
Fibroblast Growth Factors and Cellular Communication Network Factors: Intimate Interplay by the Founding Members in Cartilage


Abstract
1. Interplay by the Members of Two Distinct Families of Proteins in Cartilage
2. FGF Family
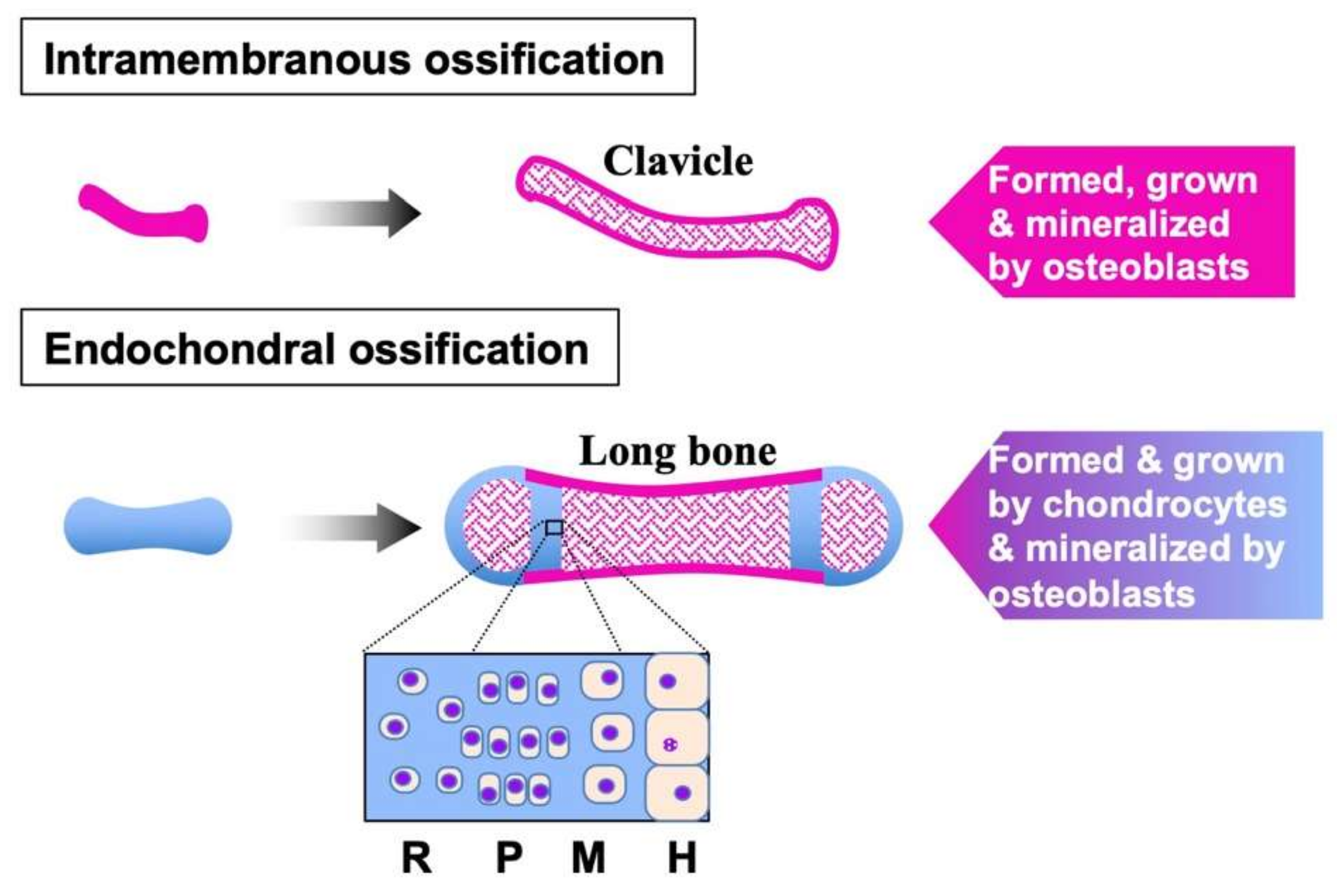
3. FGFs in Cartilage
4. CCN Family
5. CCNs in Cartilage
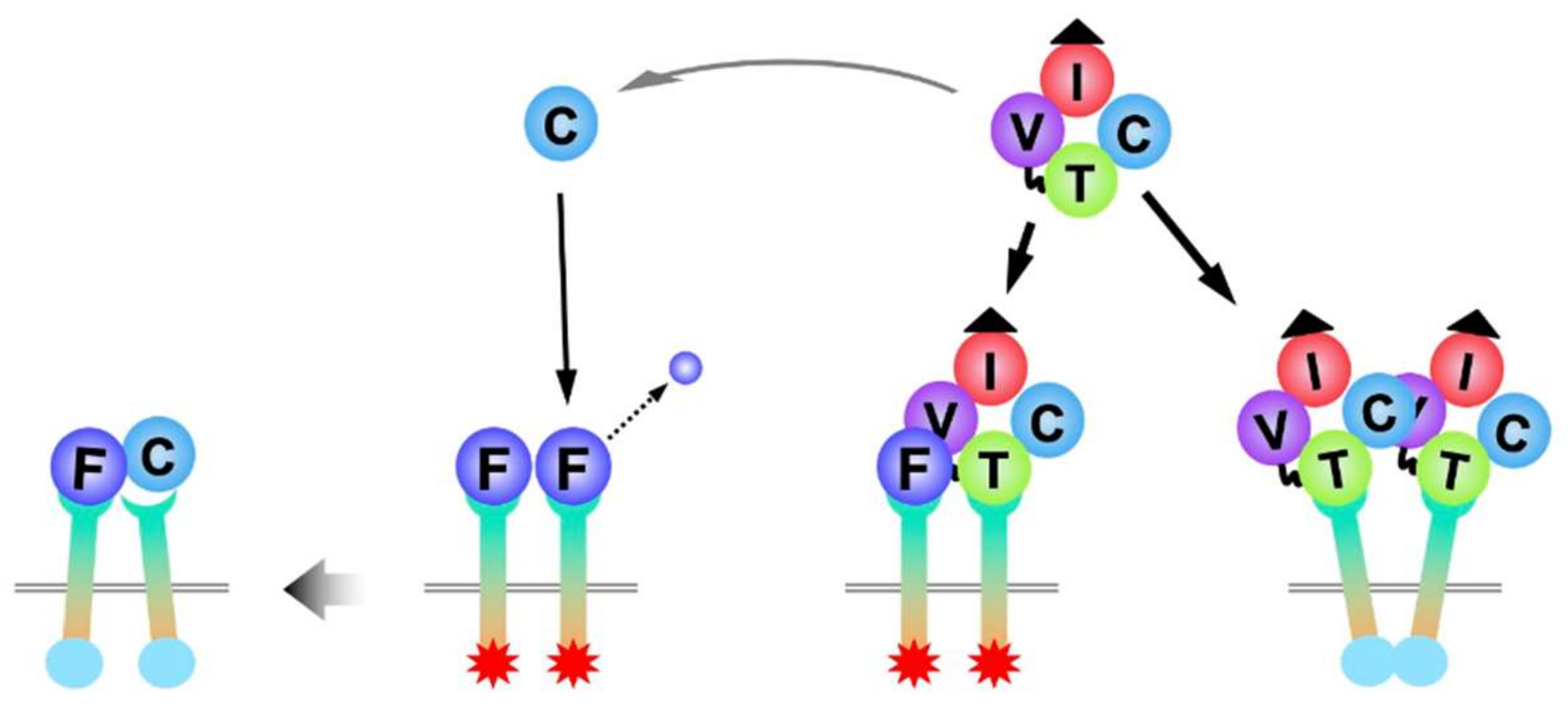
6. Molecular Interaction between FGFs and CCNs
7. FGF–CCN Collaboration in Cartilage
8. FGF–CCN Genetic Interaction in Chondrocytes
9. Mechanism of the Integrated FGF–CCN Regulation in Chondrocytes
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2, REVIEWS3005. [Google Scholar] [CrossRef] [PubMed]
- Perbal, B. CCN proteins: Multifunctional signalling regulators. Lancet 2004, 363, 62–64. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Marie, P.J. Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. 2015, 29, 1463–1486. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Marie, P.J. Fibroblast growth factors in skeletal development. Curr. Top. Dev. Biol. 2019, 133, 195–234. [Google Scholar]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef]
- Su, W.C.; Kitagawa, M.; Xue, N.; Xie, B.; Garofalo, S.; Cho, J.; Deng, C.; Horton, W.A.; Fu, X.Y. Activation of Stat1 by mutant fibro-blast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature 1997, 386, 288–292. [Google Scholar] [CrossRef]
- Kouhara, H.; Hadari, Y.R.; Spivak-Kroizman, T.; Schilling, J.; Bar-Sagi, D.; Lax, I.; Schlessinger, J. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 1997, 89, 693–702. [Google Scholar] [CrossRef]
- Tan, Y.; Rouse, J.; Zhang, A.; Cariati, S.; Cohen, P.; Comb, M.J. FGF and stress regulate CREB and ATF-1 via a pathway involving p38 MAP kinase and MAPKAP kinase-2. EMBO J. 1996, 15, 4629–4642. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Honegger, A.M.; Rotin, D.; Fischer, R.; Bellot, F.; Li, W.; Dionne, C.A.; Jaye, M.; Rubinstein, M.; Schlessinger, J. A tyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factor receptor (Flg) is a binding site for the SH2 domain of phospholipase C-gamma 1. Mol. Cell Biol. 1991, 11, 5068–5078. [Google Scholar] [PubMed]
- Jones, S. Mini-review: Endocrine actions of fibroblast growth factor 19. Mol. Pharm. 2008, 5, 42–48. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Invest. 2005, 115, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; White, K.E. FGF23 and disorders of phosphate homeostasis. Cytokine Growth Factor Rev. 2005, 16, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Invest. 2004, 113, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, M.; Schoorlemmer, J.; Williams, A.; Diwakar, S.; Wang, Q.; Huang, X.; Giza, J.; Tchetchik, D.; Kelley, K.; Vega, A.; et al. Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage-gated sodium channels. Neuron 2007, 55, 449–463. [Google Scholar] [CrossRef]
