
Cellular Conditions Responsible for Methylmercury-Mediated Neurotoxicity

{kind=link}
{kind=link}
Abstract
:1. Introduction
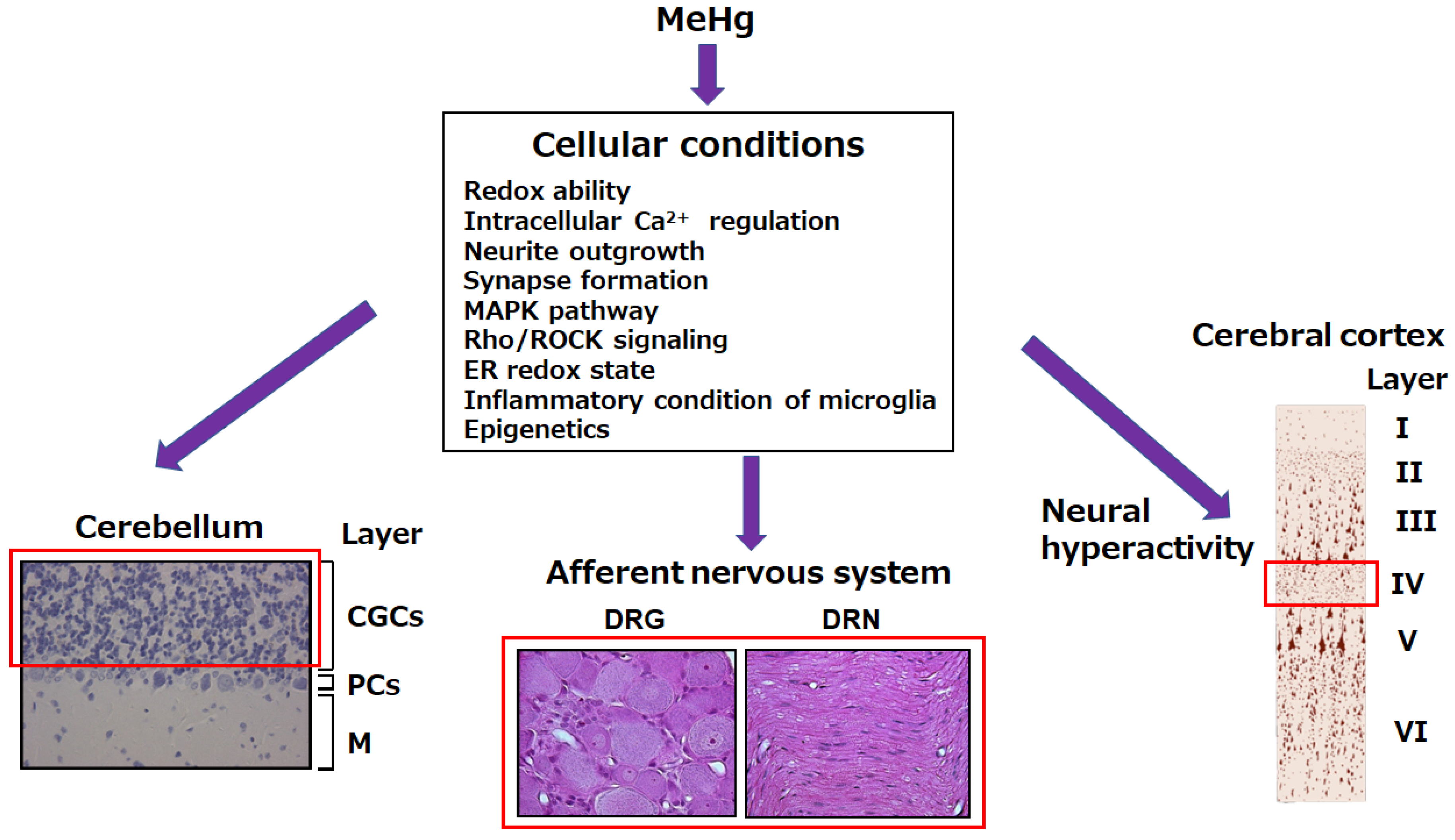
2. MeHg-Mediated Neurotoxicity in Cerebellum
2.1. Involvement of Local Redox Ability in Site-Specific Cerebellar Neurotoxicity
2.2. Involvement of Disruption of Intracellular Ca2+ Regulation in Cerebellar Neurotoxicity
2.3. Impairment of Neurite Outgrowth in the Developing Cerebellum
2.4. Cerebellar Synaptic and Neuritic Remodeling in Pregnant Rats
3. MeHg-Mediated Neurotoxicity in the Cerebral Cortex
3.1. Involvement of Local Redox Ability in Site-Specific Neurodegeneration
3.2. MeHg-Mediated Neural Hyperactivation in the Deep Layers of Cerebrocortical Neurons
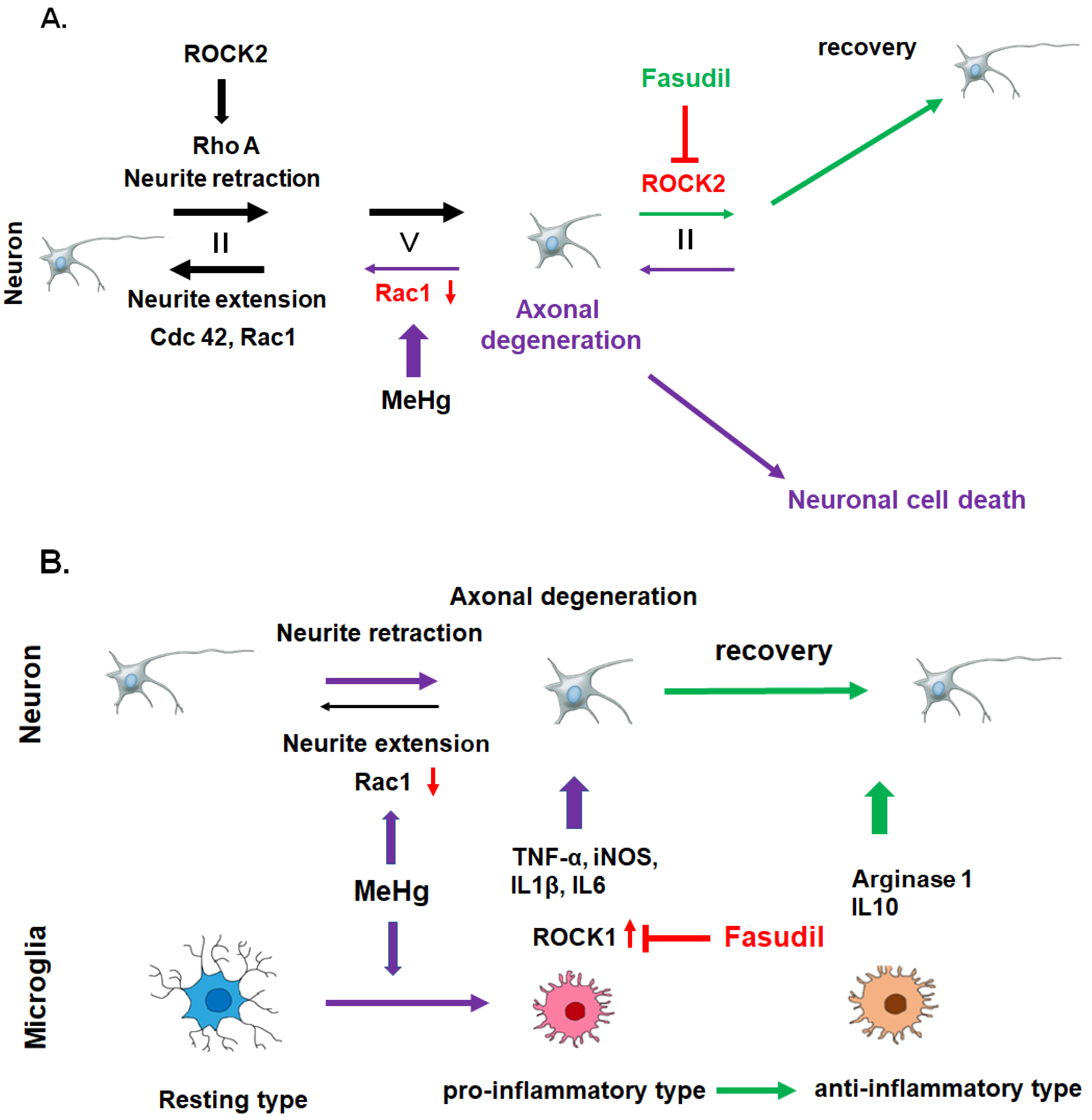
3.3. Role of the Rho-ROCK Signaling Pathway in MeHg-Mediated Axonal Degeneration
3.4. MeHg-Mediated Activation of the c-Jun N-Terminal Kinase (JNK) Pathway Causes Neurodegeneration in Cerebrocortical Neurons
3.5. Role of Endoplasmic Reticulum (ER) Stress in MeHg-Mediated Cerebrocortical Neurotoxicity
4. MeHg-Mediated Neurotoxicity in the Primary Afferent Nervous System
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MeHg | Methylmercury |
| Hg | Mercury |
| ROS | Reactive oxygen species |
| MAPK | Mitogen-activated protein kinase |
| Rho | Ras homolog gene |
| ROCK | Rho-associated coiled coil-forming protein kinase |
| Cu: Zn-SOD | Copper, zinc-superoxide dismutase |
| Mn-SOD | Manganese-superoxide dismutase |
| GPx1 | Glutathione peroxidase 1 |
| TRxR1 | Thioredoxin reductase 1 |
| PCR | Polymerase chain reaction |
| Trk | Tropomyosin receptor kinase |
| eEF1A1 | Eukaryotic elongation factor 1A1 |
| PND | Postnatal day |
| Arc | Activity-regulated cytoskeleton-associated protein |
| Sl-CCNs | shallower layers of cerebrocortical neurons |
| dl-CCNs | deep layers of cerebrocortical neurons |
| IEGs | Immediate early genes |
| BDNF | Brain-derived neurotrophic factor |
| CREB | cAMP response element binding protein |
| PKA | cAMP-dependent protein kinase |
| Rac | Ras-related C3 botulinum toxin substrate |
| Cdc42 | Cell division control protein 42 homolog |
| CNS | Central nervous system |
| JNK | c-Jun N-terminal kinase |
| MKK4 | Activated protein kinase kinase 4 |
| Vsp34 | Vacuolar sorting protein 34 |
| ER | Endoplasmic reticulum |
| UPR | Unfolded protein response |
| ERAI | ER stress-activated indicator |
| XBP1 | X-box binding protein 1 |
| IRE1 | Inositol-requiring protein 1 |
