
Enhancer Regulation of Dopaminergic Neurochemical Transmission in the Striatum

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
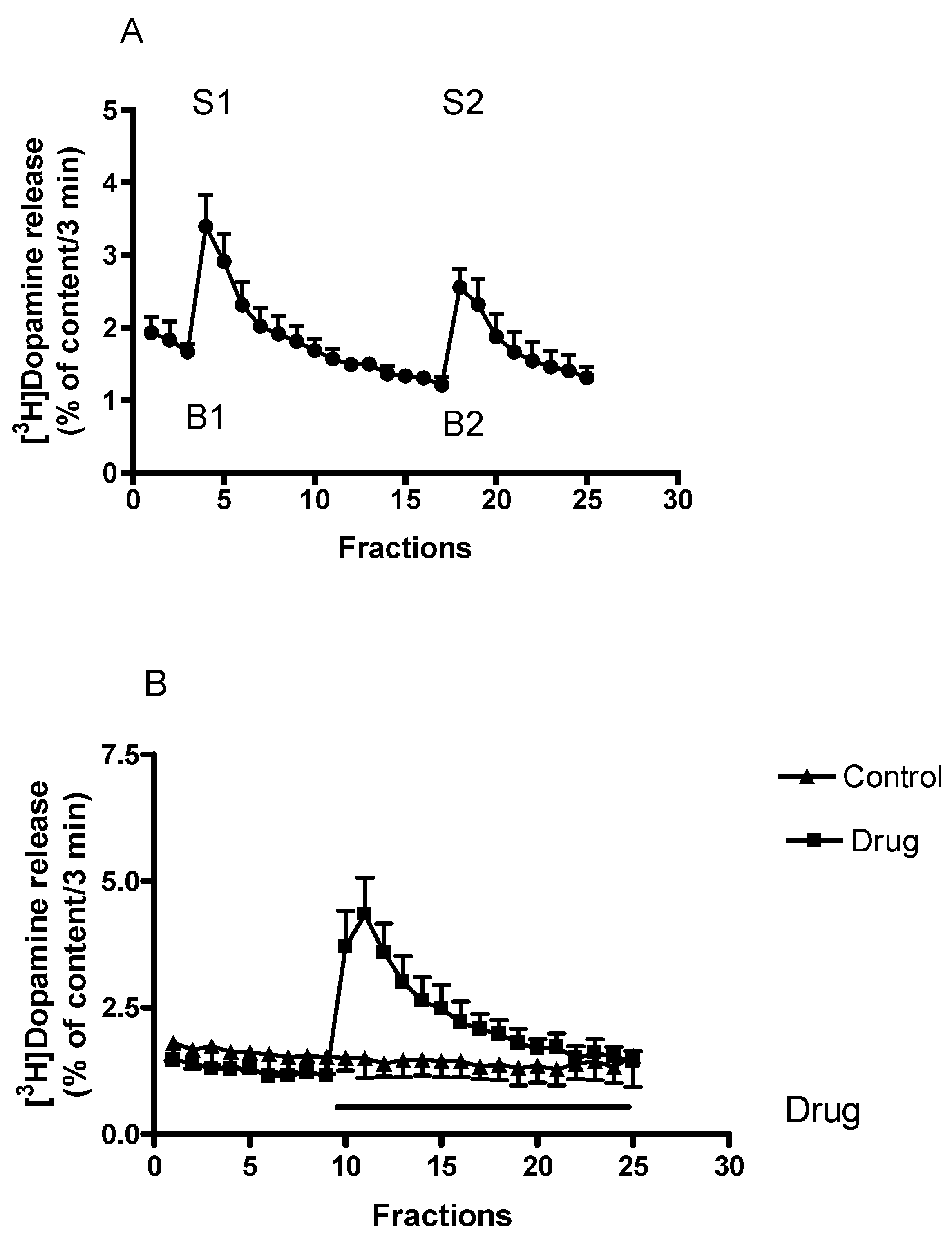
2.1. Characterization of [3H]Dopamine Release from Rat Striatum
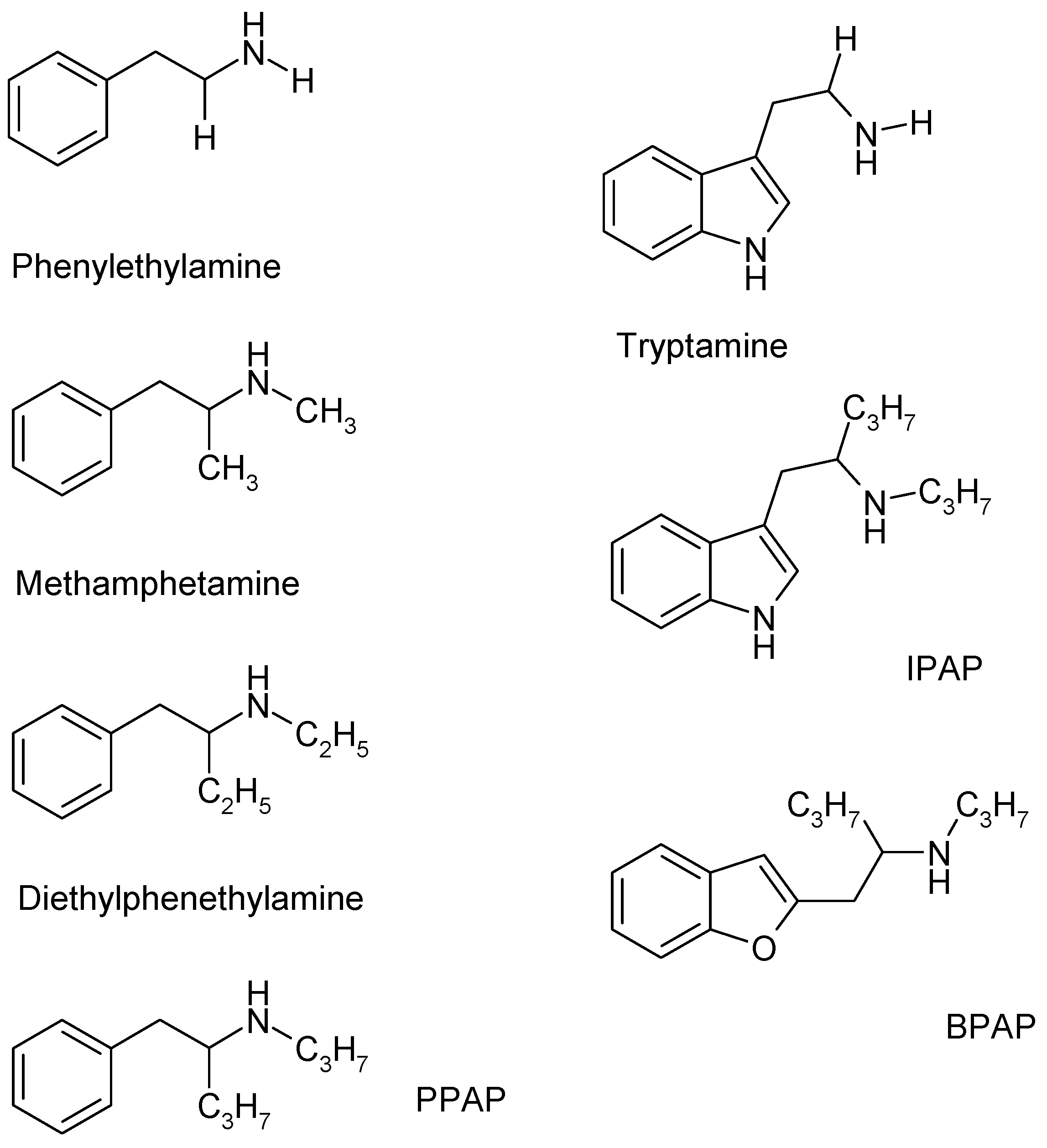