- Wang, C.; Hennessey, J.A.; Kirkton, R.D.; Graham, V.; Puranam, R.S.; Rosenberg, P.B.; Bursac, N.; Pitt, G.S. Fibroblast Growth Factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ. Res. 2011, 109, 775–782. [Google Scholar] [CrossRef]
- Lou, J.Y.; Laezza, F.; Gerber, B.R.; Xiao, M.; Yamada, K.A.; Hartmann, H.; Craig, A.M.; Nerbonne, J.M.; Ornitz, D.M. Fibroblast Growth Factor 14 is an intracellular modulator of voltage-gated sodium channels. J. Physiol. 2005, 569, 179–193. [Google Scholar] [CrossRef]
- Baldin, V.; Roman, A.M.; Bosc-Bierne, I.; Amalric, F.; Bouche, G. Translocation of bFGF to the nucleus is G1 phase cell cycle specific in bovine aortic endothelial cells. EMBO J. 1990, 9, 1511–1517. [Google Scholar] [CrossRef]
- Sherman, L.; Stocker, K.M.; Morrison, R.; Ciment, G. Basic fibroblast growth factor (bFGF) acts intracellularly to cause the transdifferentiation of avian neural crest-derived Schwann cell precursors into melanocytes. Development 1993, 118, 1313–1326. [Google Scholar] [CrossRef]
- Suh, J.M.; Jonker, J.W.; Ahmadian, M.; Goetz, R.; Lackey, D.; Osborn, O.; Huang, Z.; Liu, W.; Yoshihara, E.; van Dijk, T.H.; et al. Endocrinization of FGF1 produces a neomorphic and potent insulin sensitizer. Nature 2014, 513, 436–439. [Google Scholar] [CrossRef]
- Gasser, E.; Moutos, C.P.; Downes, M.; Evans, R.M. FGF1: A new weapon to control type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2017, 13, 599–609. [Google Scholar] [CrossRef]
- Kubota, S.; Kawaki, H.; Perbal, B.; Kawata, K.; Hattori, T.; Nishida, T. Cellular communication network factor 3 in cartilage development and maintenance. J. Cell Commun. Signal 2021, 15, 533–543. [Google Scholar] [CrossRef]
- Martin, G.R. The roles of FGFs in the early development of vertebrate limbs. Genes Dev. 1998, 12, 1571–1586. [Google Scholar] [CrossRef]
- Gonzalez, A.M.; Hill, D.J.; Logan, A.; Maher, P.A.; Baird, A. Distribution of fibroblast growth factor (FGF)-2 and FGF receptor-1 messenger RNA expression and protein presence in the mid-trimester human fetus. Pediatr. Res 1996, 39, 75–385. [Google Scholar] [CrossRef][Green Version]
- Montero, A.; Okada, Y.; Tomita, M.; Ito, M.; Tsurukami, H.; Nakamura, T.; Doetschman, T.; Coffin, J.D.; Hurley, M.M. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J. Clin. Invest. 2000, 105, 1085–1093. [Google Scholar] [CrossRef]
- Schmal, H.; Zwingmann, J.; Fehrenbach, M.; Finkenzeller, G.; Stark, G.B.; Südkamp, N.P.; Hartl, D.; Mehlhorn, A.T. bFGF influences human articular chondrocyte differentiation. Cytotherapy 2007, 9, 184–193. [Google Scholar] [CrossRef]
- Im, H.J.; Li, X.; Muddasani, P.; Kim, G.H.; Davis, F.; Rangan, J.; Forsyth, C.B.; Ellman, M.; Thonar, E.J. Basic fibroblast growth factor accelerates matrix degradation via a neuro-endocrine pathway in human adult articular chondrocytes. J. Cell Physiol. 2008, 215, 452–463. [Google Scholar] [CrossRef]
