriksson, E.; Huber, A.L.; Papp, S.J.; Nguyen, M.; Afetian, M.; et al. CRY1/2 Selectively Repress PPARδ and Limit Exercise Capacity. Cell Metab. 2017, 26, 243–255.e6. [Google Scholar] [CrossRef]
- Narkar, V.A.; Downes, M.; Yu, R.T.; Embler, E.; Wang, Y.X.; Banayo, E.; Mihaylova, M.M.; Nelson, M.C.; Zou, Y.; Juguilon, H.; et al. AMPK and PPARdelta agonists are exercise mimetics. Cell 2008, 134, 405–415. [Google Scholar] [CrossRef]
- Mahajan, M.A.; Samuels, H.H. Nuclear receptor coactivator/coregulator NCoA6(NRC) is a pleiotropic coregulator involved in transcription, cell survival, growth and development. Nucl. Recept. Signal. 2008, 6, e002. [Google Scholar] [CrossRef]
- Roh, J.-I.; Cheong, C.; Sung, Y.H.; Lee, J.; Oh, J.; Lee, B.S.; Lee, J.-E.; Gho, Y.S.; Kim, D.-K.; Park, C.B.; et al. Perturbation of NCOA6 Leads to Dilated Cardiomyopathy. Cell Rep. 2014, 8, 991–998. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Arab, H.H.; Gad, A.M.; Reda, E.; Yahia, R.; Eid, A.H. Activation of autophagy by sitagliptin attenuates cadmium-induced testicular impairment in rats: Targeting AMPK/mTOR and Nrf2/HO-1 pathways. Life Sci. 2021, 269, 119031. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Ortega, V.; Cano-Barquilla, P.; Scacchi, P.A.; Cardinali, D.P.; Esquifino, A.I. Cadmium-Induced Disruption in 24-h Expression of Clock and Redox Enzyme Genes in Rat Medial Basal Hypothalamus: Prevention by Melatonin. Front. Neurol. 2011, 2, 13. [Google Scholar] [CrossRef]
- Bailén, M.; Tabone, M.; Bressa, C.; Lominchar, M.G.M.; Larrosa, M.; González-Soltero, R. Unraveling Gut Microbiota Signatures Associated with PPARD and PARGC1A Genetic Polymorphisms in a Healthy Population. Genes 2022, 13, 289. [Google Scholar] [CrossRef] [PubMed]
- Hishida, A.; Wakai, K.; Naito, M.; Tamura, T.; Kawai, S.; Hamajima, N.; Oze, I.; Imaizumi, T.; Turin, T.C.; Suzuki, S.; et al. Polymorphisms in PPAR Genes (PPARD, PPARG, and PPARGC1A) and the Risk of Chronic Kidney Disease in Japanese: Cross-Sectional Data from the J-MICC Study. PPAR Res. 2013, 2013, 980471. [Google Scholar] [CrossRef] [PubMed]
- Hosohata, K.; Mise, N.; Kayama, F.; Iwanaga, K. Augmentation of cadmium-induced oxidative cytotoxicity by pioglitazone in renal tubular epithelial cells. Toxicol. Ind. Health 2019, 35, 530–536. [Google Scholar] [CrossRef]
- Wang, H.; Wang, A.; Wang, X.; Zeng, X.; Xing, H. AMPK/PPAR-γ/NF-κB axis participates in ROS-mediated apoptosis and autophagy caused by cadmium in pig liver. Environ. Pollut. 2022, 294, 118659. [Google Scholar] [CrossRef] [PubMed]
- Samokhvalov, V.; Zlobine, I.; Jamieson, K.L.; Jurasz, P.; Chen, C.; Lee, K.S.S.; Hammock, B.D.; Seubert, J.M. PPARδ signaling mediates the cytotoxicity of DHA in H9c2 cells. Toxicol. Lett. 2014, 232, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Hatae, T.; Wada, M.; Yokoyama, C.; Shimonishi, M.; Tanabe, T. Prostacyclin-dependent apoptosis mediated by PPAR delta. J. Biol. Chem. 2001, 276, 46260–46267. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.J.; Deng, Z.; Hamblin, M.; Xiang, Y.; Huang, H.; Zhang, J.; Jiang, X.; Wang, Y.; Chen, Y.E. Peroxisome proliferator-activated receptor delta regulation of miR-15a in ischemia-induced cerebral vascular endothelial injury. J. Neurosci. 2010, 30, 6398–6408. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, R.; Solary, E.; O’Connor, P.; Kohn, K.W.; Pommier, Y. Induction of a Common Pathway of Apoptosis by Staurosporine. Exp. Cell Res. 1994, 211, 314–321. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession Number | Description | Fold Change |