References
- Harada, M.; Akagi, H.; Tsuda, T.; Kizaki, T.; Ohno, H. Methylmercury level in umbilical cords from patients with congenital Minamata disease. Sci. Total Environ. 1999, 234, 59–62. [Google Scholar] [CrossRef]
- Eto, K. Pathology of Minamata disease. Toxicol. Pathol. 1997, 25, 614–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eto, K.; Tokunaga, H.; Nagashima, K.; Takeuchi, T. An autopsy case of minamata disease (methylmercury poisoning)—Pathological viewpoints of peripheral nerves. Toxicol. Pathol. 2002, 30, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Usuki, F.; Yasutake, A.; Umehara, F.; Tokunaga, H.; Matsumoto, M.; Eto, K.; Ishiura, S.; Higuchi, I. In vivo protection of a water-soluble derivative of vitamin E, Trolox, against methylmercury-intoxication in the rat. Neurosci. Lett. 2001, 304, 199–203. [Google Scholar] [CrossRef]
- Fujimura, M.; Usuki, F. Low in situ expression of antioxidative enzymes in rat cerebellar granular cells susceptible to methylmercury. Arch. Toxicol. 2014, 88, 109–113. [Google Scholar] [CrossRef]
- Fujimura, M.; Usuki, F. In situ different antioxidative systems contribute to the site-specific methylmercury neurotoxicity in mice. Toxicology 2017, 392, 55–63. [Google Scholar] [CrossRef]
- Shinoda, Y.; Ehara, S.; Tatsumi, S.; Yoshida, E.; Takahashi, T.; Eto, K.; Kaji, T.; Fujiwara, Y. Methylmercury-induced neural degeneration in rat dorsal root ganglion is associated with the accumulation of microglia/macrophages and the proliferation of Schwann cells. J. Toxicol. Sci. 2019, 44, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Karri, V.; Professor, M.S.; Kumar, V. A systems toxicology approach to compare the heavy metal mixtures (Pb, As, MeHg) impact in neurodegenerative diseases. Food Chem. Toxicol. 2020, 139, 111257. [Google Scholar] [CrossRef]
- Fujimura, M.; Usuki, F. Methylmercury-mediated oxidative stress and activation of the cellular protective system. Antioxidants 2020, 9, 1004. [Google Scholar] [CrossRef]
- Palacios, E.R.; Houghton, C.; Chadderton, P. Accounting for uncertainty: Inhibition for neural inference in the cerebellum. Proc. Biol. Sci. 2021, 288, 20210276. [Google Scholar] [CrossRef]
- O’Donoghue, J.L.; Watson, G.E.; Brewer, R.; Zareba, G.; Eto, K.; Takahashi, H.; Marumoto, M.; Love, T.; Harrington, D.; Myers, G.J. Neuropathology associated with exposure to different concentrations and species of mercury: A review of autopsy cases and the literature. Neurotoxicology 2020, 78, 88–98. [Google Scholar] [CrossRef]