- El-Seoudi, A.; Abd El Kader, T.; Nishida, T.; Eguchi, T.; Aoyama, E.; Takigawa, M.; Kubota, S. Catabolic effects of FGF-1 on chondrocytes and its possible role in osteoarthritis. J. Cell Commun. Signal 2017, 11, 255–263. [Google Scholar] [CrossRef]
- Baujat, G.; Legeai-Mallet, L.; Finidori, G.; Cormier-Daire, V.; Le Merrer, M. Achondroplasia. Best Practice & Research. Clin. Rheumatol. 2008, 22, 3–18. [Google Scholar]
- Bonaventure, J.; Rousseau, F.; Legeai-Mallet, L.; Le Merrer, M.; Munnich, A.; Maroteaux, P. Common mutations in the fibroblast growth factor receptor 3 (FGFR 3) gene account for achondroplasia, hypochondroplasia, and thanatophoric dwarfism. Am. J. Med. Genet. 1996, 63, 148–154. [Google Scholar] [CrossRef]
- Xiao, L.; Williams, D.; Hurley, M.M. Inhibition of FGFR Signaling Partially Rescues Osteoarthritis in Mice Overexpressing High Molecular Weight FGF2 Isoforms. Endocrinology 2020, 161, bqz016. [Google Scholar] [CrossRef]
- Hung, I.H.; Yu, K.; Lavine, K.J.; Ornitz, D.M. FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascu- larization in the developing stylopod. Dev. Biol. 2007, 307, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lavine, K.J.; Hung, I.H.; Ornitz, D.M. FGF18 is required for early chondrocyte proliferation, hypertrophy and vascular invasion of the growth plate. Dev. Biol. 2007, 302, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Gigout, A.; Guehring, H.; Froemel, D.; Meurer, A.; Ladel, C.; Reker, D.; Bay-Jensen, A.C.; Karsdal, M.A.; Lindemann, S. Sprifermin (rhFGF18) enables proliferation of chondrocytes producing a hyaline cartilage matrix. Osteoarthr. Cartil. 2017, 25, 1858–1867. [Google Scholar] [CrossRef] [PubMed]
- Meloni, G.R.; Farran, A.; Mohanraj, B.; Guehring, H.; Cocca, R.; Rabut, E.; Mauck, R.L.; Dodge, G.R. Recombinant human FGF18 preserves depth-dependent mechanical inhomogeneity in articular cartilage. Eur. Cell Mater. 2019, 38, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Bork, P. The modular architecture of a new family of growth regulators related to connective tissue growth factor. FEBS Lett. 1993, 327, 125–130. [Google Scholar] [CrossRef]
- Simmons, D.L.; Levy, D.B.; Yannoni, Y.; Erikson, R.L. Identification of a phorbol ester-repressible v-src-inducible gene. Proc. Natl. Acad. Sci. USA 1989, 86, 1178–1182. [Google Scholar] [CrossRef]
- Bradham, D.M.; Igarashi, A.; Potter, R.L.; Grotendorst, G.R. Connective tissue growth factor: A cysteine-rich mitogen secreted by human vascular endothelial cells is related to the SRC-induced immediate early gene product CEF-10. J. Cell Biol. 1991, 114, 1285–1294. [Google Scholar] [CrossRef]
- Joliot, V.; Martinerie, C.; Dambrine, G.; Plassiart, G.; Brisac, M.; Crochet, J.; Perbal, B. Proviral rearrangements and overexpression of a new cellular gene (nov) in myeloblastosis-associated virus type 1-induced nephroblastomas. Mol. Cell Biol. 1992, 12, 10–21. [Google Scholar]