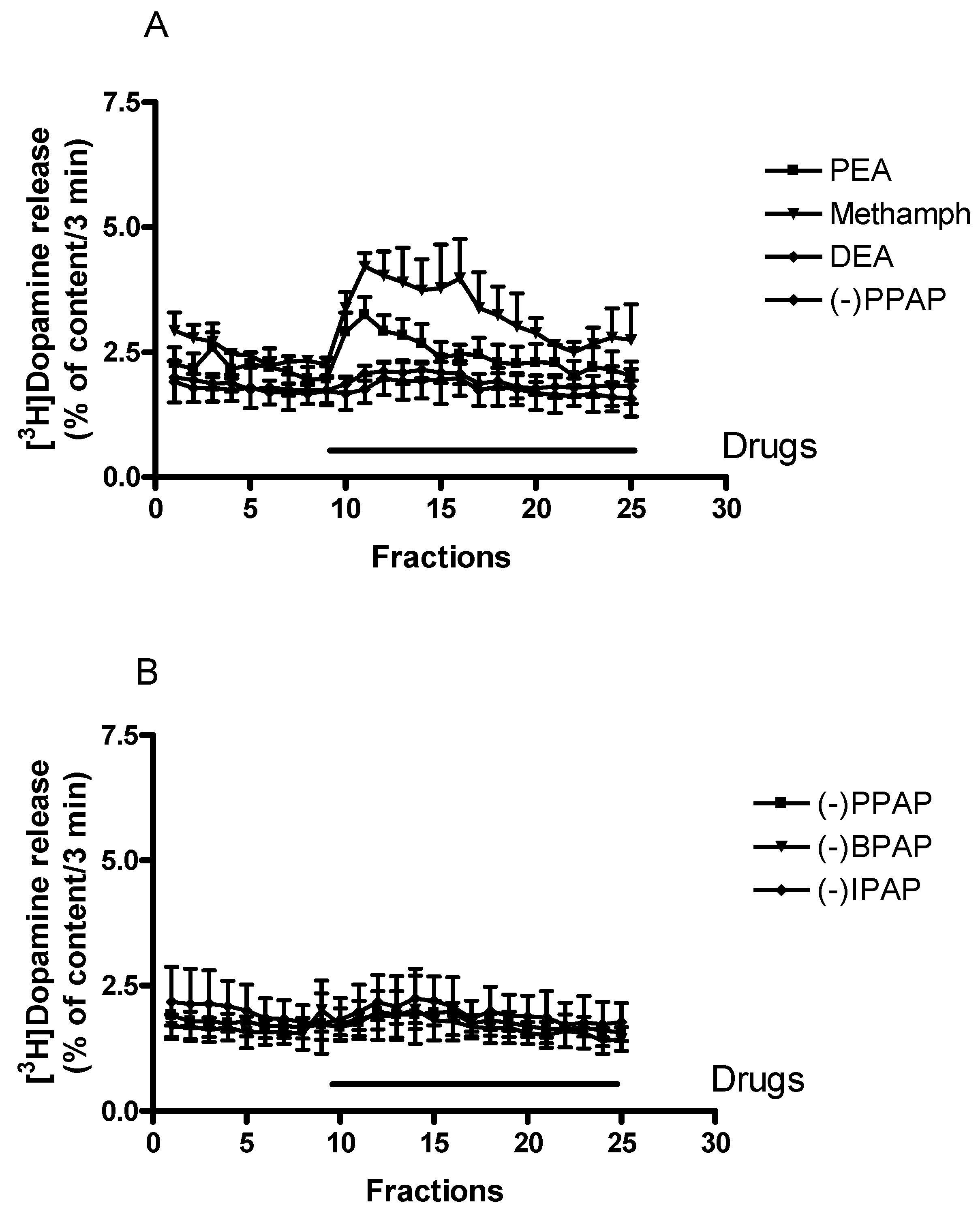
2.2. Effect of Trace Amines, Releaser, and Enhancer Compounds on Non-Vesicular Release of [3H]Dopamine from Rat Striatum
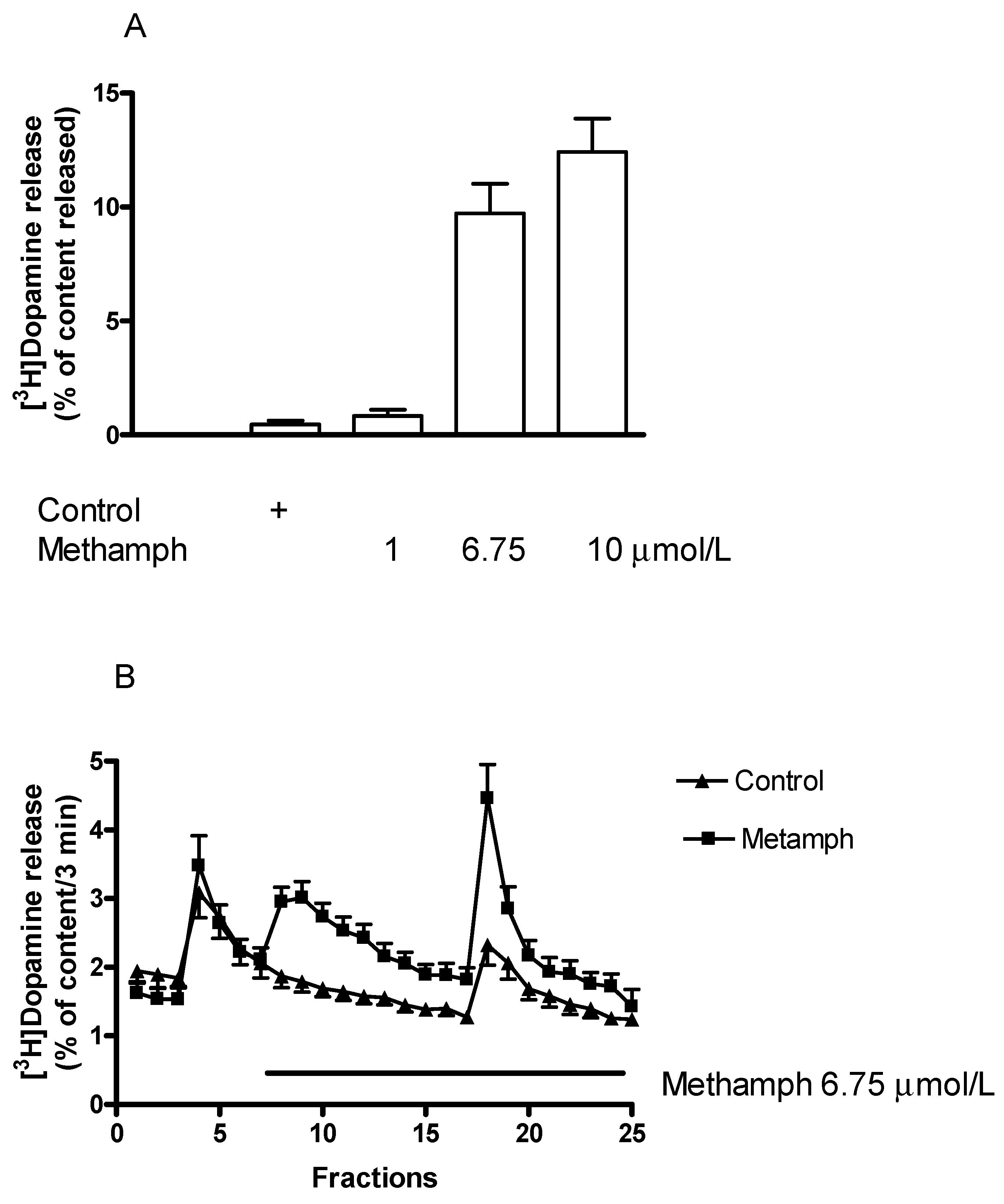
2.3. Effects of the Catecholamine Releaser (±)Methamphetamine on Vesicular and Non-Vesicular [3H]Dopamine Release in Rat Striatum