|---|---|---|---|
| RYR2 | NM_001035 | Ryanodine receptor 2 | 5.75 |
| CDHR2 | NM_017675 | Cadherin related family member 2 | 5.31 |
| PCDH19 | NM_020766 | Protocadherin 19 | 4.92 |
| TMEM184A | NM_001097620 | Transmembrane protein 184A | 4.91 |
| GLYAT | NM_005838 | Glycine-N-acyltransferase | 4.89 |
| AUTS2 | NM_001127231 | Activator of transcription and developmental regulator AUTS2 | 4.86 |
| MSR1 | NM_002445 | Macrophage scavenger receptor 1 | 4.80 |
| CRLF2 | NM_001012288 | Cytokine receptor like factor 2 | 4.77 |
| SLC23A3 | NM_144712 | Solute carrier family 23 member 3 | 4.76 |
| IL27 | NM_145659 | Interleukin 27 | 4.71 |
| PSG2 | NM_031246 | Pregnancy specific beta-1-glycoprotein 2 | 4.70 |
| PITPNM3 | NM_031220 | PITPNM family member 3 | 4.46 |
| ITPK1 | NM_014216 | Inositol-tetrakisphosphate 1-kinase | 4.42 |
| ACTN2 | NM_001278344 | Actinin alpha 2 | 4.39 |
| BTBD16 | NM_001318189 | BTB domain containing 16 | 4.38 |
| PALD1 | NM_014431 | Phosphatase domain containing paladin 1 | 4.38 |
| RGS11 | NM_003834 | Regulator of G protein signaling 11 | 4.34 |
| SMCO1 | NM_001077657 | Single-pass membrane protein with coiled-coil domains 1 | 4.29 |
| ZNF488 | NM_153034 | Zinc finger protein 488 | 4.21 |
| GAL3ST1 | NM_004861 | Pentatricopeptide repeat domain 2 | 4.20 |
| CACNG7 | NM_031896 | Calcium voltage-gated channel auxiliary subunit gamma 7 | 4.19 |
| SERPINF1 | NM_002615 | Serpin family F member 1 | 4.19 |
| ESPNL | NM_194312 | Espin like | 4.19 |
| LONRF2 | NM_198461 | LON peptidase N-terminal domain and ring finger 2 | 4.19 |
| PDE4C | NM_001330172 | Phosphodiesterase 4C | 4.18 |
| PTPN11 | NM_002834 | Transmembrane protein 225B | 4.16 |
| PRDM11 | NM_001359633 | PR/SET domain 11 | 4.11 |
| TFF2 | NM_005423 | Trefoil factor 2 | 4.11 |
| PDZD4 | NM_032512 | PDZ domain containing 4 | 4.02 |
| CHRNE | NM_000080 | Cholinergic receptor nicotinic epsilon subunit | 4.01 |
| LRRC3 | NM_030891 | Leucine rich repeat containing 3 | 4.00 |
| IL9R | NM_176786 | Interleukin 9 receptor | 3.94 |
| PANX2 | NM_001160300 | Pannexin 2 | 3.88 |
| CPA4 | NM_016352 | Carboxypeptidase A4 | 3.76 |
| PLCB1 | NM_182734 | Phospholipase C beta 1 | 3.65 |
| TLDC2 | NM_080628 | TBC/LysM-associated domain containing 2 | 3.63 |
| PPCDC | NM_134260 | Phosphopantothenoylcysteine decarboxylase | 3.61 |
| RGMB | NM_001366509 | Repulsive guidance molecule BMP co-receptor b | 3.51 |
| PDE4DIP | NM_001350522 | Phosphodiesterase 4D interacting protein | 3.51 |
| RNF125 | NM_017831 | Ring finger protein 125 | 3.50 |
| JRK | NM_001279352 | Jrk helix-turn-helix protein | 3.50 |
| TPRX1 | NM_198479 | Tetrapeptide repeat homeobox 1 | 3.44 |
| HAAO | NM_012205 | 3-Hydroxyanthranilate 3,4-dioxygenase | 3.41 |
| SLC43A1 | NM_003627 | Solute carrier family 43 member 1 | 3.35 |
| CCL19 | NM_006274 | C-C motif chemokine ligand 19 | 3.29 |
| RBM20 | NM_001134363 | RNA binding motif protein 20 | 3.23 |
| FSIP1 | NM_152597 | Fibrous sheath interacting protein 1 | 3.19 |
| MPZL3 | NM_198275 | Myelin protein zero like 3 | 3.18 |
| SMKR1 | NM_001195243 | Small lysine rich protein 1 | 3.12 |
| MLXIPL | NM_032951 | MLX interacting protein like | 3.11 |
| PLXNC1 | NM_005761 | Plexin C1 | 3.11 |
| OR6T1 | NM_001005187 | Olfactory receptor family 6 subfamily T member 1 | 3.10 |
| EGF | NM_001178130 | Epidermal growth factor | 3.06 |
| Gene | Accession Number | Description | Fold Change |
|---|---|---|---|
| TTLL9 | NM_001035 | Tubulin tyrosine ligase like 9 | 0.10 |