- Kawaki, H.; Kubota, S.; Suzuki, A.; Yamada, T.; Matsumura, T.; Mandai, T.; Yao, M.; Maeda, T.; Lyons, K.M.; Takigawa, M. Functional requirement of CCN2 for intramembranous bone formation in embryonic mice. Biochem. Biophys. Res. Commun. 2008, 366, 450–456. [Google Scholar] [CrossRef]
- Kubota, S.; Takigawa, M. The CCN family acting throughout the body: Recent research developments. Biomol. Concepts 2013, 4, 477–494. [Google Scholar] [CrossRef]
- Lau, L.F. Cell surface receptors for CCN proteins. J. Cell Commun. Signal 2016, 10, 121–127. [Google Scholar] [CrossRef]
- Pennica, D.; Swanson, T.A.; Welsh, J.W.; Roy, M.A.; Lawrence, D.A.; Lee, J.; Brush, J.; Taneyhill, L.A.; Deuel, B.; Lew, M.; et al. WISP genes are members of the connective tissue growth factor family that are up-regulated in wnt-1-transformed cells and aberrantly expressed in human colon tumors. Proc. Natl. Acad. Sci. USA 1998, 95, 14717–14722. [Google Scholar] [CrossRef] [PubMed]
- Brigstock, D.R. The connective tissue growth factor/cysteine-rich 61/nephroblastoma overexpressed (CCN) family. Endocr. Rev. 1999, 20, 189–206. [Google Scholar] [PubMed]
- Brigstock, D.R.; Goldschmeding, R.; Katsube, K.I.; Lam, S.C.; Lau, L.F.; Lyons, K.; Naus, C.; Perbal, B.; Riser, B.; Takigawa, M.; et al. Proposal for a unified CCN nomenclature. Mol. Pathol. 2003, 56, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Segarini, P.; Raoufi, F.; Bradham, D.; Leask, A. Connective tissue growth factor is secreted through the Golgi and is degraded in the endosome. Exp. Cell Res. 2001, 271, 109–117. [Google Scholar] [CrossRef]
- Sabbah, M.; Prunier, C.; Ferrand, N.; Megalophonos, V.; Lambein, K.; De Wever, O.; Nazaret, N.; Lachuer, J.; Dumont, S.; Redeuilh, G. CCN5, a novel transcriptional repressor of the transforming growth factor β signaling pathway. Mol. Cell Biol. 2011, 31, 1459–1469. [Google Scholar] [CrossRef]
- Wahab, N.A.; Brinkman, H.; Mason, R.M. Uptake and intracellular transport of the connective tissue growth factor: A potential mode of action. Biochem. J. 2001, 359, 89–97. [Google Scholar] [CrossRef]
- Perbal, B. Nuclear localisation of NOVH protein: A potential role for NOV in the regulation of gene expression. Mol. Pathol. 1999, 52, 84–91. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. All in the CCN family: Essential matricellular signaling modulators emerge from the bunker. J. Cell Sci. 2006, 119, 4803–4810. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 945–963. [Google Scholar] [CrossRef]