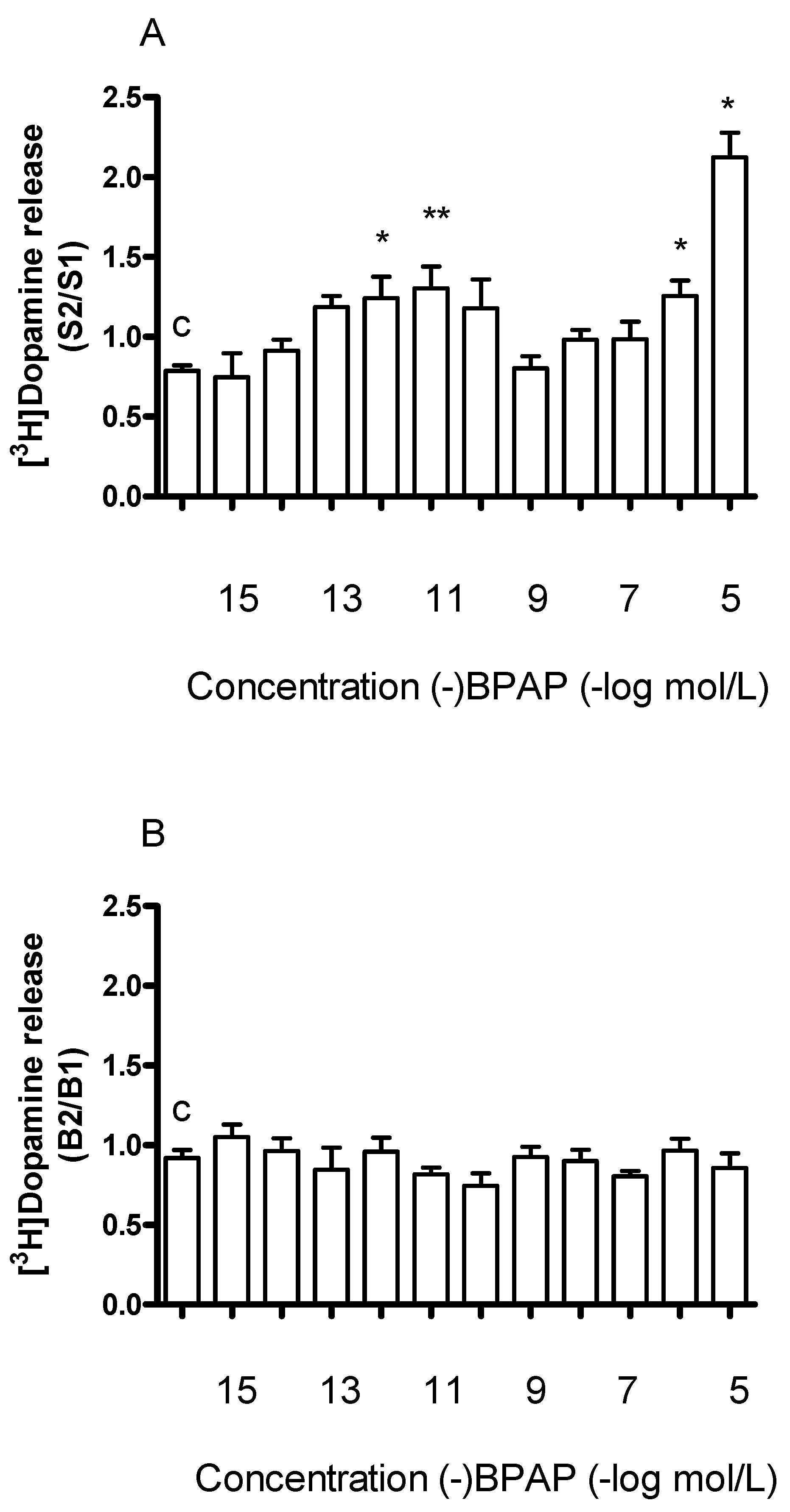
2.4. Effects of the Catecholamine Activity Enhancer (−)BPAP on Vesicular and Non-Vesicular [3H]Dopamine Release in Rat Striatum
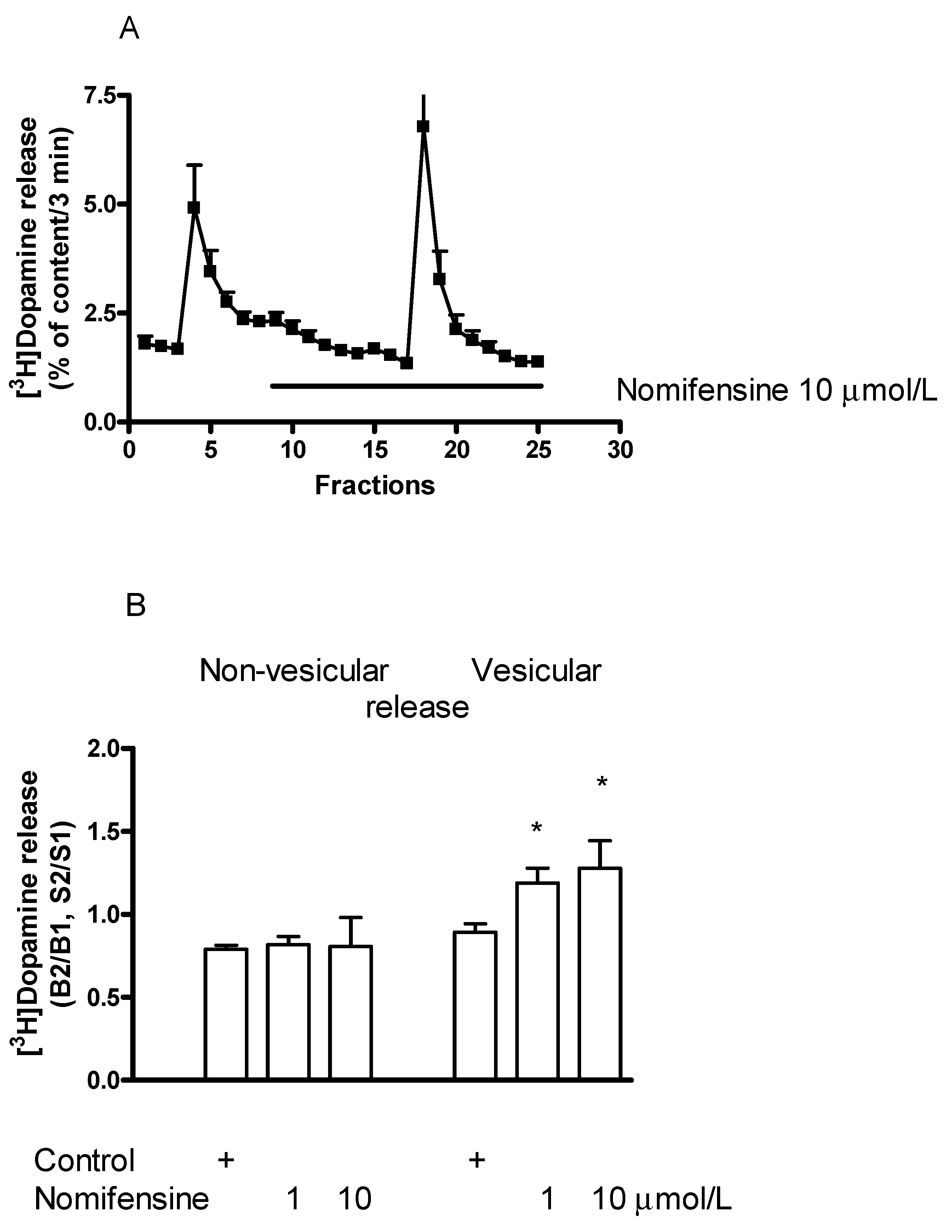
2.5. Effect of the Dopamine Transporter Inhibitor