- Ihara, H.; Kasamatsu, S.; Kitamura, A.; Nishimura, A.; Tsutsuki, H.; Ida, T.; Ishizaki, K.; Toyama, T.; Yoshida, E.; Abdul Hamid, H.; et al. Exposure to electrophiles impairs reactive persulfide-dependent redox signaling in neuronal cells. Chem. Res. Toxicol. 2017, 30, 1673–1684. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Ando, Y.; Nakamura, M.; Obayashi, K.; Terazaki, H.; Haraoka, K.; Guo, S.X.; Ueda, M.; Uchino, M. Inhibitory effect of α-tocopherol on methylmercury-induced oxidative steress. Environ. Health Prev. Med. 2004, 9, 111–117. [Google Scholar] [CrossRef]
- Tucker, E.K.; Nowak, R.A. Methylmercury alters proliferation, migration, and antioxidant capacity in human HTR8/SV-neo trophoblast cells. Reprod. Toxicol. 2018, 78, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Limke, T.L.; Bearss, J.J.; Atchison, W.D. Acute exposure to methylmercury causes Ca2+ dysregulation and neuronal death in rat cerebellar granule cells through an M3 muscarinic receptor-linked pathway. Toxicol. Sci. 2004, 80, 60–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, J.R.; Marty, M.S.; Atchison, W.D. Comparative sensitivity of rat cerebellar neurons to dysregulation of divalent cation homeostasis and cytotoxicity caused by methylmercury. Toxicol. Appl. Pharmacol. 2005, 208, 222–232. [Google Scholar] [CrossRef]
- Neustadt, A.; Frostholm, A.; Rotter, A. Topographical distribution of muscarinic cholinergic receptors in the cerebellar cortex of the mouse, rat, guinea pig, and rabbit: A species comparison. J. Comp. Neurol. 1988, 272, 317–330. [Google Scholar] [CrossRef]
- Yuan, Y.; Atchison, W.D. Multiple sources of Ca2+ contribute to methylmercury-induced increased frequency of spontaneous inhibitory synaptic responses in cerebellar slices of rat. Toxicol. Sci. 2016, 150, 117–130. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, M.; Cheng, J.; Zhao, W. Perinatal exposure to low-dose methylmercury induces dysfunction of motor coordination with decreases in synaptophysin expression in the cerebellar granule cells of rats. Brain Res. 2012, 1464, 1–7. [Google Scholar] [CrossRef]
- Fujimura, M.; Usuki, F.; Cheng, J.; Zhao, W. Prenatal low-dose methylmercury exposure impairs neurite outgrowth and synaptic protein expression and suppresses TrkA pathway activity and eEF1A1 expression in the rat cerebellum. Toxicol. Appl. Pharmacol. 2016, 298, 1–8. [Google Scholar] [CrossRef]