- Perbal, B.; Tweedie, S.; Bruford, E. The official unified nomenclature adopted by the HGNC calls for the use of the acronyms, CCN1-6, and discontinuation in the use of CYR61, CTGF, NOV and WISP 1-3 respectively. J. Cell Commun. Signal. 2018, 12, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Kawaki, H.; Kubota, S.; Suzuki, A.; Lazar, N.; Yamada, T.; Matsumura, T.; Ohgawara, T.; Maeda, T.; Perbal, B.; Lyons, K.M.; et al. Cooperative regulation of chondrocyte differentiation by CCN2 and CCN3 shown by a comprehensive analysis of the CCN family proteins in cartilage. J. Bone Miner. Res. 2008, 23, 1751–1764. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Le, A.T.; Yeger, H.; Perbal, B.; Alman, B.A. NOV (CCN3) regulation in the growth plate and CCN family member expression in cartilage neoplasia. J. Pathol. 2003, 201, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Omoto, S.; Nishida, K.; Yamaai, Y.; Shibahara, M.; Nishida, T.; Doi, T.; Asahara, H.; Nakanishi, T.; Inoue, H.; Takigawa, M. Expression and localization of connective tissue growth factor (CTGF/Hcs24/CCN2) in osteoarthritic cartilage. Osteoarthr. Cartil. 2004, 12, 771–778. [Google Scholar] [CrossRef]
- Nishida, T.; Kubota, S.; Kojima, S.; Kuboki, T.; Nakao, K.; Kushibiki, T.; Tabata, Y.; Takigawa, M. Regeneration of defects in articular cartilage in rat knee joints by CCN2 (connective tissue growth factor). J. Bone Miner. Res. 2004, 19, 1308–1319. [Google Scholar] [CrossRef]
- Abd El Kader, T.; Kubota, S.; Nishida, T.; Hattori, T.; Aoyama, E.; Janune, D.; Hara, E.S.; Ono, M.; Tabata, Y.; Kuboki, T.; et al. The regenerative effects of CCN2 independent modules on chondrocytes in vitro and osteoarthritis models in vivo. Bone 2014, 59, 180–188. [Google Scholar] [CrossRef]
- Heath, E.; Tahri, D.; Andermarcher, E.; Schofield, P.; Fleming, S.; Boulter, C.A. Abnormal skeletal and cardiac development, cardiomyopathy, muscle atrophy and cataracts in mice with a targeted disruption of the Nov (Ccn3) gene. BMC Dev. Biol. 2008, 8, 18. [Google Scholar] [CrossRef]
- Itoh, S.; Hattori, T.; Tomita, N.; Aoyama, E.; Yutani, Y.; Yamashiro, T.; Takigawa, M. CCN family member 2/connective tissue growth factor (CCN2/CTGF) has anti-aging effects that protect articular cartilage from age-related degenerative changes. PLoS ONE 2013, 8, e71156. [Google Scholar] [CrossRef]
- Maeda, A.; Ono, M.; Holmbeck, K.; Li, L.; Kilts, T.M.; Kram, V.; Noonan, M.L.; Yoshioka, Y.; McNerny, E.M.; Tantillo, M.A.; et al. WNT1-induced Secreted Protein-1 (WISP1), a Novel Regulator of Bone Turnover and Wnt Signaling. J. Biol. Chem. 2015, 290, 14004–14018. [Google Scholar] [CrossRef]
- Jiang, J.; Zhao, G.; Lyons, K.M. Characterization of bone morphology in CCN5/WISP5 knockout mice. J. Cell Commun. Signal 2018, 12, 265–270. [Google Scholar] [CrossRef]
- Kutz, W.E.; Gong, Y.; Warman, M.L. WISP3, the gene responsible for the human skeletal disease progressive pseudorheumatoid dysplasia, is not essential for skeletal function in mice. Mol. Cell Biol. 2005, 25, 414–421. [Google Scholar] [CrossRef]