Nomifensine on [3H]Dopamine Release in Rat Striatum
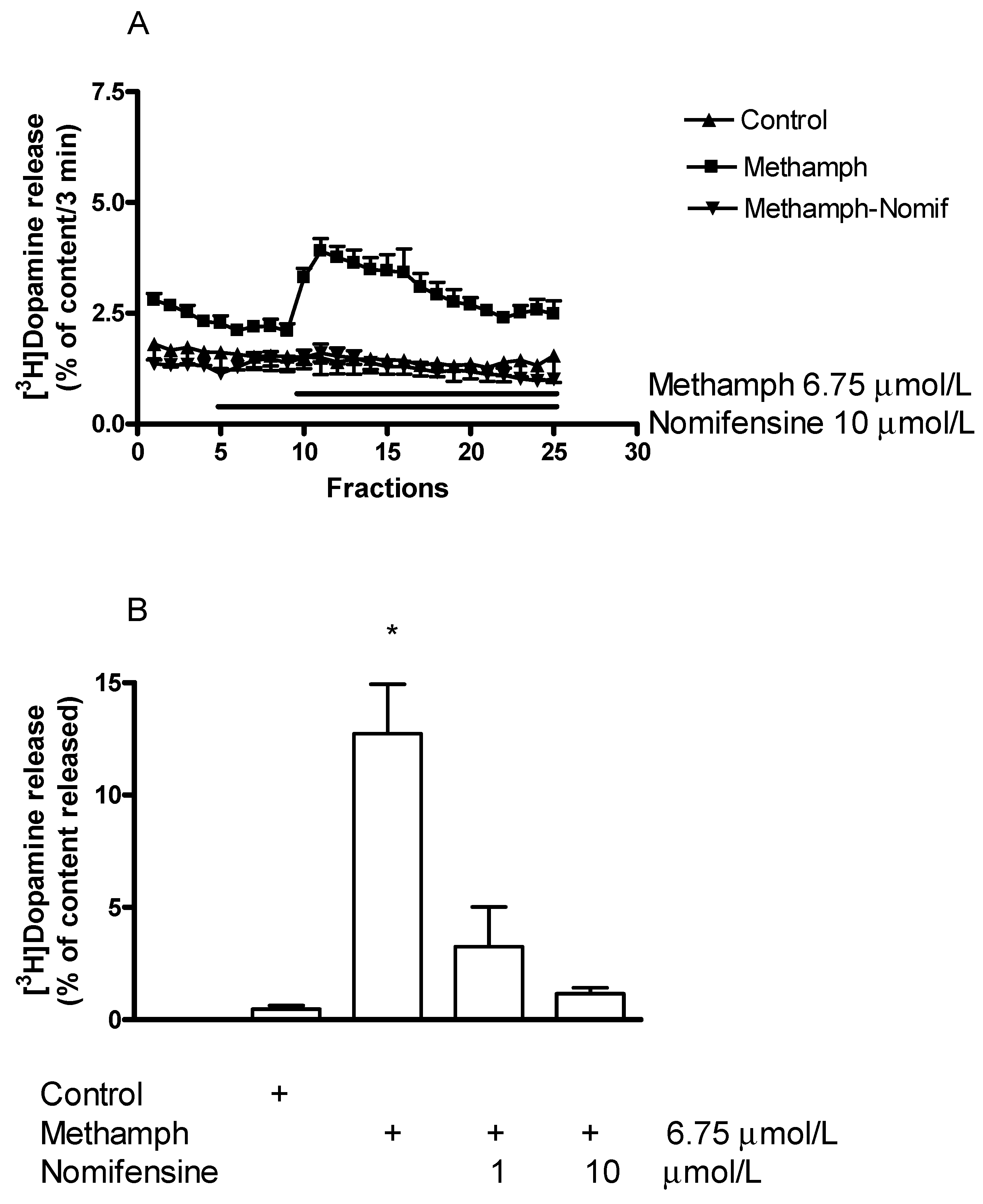
2.5.1. Effect of Nomifensine on (±)Methamphetamine-Induced [3H]Dopamine Release
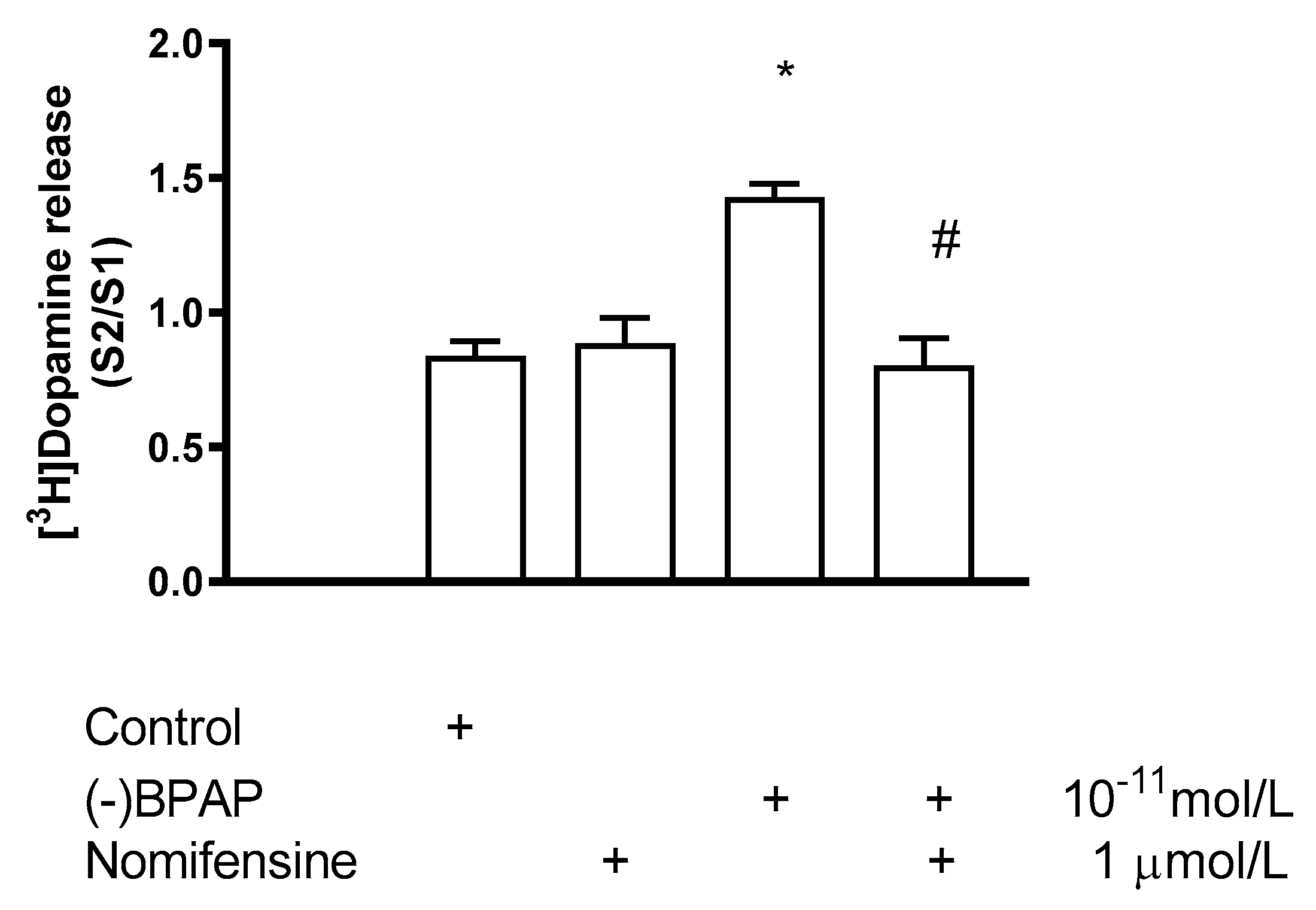
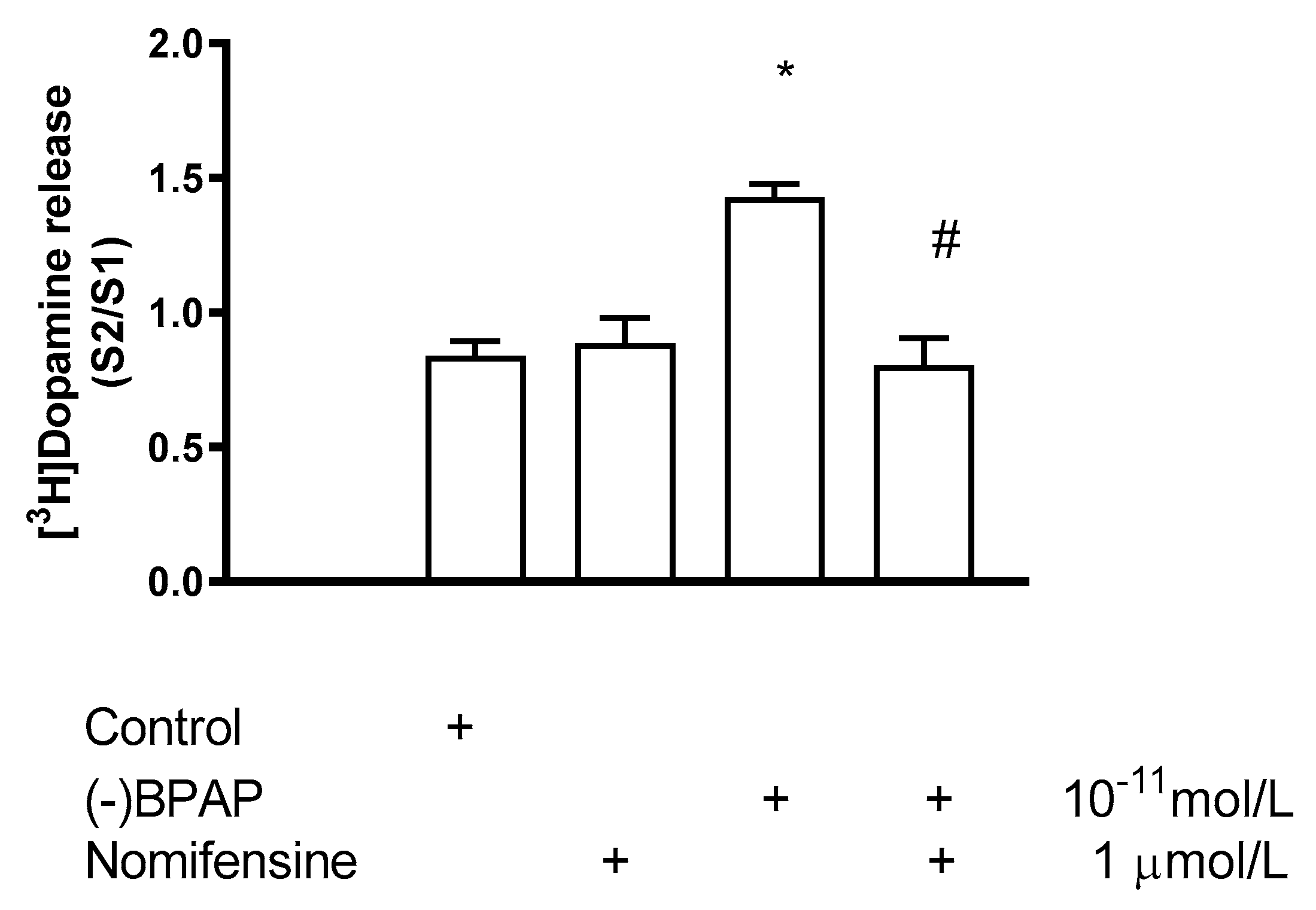
2.5.2. Effect of Nomifensine on (−)BPAP-Induced [3H]Dopamine Release