- Go, S.; Kurita, H.; Hatano, M.; Matsumoto, K.; Nogawa, H.; Fujimura, M.; Inden, M.; Hozumi, I. DNA methyltransferase- and histone deacetylase-mediated epigenetic alterations induced by low-level methylmercury exposure disrupt neuronal devel-opment. Arch. Toxicol. 2021, 95, 1227–1239. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, M.; Usuki, F. Pregnant rats exposed to low level methylmercury exhibit cerebellar synaptic and neuritic remodeling during the perinatal period. Arch. Toxicol. 2020, 94, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Hoekzema, E.; Barba-Müller, E.; Pozzobon, C.; Picado, M.; Lucco, F.; García-García, D.; Soliva, J.C.; Tobeña, A.; Desco, M.; Crone, E.A.; et al. Pregnancy leads to long-lasting changes in human brain structure. Nat. Neurosci. 2017, 20, 287–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, M.; Usuki, F. Site-specific neural hyperactivity via the activation of MAPK and PKA/CREB pathways triggers neuronal degeneration in methylmercury-intoxicated mice. Toxicol. Lett. 2017, 271, 66–73. [Google Scholar] [CrossRef]
- Meinerz, D.F.; Branco, V.; Aschner, M.; Carvalho, C.; Rocha, J.B.T. Diphenyl diselenide protects against methylmercury-induced inhibition of thioredoxin reductase and glutathione peroxidase in human neuroblastoma cells: A comparison with ebselen. J. Appl. Toxicol. 2017, 37, 1073–1081. [Google Scholar] [CrossRef]
- Allen, J.W.; Mutkus, L.A.; Aschner, M. Methylmercury-mediated inhibition of 3H-D-aspartate transport in cultured astrocytes is reversed by the antioxidant catalase. Brain Res. 2001, 902, 92–100. [Google Scholar] [CrossRef]
- Cheng, J.; Fujimura, M.; Zhao, W.; Wang, W. Neurobehavioral effects, c-Fos/Jun expression and tissue distribution in rat offspring prenatally co-exposed to MeHg and PFOA: PFOA impairs Hg retention. Chemosphere 2013, 91, 758–764. [Google Scholar] [CrossRef]
- Flores, V.L.; Parmet, T.; Mukherjee, N.; Nelson, S.; Katz, D.B.; Levitan, D. The role of the gustatory cortex in incidental experience-evoked enhancement of later taste learning. Learn. Mem. 2018, 25, 587–600. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, M.; Usuki, F. Methylmercury induces oxidative stress and subsequent neural hyperactivity leading to cell death through the p38 MAPK-CREB pathway in differentiated SH-SY5Y cells. Neurotoxicology 2018, 67, 226–233. [Google Scholar] [CrossRef]
- Borrajo, A.; Rodriguez-Perez, A.I.; Villar-Cheda, B.; Guerra, M.J.; Labandeira-Garcia, J.L. Inhibition of the microglial response is essential for the neuroprotective effects of Rho-kinase inhibitors on MPTP-induced dopaminergic cell death. Neuropharmacology 2014, 85, 1–8. [Google Scholar] [CrossRef]