- Nishida, T.; Kubota, S.; Nakanishi, T.; Kuboki, T.; Yosimichi, G.; Kondo, S.; Takigawa, M. CTGF/Hcs24, a hypertrophic chondrocyte-specific gene product, stimulates proliferation and differentiation, but not hypertrophy of cultured articular chondrocytes. J. Cell Physiol. 2002, 192, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Janune, D.; Kubota, S.; Nishida, T.; Kawaki, H.; Perbal, B.; Iida, S.; Takigawa, M. Novel effects of CCN3 that may direct the differentiation of chondrocytes. FEBS Lett. 2011, 585, 3033–3040. [Google Scholar] [CrossRef]
- Leu, S.J.; Lam, S.C.; Lau, L.F. Pro-angiogenic activities of CYR61 (CCN1) mediated through integrins alphavbeta3 and alpha6beta1 in human umbilical vein endothelial cells. J. Biol. Chem. 2002, 277, 46248–46255. [Google Scholar] [CrossRef] [PubMed]
- Shimo, T.; Nakanishi, T.; Nishida, T.; Asano, M.; Kanyama, M.; Kuboki, T.; Tamatani, T.; Tezuka, K.; Takemura, M.; Matsumura, T.; et al. Connective tissue growth factor induces the proliferation, migration, and tube formation of vascular endothelial cells in vitro, and angiogenesis in vivo. J. Biochem. 1999, 126, 137–145. [Google Scholar] [CrossRef]
- Lin, C.G.; Chen, C.C.; Leu, S.J.; Grzeszkiewicz, T.M.; Lau, L.F. Integrin-dependent functions of the angiogenic inducer NOV (CCN3): Implication in wound healing. J. Biol. Chem. 2005, 280, 8229–8237. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Nishida, T.; Shimo, T.; Kobayashi, K.; Kubo, T.; Tamatani, T.; Tezuka, K.; Takigawa, M. Effects of CTGF/Hcs24, a product of a hypertrophic chondrocyte-specific gene, on the proliferation and differentiation of chondrocytes in culture. Endocrinology 2000, 141, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Hong, D.; Iborra, F.; Sarno, S.; Enver, T. NOV (CCN3) functions as a regulator of human hematopoietic stem or progenitor cells. Science 2007, 316, 590–593. [Google Scholar] [CrossRef]
- Ishihara, J.; Umemoto, T.; Yamato, M.; Shiratsuchi, Y.; Takaki, S.; Petrich, B.G.; Nakauchi, H.; Eto, K.; Kitamura, T.; Okano, T. Nov/CCN3 regulates long-term repopulating activity of murine hematopoietic stem cells via integrin alphavbeta3. Int. J. Hematol. 2014, 99, 393–406. [Google Scholar] [CrossRef]
- Kolesnikova, T.V.; Lau, L.F. Human CYR61-mediated enhancement of bFGF-induced DNA synthesis in human umbilical vein endothelial cells. Oncogene 1998, 16, 747–754. [Google Scholar] [CrossRef]
- Vincent, T.; Hermansson, M.; Bolton, M.; Wait, R.; Saklatvala, J. Basic FGF mediates an immediate response of articular cartilage to mechanical injury. Proc. Natl. Acad. Sci. USA 2002, 99, 8259–8264. [Google Scholar] [CrossRef]
- Tang, X.; Muhammad, H.; McLean, C.; Miotla-Zarebska, J.; Fleming, J.; Didangelos, A.; Önnerfjord, P.; Leask, A.; Saklatvala, J.; Vincent, T.L. Connective tissue growth factor contributes to joint homeostasis and osteoarthritis severity by controlling the matrix sequestration and activation of latent TGFβ. Ann. Rheum. Dis. 2018, 77, 1372–1380. [Google Scholar] [CrossRef]