2.6. Effect of the TAAR1 Antagonist EPPTB on [3H]Dopamine Release in Rat Striatum
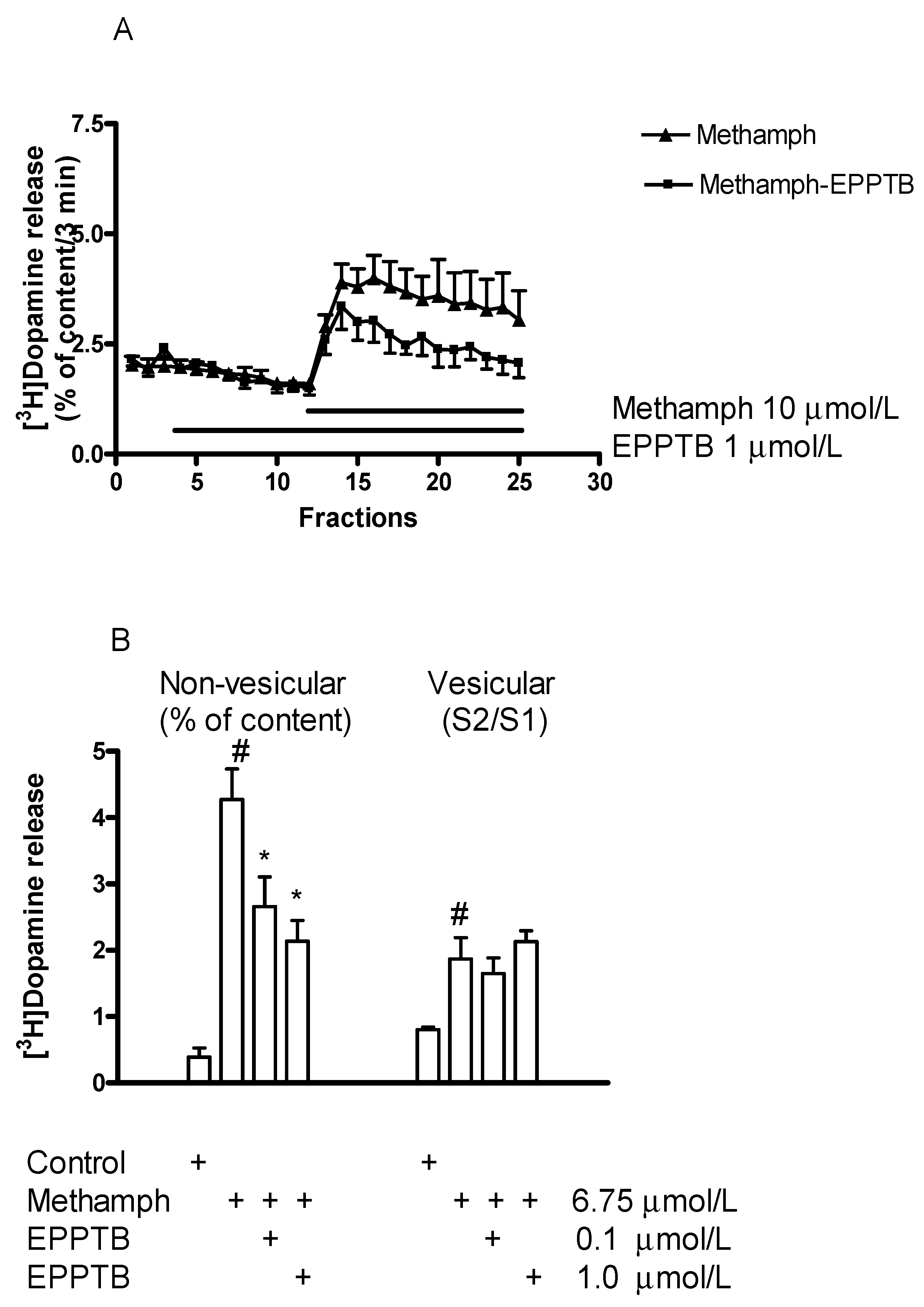
2.6.1. Effect of EPPTB on (±)Methamphetamine-Induced [3H]Dopamine Release
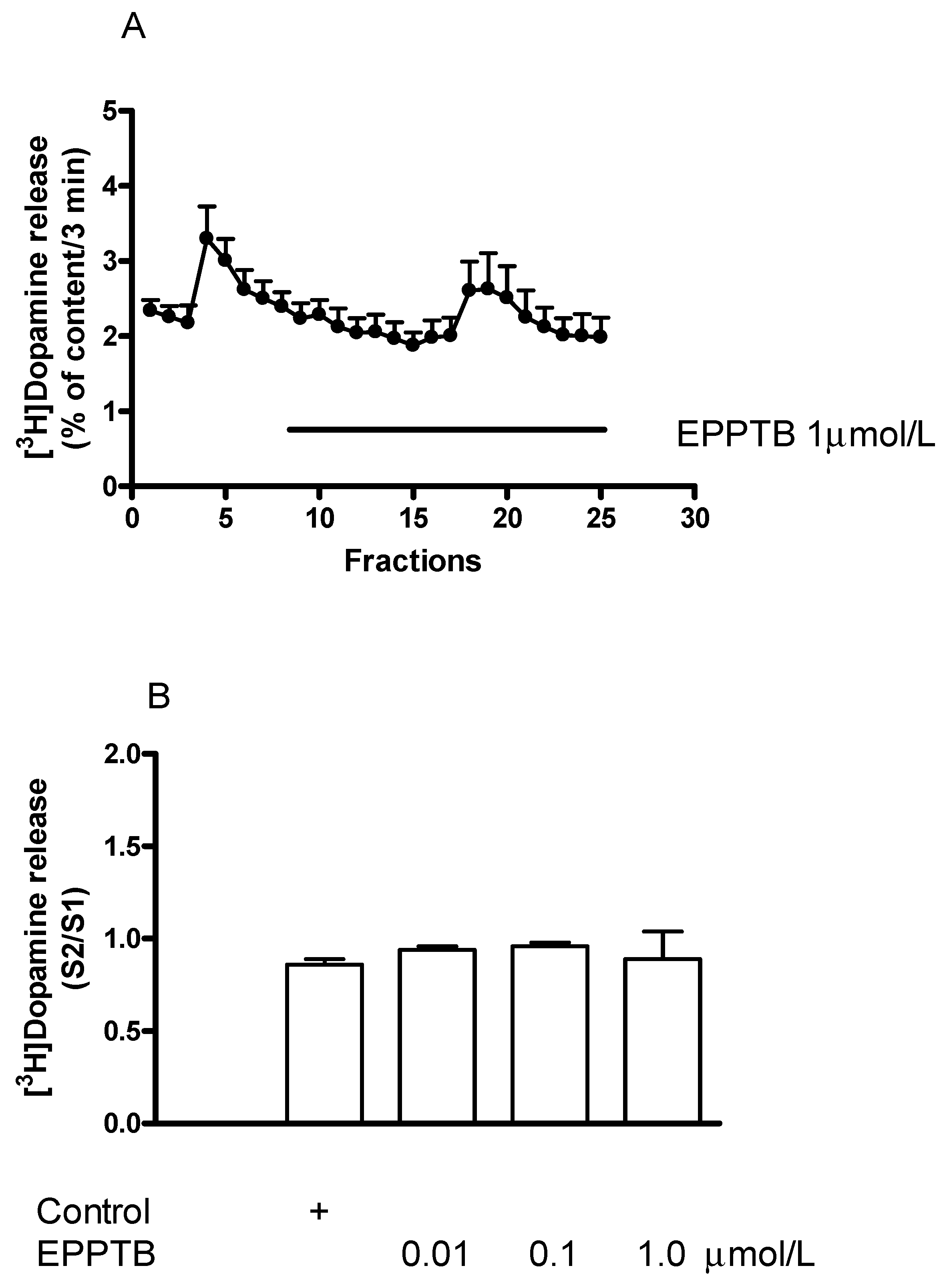
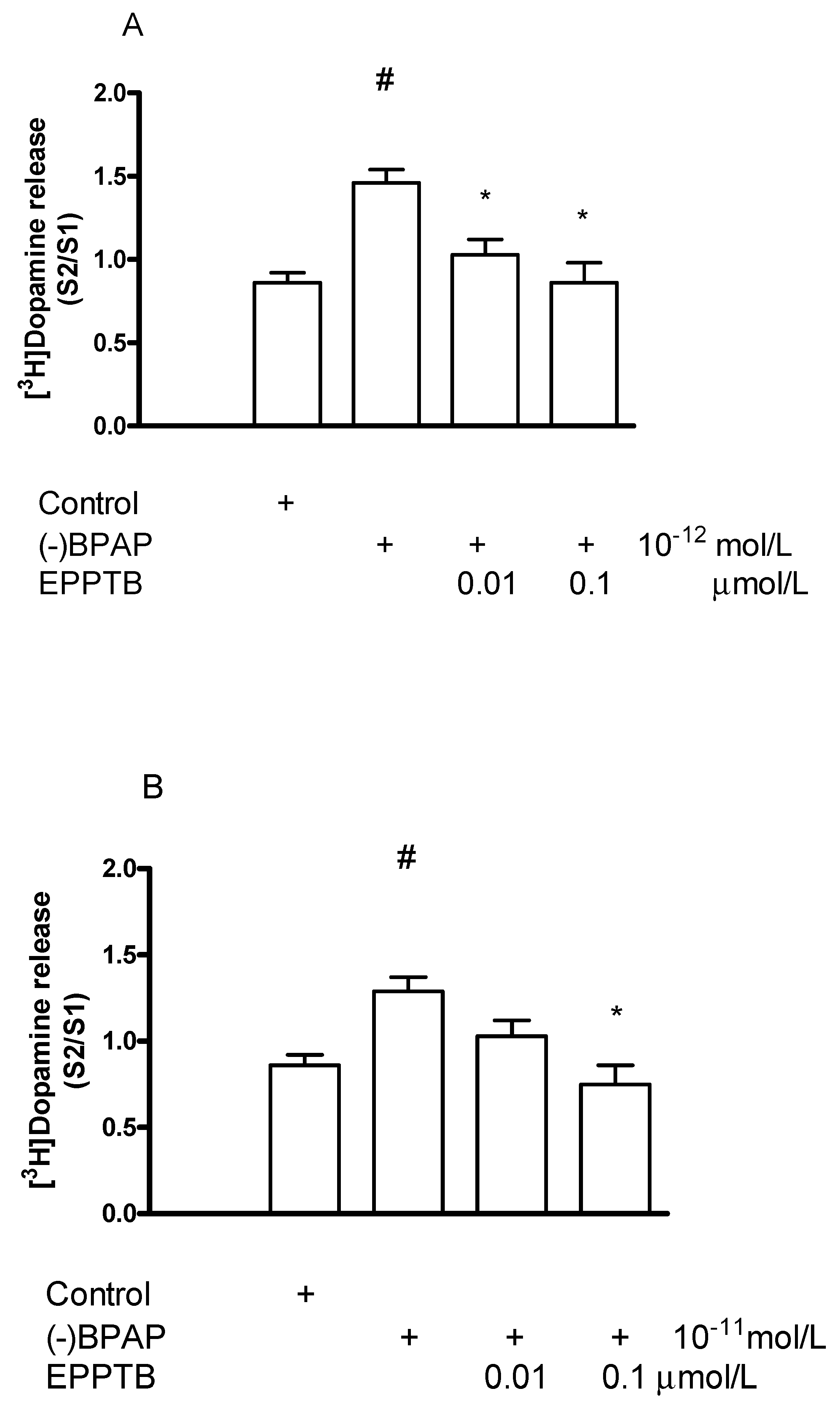