- Iizuka, M.; Kimura, K.; Wang, S.; Kato, K.; Amano, M.; Kaibuchi, K.; Mizoguchi, A. Distinct distribution and localization of Rho-kinase in mouse epithelial, muscle and neural tissues. Cell Struct. Funct. 2012, 37, 155–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, M.; Usuki, F.; Sawada, M.; Rostene, W.; Godefroy, D.; Takashima, A. Methylmercury exposure downregulates the expression of Racl and leads to neuritic degeneration and ultimately apoptosis in cerebrocortical neurons. Neurotoxicology 2009, 30, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, M.; Usuki, F. Differing effects of toxicants (methylmercury, inorganic mercury, lead, amyloid β and rotenone) on cultured rat cerebrocortical neurons: Differential expression of Rho proteins associated with neurotoxicity. Toxicol. Sci. 2012, 126, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, M.; Usuki, F.; Kawamura, M.; Izumo, S. Inhibition of the Rho/ROCK pathway prevents neuronal degeneration in vitro and in vivo following methylmercury exposure. Toxicol. Appl. Pharmacol. 2011, 250, 1–9. [Google Scholar] [CrossRef]
- Fujimura, M.; Usuki, F.; Nakamura, A. Fasudil, a Rho-associated coiled coil-forming protein kinase inhibitor, recovers methylmercury-induced axonal degeneration by changing microglial phenotype in rats. Toxicol. Sci. 2019, 168, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.K.; Kim, J.G.; Kim, H.J.; Cho, J.Y.; Jeong, H.; Park, Y.; Islam, R.; Cap, C.K.; Park, J.B. Regulation of RhoA GTPase and novel target proteins for ROCK. Small GTPases 2020, 11, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, J.; Guo, D.; Zhou, X.; Jiang, W.; Wang, J.; Tang, J.; Zou, Y.; Bi, M.; Li, Q. Fasudil alleviates brain damage in rats after carbon monoxide poisoning through regulating neurite outgrowth inhibitor/oligodendrocytemyelin glycoprotein signalling pathway. Basic Clin. Pharmacol. Toxicol. 2019, 125, 152–165. [Google Scholar]
- Jacob, S.; Thangarajan, S. Fisetin impedes developmental methylmercury neurotoxicity via downregulating apoptotic signal-ling pathway and upregulating Rho GTPase signalling pathway in hippocampus of F1 generation rats. Int. J. Dev. Neurosci. 2018, 69, 88–96. [Google Scholar] [CrossRef]
- Shinozaki, Y.; Danjo, Y.; Koizumi, S. Microglial ROCK is essential for chronic methylmercury-induced neurodegeneration. J. Neurochem. 2019, 151, 64–78. [Google Scholar] [CrossRef]