- Guillon-Munos, A.; Oikonomopoulou, K.; Michel, N.; Smith, C.R.; Petit-Courty, A.; Canepa, S.; Reverdiau, P.; Heuzé-Vourc’h, N.; Diamandis, E.P.; Courty, Y. Kallikrein-related peptidase 12 hydrolyzes matricellular proteins of the CCN family and modifies interactions of CCN1 and CCN5 with growth factors. J. Biol. Chem. 2011, 286, 25505–25518. [Google Scholar] [CrossRef]
- Nishida, T.; Kubota, S.; Aoyama, E.; Janune, D.; Maeda, A.; Takigawa, M. Effect of CCN2 on FGF2-induced proliferation and MMP9 and MMP13 productions by chondrocytes. Endocrinology 2011, 11, 4232–4241. [Google Scholar] [CrossRef]
- Abd El Kader, T.; Kubota, S.; Anno, K.; Tanaka, S.; Nishida, T.; Furumatsu, T.; Aoyama, E.; Kuboki, T.; Takigawa, M. Direct interaction between CCN family protein 2 and fibroblast growth factor 1. J. Cell Commun. Signal 2014, 8, 157–163. [Google Scholar] [CrossRef]
- Aoyama, E.; Kubota, S.; Takigawa, M. CCN2/CTGF binds to fibroblast growth factor receptor 2 and modulates its signaling. FEBS Lett. 2012, 586, 4270–4275. [Google Scholar] [CrossRef]
- Kaasbøll, O.J.; Gadicherla, A.K.; Wang, J.H.; Monsen, V.T.; Hagelin, E.M.V.; Dong, M.Q.; Attramadal, H. Connective tissue growth factor (CCN2) is a matricellular preproprotein controlled by proteolytic activation. J. Biol. Chem. 2018, 293, 17953–17970. [Google Scholar] [CrossRef]
- Kubota, S.; Eguchi, T.; Shimo, T.; Nishida, T.; Hattori, T.; Kondo, S.; Nakanishi, T.; Takigawa, M. Novel mode of processing and secretion of connective tissue growth factor/ecogenin (CTGF/Hcs24) in chondrocytic HCS-2/8 cells. Bone 2001, 29, 155–161. [Google Scholar] [CrossRef]
- Keenan, C.M.; Ramos-Mucci, L.; Kanakis, I.; Milner, P.I.; Leask, A.; Abraham, D.; Bou-Gharios, G.; Poulet, B. Post-traumatic osteoarthritis development is not modified by postnatal chondrocyte deletion of Ccn2. Dis. Models Mech. 2020, 13, dmm044719. [Google Scholar] [CrossRef]
- Burt, P.M.; Xiao, L.; Doetschman, T.; Hurley, M.M. Ablation of low-molecular-weight FGF2 isoform accelerates murine osteoarthritis while loss of high-molecular-weight FGF2 isoforms offers protection. J. Cell Physiol. 2019, 234, 4418–4431. [Google Scholar] [CrossRef]
- Elseoudi, A.; Nishida, T.; Mizukawa, T.; Hattori, T.; Kawata, K.; Taha, E.A.; Takigawa, M.; Kubota, S. Bipartite regulation of cellular communication network factor 2 and fibroblast growth factor 1 genes by fibroblast growth factor 1 through histone deacetylase 1 and fork head box protein A1. J. Cell Commun. Signal 2021, 15, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.W.; Brosens, J.J.; Gomes, A.R.; Koo, C.Y. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Uchii, M.; Tamura, T.; Suda, T.; Kakuni, M.; Tanaka, A.; Miki, I. Role of fibroblast growth factor 8 (FGF8) in animal models of osteoarthritis. Arthritis Res. Ther. 2008, 10, R90. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Cui, Y.; Zhang, D.; Xie, J.; Zhou, X. The role of fibroblast growth factor 8 in cartilage development and disease. J. Cell Mol. Med. 2022, 26, 990–999. [Google Scholar] [CrossRef]