2.6.2. Effect of EPPTB on (−)BPAP-Induced [3H]Dopamine Release
2.7. The Role of the Protein Kinase-Mediated Phosphorylation in the Dopamine Releaser and Enhancer Drug Actions
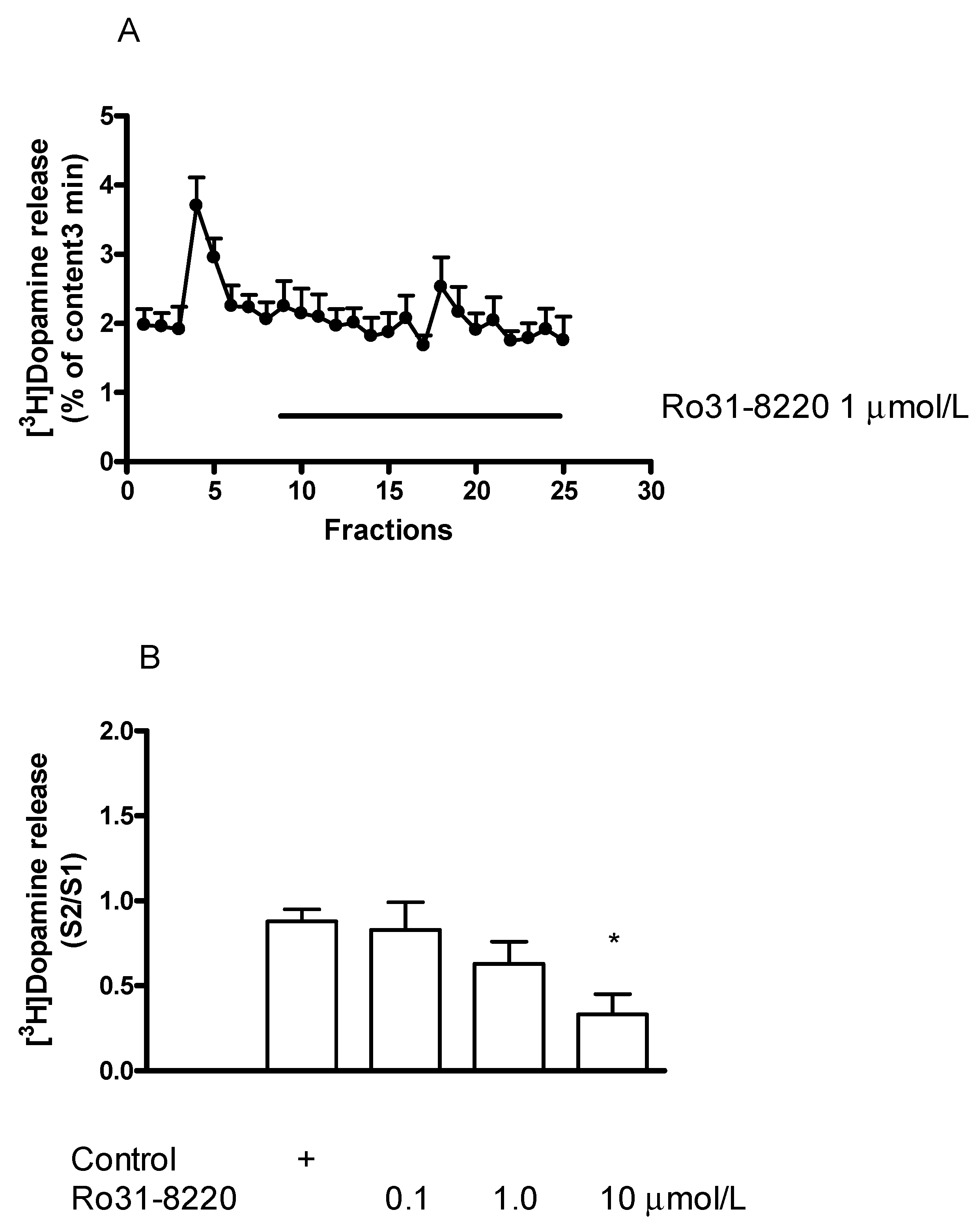
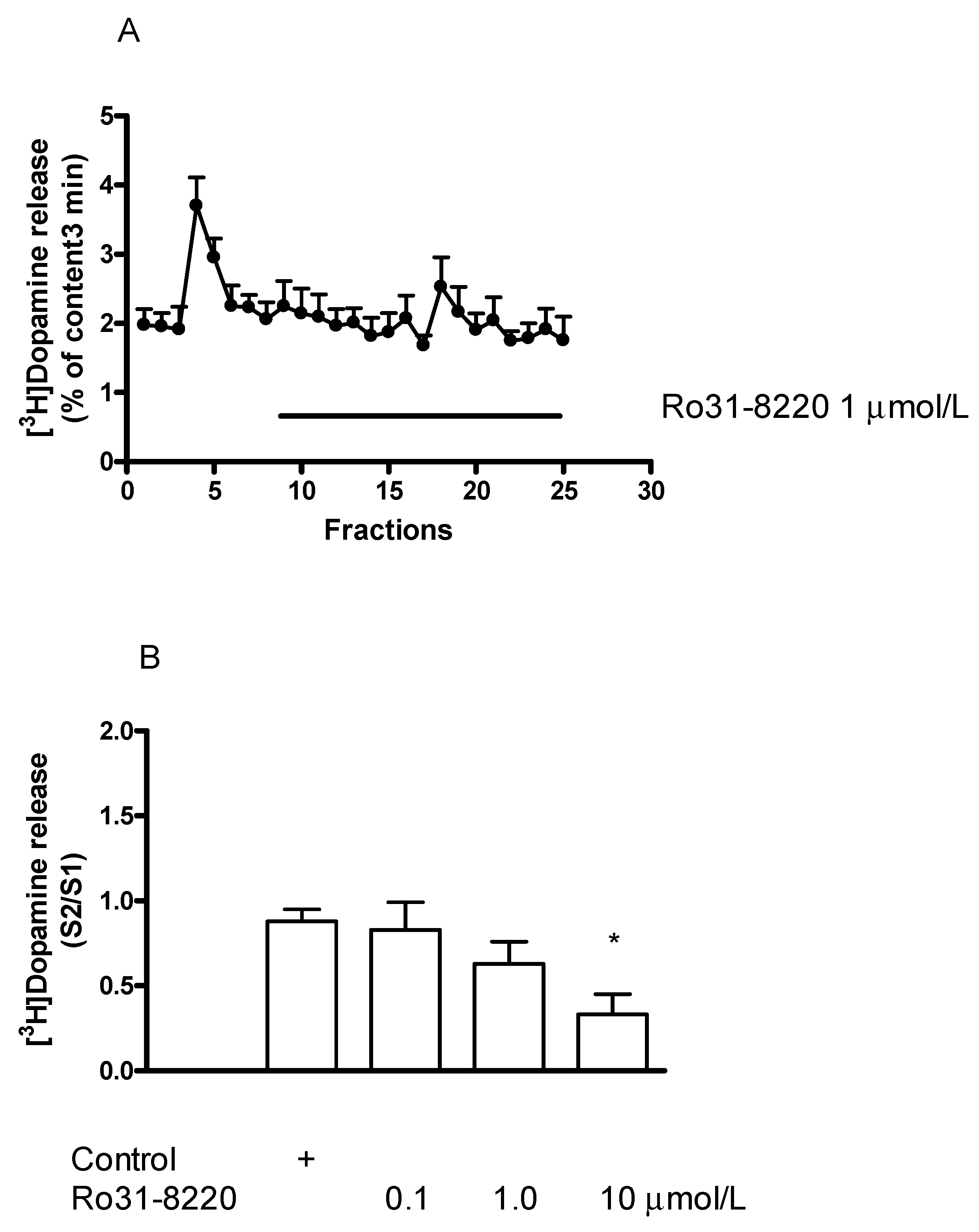