- Pernègre, C.; Duquette, A.; Leclerc, N. Tau secretion: Good and bad for neurons. Front. Neurosci. 2019, 13, 649. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, M.; Usuki, F.; Sawada, M.; Takashima, A. Methylmercury induces neuropathological changes with tau hyperphos-phorylation mainly through the activation of the c-jun-N-terminal kinase pathway in the cerebral cortex, but not in the hip-pocampus of the mouse brain. Neurotoxicology 2009, 30, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Miao, Y.; Muhammad, I.; Tian, E.; Hu, W.; Wang, J.; Wang, B.; Li, R.; Li, J. Colistin-induced autophagy and apoptosis involves the JNK-Bcl2-Bax signaling pathway and JNK-p53-ROS positive feedback loop in PC-12 cells. Chem. Biol. Interact. 2017, 277, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Funderburk, S.F.; Wang, Q.J.; Yue, Z. The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends Cell Biol. 2010, 20, 355–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.; Ruan, S.; Huang, D.; Meng, X.; Li, W.; Wang, B.; Zou, F. MeHg-induced autophagy via JNK/Vps34 complex pathway promotes autophagosome accumulation and neuronal cell death. Cell Death Dis. 2019, 10, 399. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.W.; Wang, Y.; Wang, T.; Zhang, K.B.; Jiang, C.Y.; Hu, F.F.; Yuan, Y.; Bian, J.C.; Liu, X.Z.; Gu, J.H.; et al. Cadmium-induced autophagy promotes survival of rat cerebral cortical neurons by activating class III phosphoinositide 3-kinase/beclin-1/B-cell lymphoma 2 signaling pathways. Mol. Med. Rep. 2015, 12, 2912–2918. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Cao, R.; Cai, T.; Aschner, M.; Zhao, F.; Yao, T.; Chen, Y.; Cao, Z.; Luo, W.; Chen, J. The role of autophagy dysregula-tion in manganese-induced dopaminergic neurodegeneration. Neurotox. Res. 2013, 24, 478–490. [Google Scholar] [CrossRef] [Green Version]
- Usuki, F.; Fujita, E.; Sasagawa, N. Methylmercury activates ASK1/JNK signaling pathways, leading to apoptosis due to both mitochondria- and endoplasmic reticulum (ER)-generated processes in myogenic cell lines. Neurotoxicology 2008, 29, 22–30. [Google Scholar] [CrossRef]
- Liu, W.; Yang, T.; Xu, Z.; Xu, B.; Deng, Y. Methyl-mercury induces apoptosis through ROS-mediated endoplasmic reticulum stress and mitochondrial apoptosis pathways activation in rat cortical neurons. Free Radic. Res. 2019, 53, 26–44. [Google Scholar] [CrossRef]