- Iwafuchi, M.; Cuesta, I.; Donahue, G.; Takenaka, N.; Osipovich, A.B.; Magnuson, M.A.; Roder, H.; Seeholzer, S.H.; Santisteban, P.; Zaret, K.S. Gene network transitions in embryos depend upon interactions between a pioneer transcription factor and core histones. Nat. Genet. 2020, 52, 418–427. [Google Scholar] [CrossRef]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef]
- Henninger, J.E.; Oksuz, O.; Shrinivas, K.; Sagi, I.; LeRoy, G.; Zheng, M.M.; Andrews, J.O.; Zamudio, A.V.; Lazaris, C.; Hannett, N.M.; et al. RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 2021, 184, 207–225. [Google Scholar] [CrossRef]
- Onoguchi-Mizutani, R.; Akimitsu, N. Long noncoding RNA and phase separation in cellular stress response. J. Biochem. 2022, 171, 269–276. [Google Scholar] [CrossRef]
- Lu, H.; Yu, D.; Hansen, A.S.; Ganguly, S.; Liu, R.; Heckert, A.; Darzacq, X.; Zhou, Q. Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 2018, 558, 318–323. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | FGF Family | CCN Family |
|---|---|---|
| Family members | 22 | 6 |
| Molecular weights | 17–34 kD | 1 26–42 kD |
| Molecular structure | Single core region | Four modules linked tandem |
| Receptors | Specific | Multiple and diverse |
| Function | Specific | Context-dependent |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubota, S.; Aoyama, E.; Takigawa, M.; Nishida, T. Fibroblast Growth Factors and Cellular Communication Network Factors: Intimate Interplay by the Founding Members in Cartilage. Int. J. Mol. Sci. 2022, 23, 8592. https://doi.org/10.3390/ijms23158592
Kubota S, Aoyama E, Takigawa M, Nishida T. Fibroblast Growth Factors and Cellular Communication Network Factors: Intimate Interplay by the Founding Members in Cartilage. International Journal of Molecular Sciences. 2022; 23(15):8592. https://doi.org/10.3390/ijms23158592
Chicago/Turabian StyleKubota, Satoshi, Eriko Aoyama, Masaharu Takigawa, and Takashi Nishida. 2022. "Fibroblast Growth Factors and Cellular Communication Network Factors: Intimate Interplay by the Founding Members in Cartilage" International Journal of Molecular Sciences 23, no. 15: 8592. https://doi.org/10.3390/ijms23158592
APA StyleKubota, S., Aoyama, E., Takigawa, M., & Nishida, T. (2022). Fibroblast Growth Factors and Cellular Communication Network Factors: Intimate Interplay by the Founding Members in Cartilage. International Journal of Molecular Sciences, 23(15), 8592. https://doi.org/10.3390/ijms23158592