2.7.1. Effect of the Protein Kinase C Inhibitor Ro31-8220 on [3H]Dopamine Release in Rat Striatum: An Inhibition
2.7.2. Effect of Ro31-8220 on (±)Methamphetamine-Induced [3H]Dopamine Release
2.7.3. Effect of Ro31-8220 on (−)BPAP-Stimulated [3H]Dopamine Release
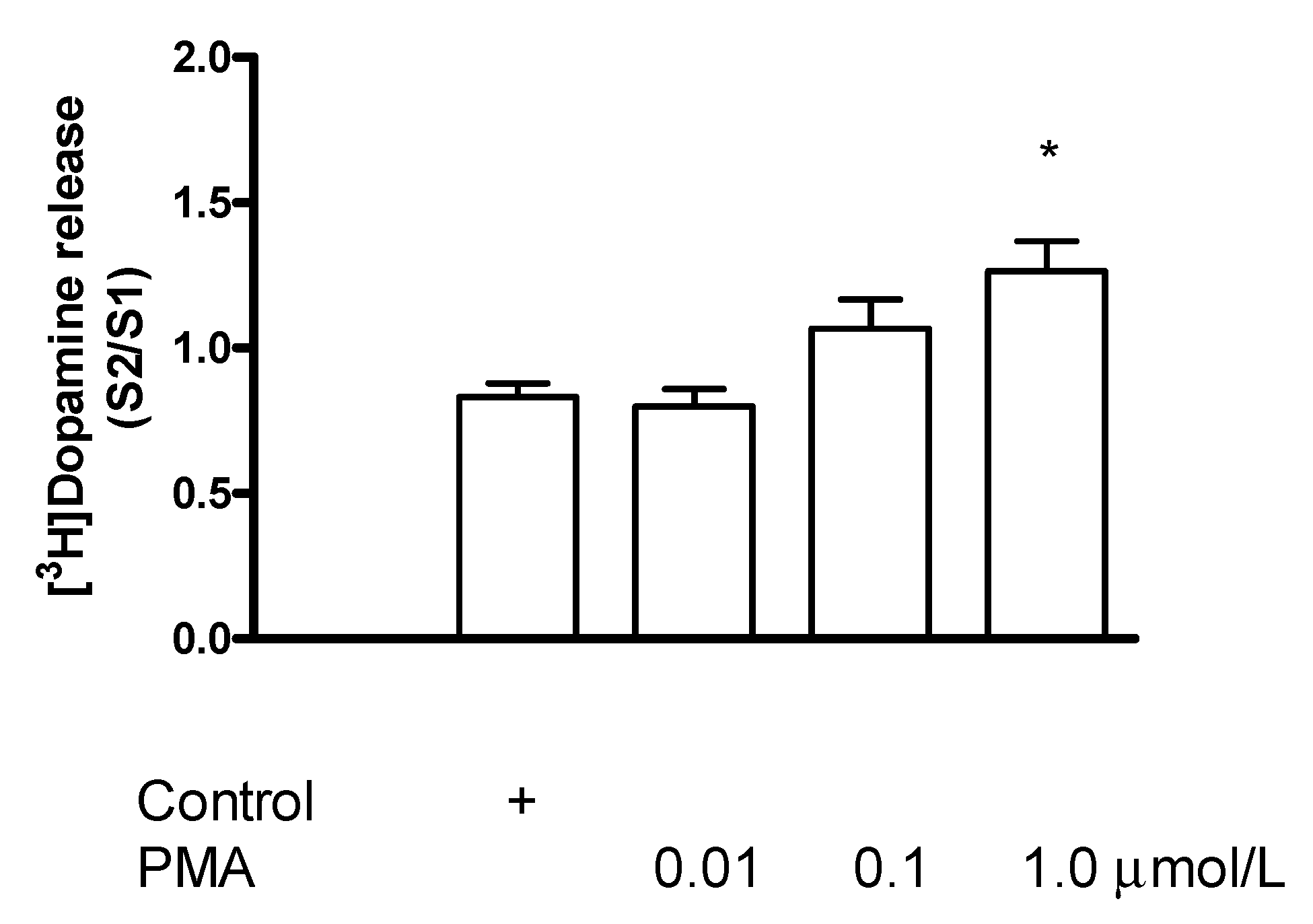