- Hiraoka, H.; Nomura, R.; Takasugi, N.; Akai, R.; Iwawaki, T.; Kumagai, Y.; Fujimura, M.; Uehara, T. Spatiotemporal analysis of the UPR transition induced by methylmercury in the mouse brain. Arch. Toxicol. 2021, 95, 1241–1250. [Google Scholar] [CrossRef]
- Iwawaki, T.; Akai, R.; Kohno, K.; Miura, M. A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat. Med. 2004, 10, 98–102. [Google Scholar] [CrossRef]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igata, A. Neurological aspects of methylmercury poisoning in Minamata. In Recent Advances in Minamata Disease Studies; Tsu-baki, T., Takahashi, H., Eds.; Kodansha Ltd.: Tokyo, Japan, 1986; pp. 41–57. [Google Scholar]
- Usuki, F.; Yasutake, A.; Umehara, F.; Higuchi, I. Beneficial effects of mild lifelong dietary restriction on skeletal muscle: Prevention of age-related mitochondrial damage, morphological changes, and vulnerability to a chemical toxin. Acta Neuropathol. 2004, 108, 1–9. [Google Scholar] [PubMed]
- Gnanashanmugam, G.; Balakrishnan, R.; Somasundaram, S.P.; Parimalam, N.; Rajmohan, P.; Pranesh, M.B. Mercury toxicity following unauthorized siddha medicine intake—A mimicker of acquired neuromyotonia—Report of 32 cases. Ann. Indian. Acad. Neurol. 2018, 21, 49–56. [Google Scholar] [PubMed]
- Khodashenas, E.; Aelami, M.; Balali-Mood, M. Mercury poisoning in two 13-year-old twin sisters. J. Res. Med. Sci. 2015, 20, 308–311. [Google Scholar]
- Fujimura, M.; Usuki, F.; Nakamura, A. Methylmercury induces hyperalgesia and allodynia through spinal cord dorsal horn neuronal activation and subsequent somatosensory cortical circuit formation in rats. Arch. Toxicol. 2021, 95, 2151–2162. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujimura, M.; Usuki, F. Cellular Conditions Responsible for Methylmercury-Mediated Neurotoxicity. Int. J. Mol. Sci. 2022, 23, 7218. https://doi.org/10.3390/ijms23137218
Fujimura M, Usuki F. Cellular Conditions Responsible for Methylmercury-Mediated Neurotoxicity. International Journal of Molecular Sciences. 2022; 23(13):7218. https://doi.org/10.3390/ijms23137218
Chicago/Turabian StyleFujimura, Masatake, and Fusako Usuki. 2022. "Cellular Conditions Responsible for Methylmercury-Mediated Neurotoxicity" International Journal of Molecular Sciences 23, no. 13: 7218. https://doi.org/10.3390/ijms23137218
APA StyleFujimura, M., & Usuki, F. (2022). Cellular Conditions Responsible for Methylmercury-Mediated Neurotoxicity. International Journal of Molecular Sciences, 23(13), 7218. https://doi.org/10.3390/ijms23137218