| TNF | NM_000594 | Tumor necrosis factor | 0.23 |
| SAA1 | NM_000331 | Serum amyloid A1 | 0.24 |
| CCL20 | NM_004591 | C-C motif chemokine ligand 20 | 0.25 |
| FLG | NM_002016 | Filaggrin | 0.26 |
| PPARD | NM_006238 | Peroxisome proliferator activated receptor delta | 0.32 |
| LPAR3 | NM_012152 | Lysophosphatidic acid receptor 3 | 0.35 |
| PCOTH | NM_001348114 | Pro-X-Gly collagen triple helix like repeat containing | 0.36 |
| HACD3 | NM_016395 | 3-Hydroxyacyl-CoA dehydratase 3 | 0.38 |
| ANKS1B | NM_001352196 | Ankyrin repeat and sterile alpha motif domain containing 1B | 0.39 |
| TRAK2 | NM_015049 | Trafficking kinesin protein 2 | 0.40 |
| ST3GAL6 | NM_006100 | ST3 beta-galactoside alpha-2,3-sialyltransferase 6 | 0.40 |
| LTB | NM_014216 | Lymphotoxin beta | 0.40 |
| UHMK1 | NM_175866 | U2AF homology motif kinase 1 | 0.40 |
| DDR1 | NM_001202522 | Discoidin domain receptor tyrosine kinase 1 | 0.41 |
| KCNS3 | NM_014431 | Potassium voltage-gated channel modifier subfamily S member 3 | 0.42 |
| SAA2 | NM_030754 | Serum amyloid A2 | 0.42 |
| FSIP1 | NM_152597 | Fibrous sheath interacting protein 1 | 0.43 |
| EFHC1 | NM_153034 | EF-hand domain containing 1 | 0.43 |
| GAL3ST1 | NM_004861 | Galactose-3-O-sulfotransferase 1 | 0.43 |
| IL18R1 | NM_003855 | Interleukin 18 receptor 1 | 0.44 |
| SGCB | NM_000232 | Sarcoglycan beta | 0.44 |
| PCDH20 | NM_022843 | Protocadherin 20 | 0.44 |
| CA9 | NM_001216 | Carbonic anhydrase 9 | 0.44 |
| ZDHHC11B | NM_001351303 | Zinc finger DHHC-type containing 11B | 0.44 |
| PTPN11 | NM_002834 | Protein tyrosine phosphatase non-receptor type 11 | 0.45 |
| CXADR | NM_001338 | CXADR Ig-like cell adhesion molecule | 0.46 |
| YES1 | NM_005423 | YES proto-oncogene 1, Src family tyrosine kinase | 0.46 |
| LOC391322 | NM_032512 | D-dopachrome tautomerase-like | 0.47 |
| CCDC146 | NM_020879 | Coiled-coil domain containing 146 | 0.47 |
| EIF1AY | NM_004681 | Eukaryotic translation initiation factor 1A Y-linked | 0.47 |
| PTX3 | NM_176786 | Pentraxin 3 | 0.47 |
| PTGFRN | NM_001160300 | Prostaglandin F2 receptor inhibitor | 0.47 |
| CSF2 | NM_016352 | Colony stimulating factor 2 | 0.48 |
| ANKRD37 | NM_181726 | Ankyrin repeat domain 37 | 0.48 |
| ENTPD2 | NM_001246 | Ectonucleoside triphosphate diphosphohydrolase 2 | 0.49 |
| RORA | NM_134260 | RAR related orphan receptor A | 0.49 |
| PACC1 | NM_018252 | Proton activated chloride channel 1 | 0.50 |
| CD83 | NM_004233 | CD83 molecule | 0.50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mori, C.; Lee, J.-Y.; Tokumoto, M.; Satoh, M. Cadmium Toxicity Is Regulated by Peroxisome Proliferator-Activated Receptor δ in Human Proximal Tubular Cells. Int. J. Mol. Sci. 2022, 23, 8652. https://doi.org/10.3390/ijms23158652
Mori C, Lee J-Y, Tokumoto M, Satoh M. Cadmium Toxicity Is Regulated by Peroxisome Proliferator-Activated Receptor δ in Human Proximal Tubular Cells. International Journal of Molecular Sciences. 2022; 23(15):8652. https://doi.org/10.3390/ijms23158652
Chicago/Turabian StyleMori, Chikage, Jin-Yong Lee, Maki Tokumoto, and Masahiko Satoh. 2022. "Cadmium Toxicity Is Regulated by Peroxisome Proliferator-Activated Receptor δ in Human Proximal Tubular Cells" International Journal of Molecular Sciences 23, no. 15: 8652. https://doi.org/10.3390/ijms23158652
APA StyleMori, C., Lee, J.-Y., Tokumoto, M., & Satoh, M. (2022). Cadmium Toxicity Is Regulated by Peroxisome Proliferator-Activated Receptor δ in Human Proximal Tubular Cells. International Journal of Molecular Sciences, 23(15), 8652. https://doi.org/10.3390/ijms23158652