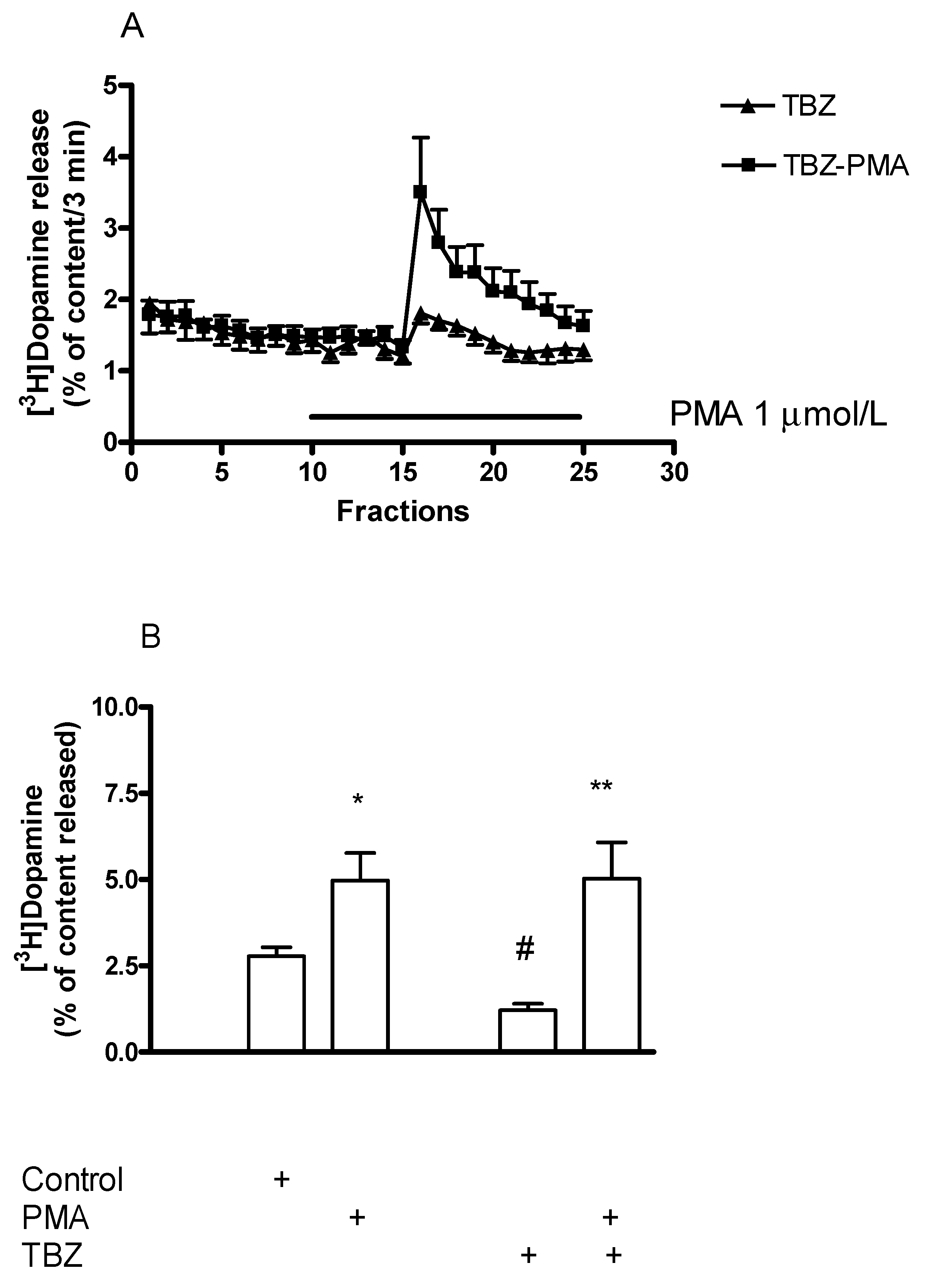
2.7.4. Effect of the Protein Kinase C Activator Phorbol 12-Myristate 13-Acetate on [3H]Dopamine Release in Rat Striatum: A Stimulation
2.7.5. Effect of Phorbol 12-Myristate 13-Acetate on (−)BPAP-Stimulated [3H]Dopamine Release
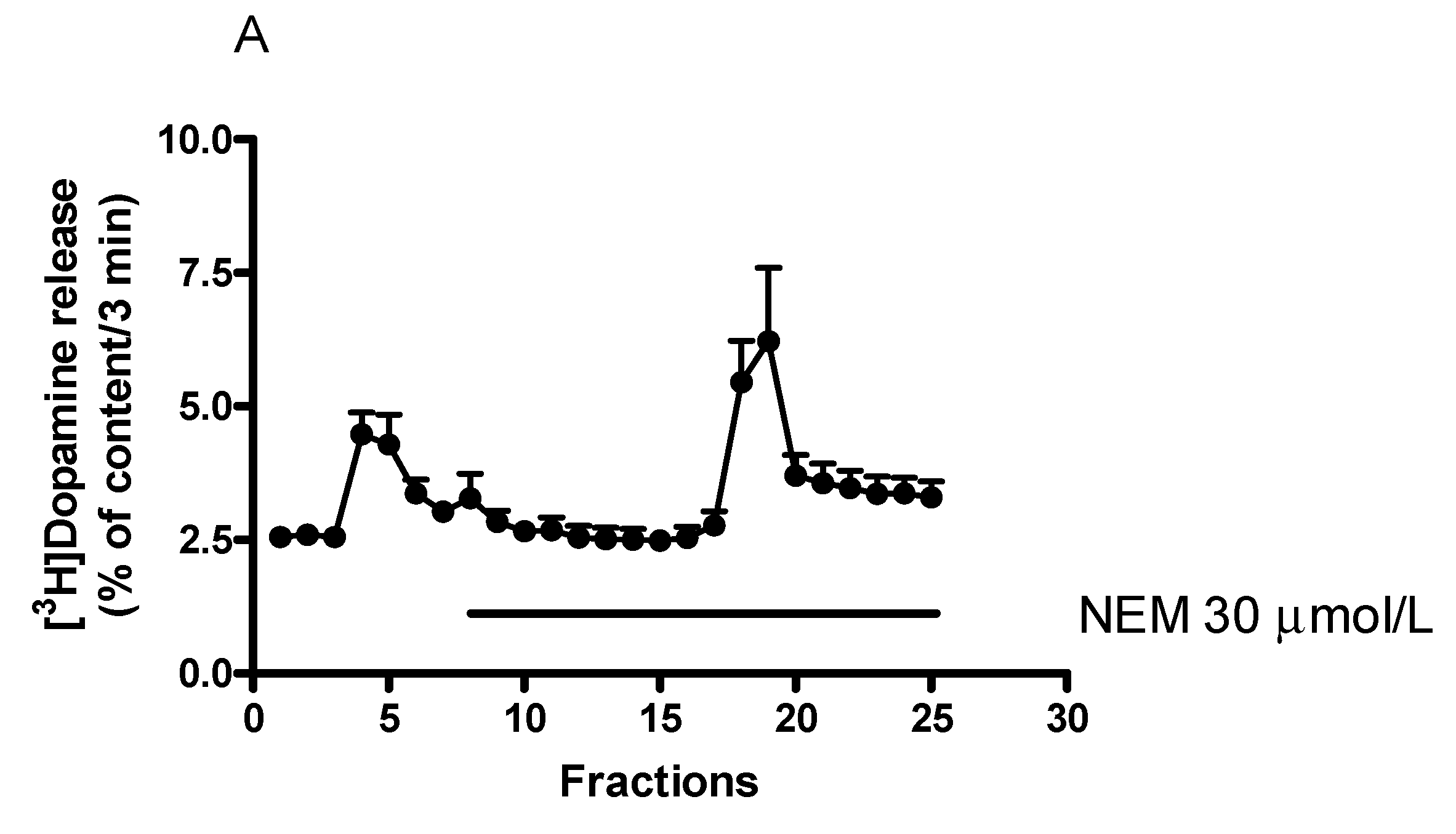
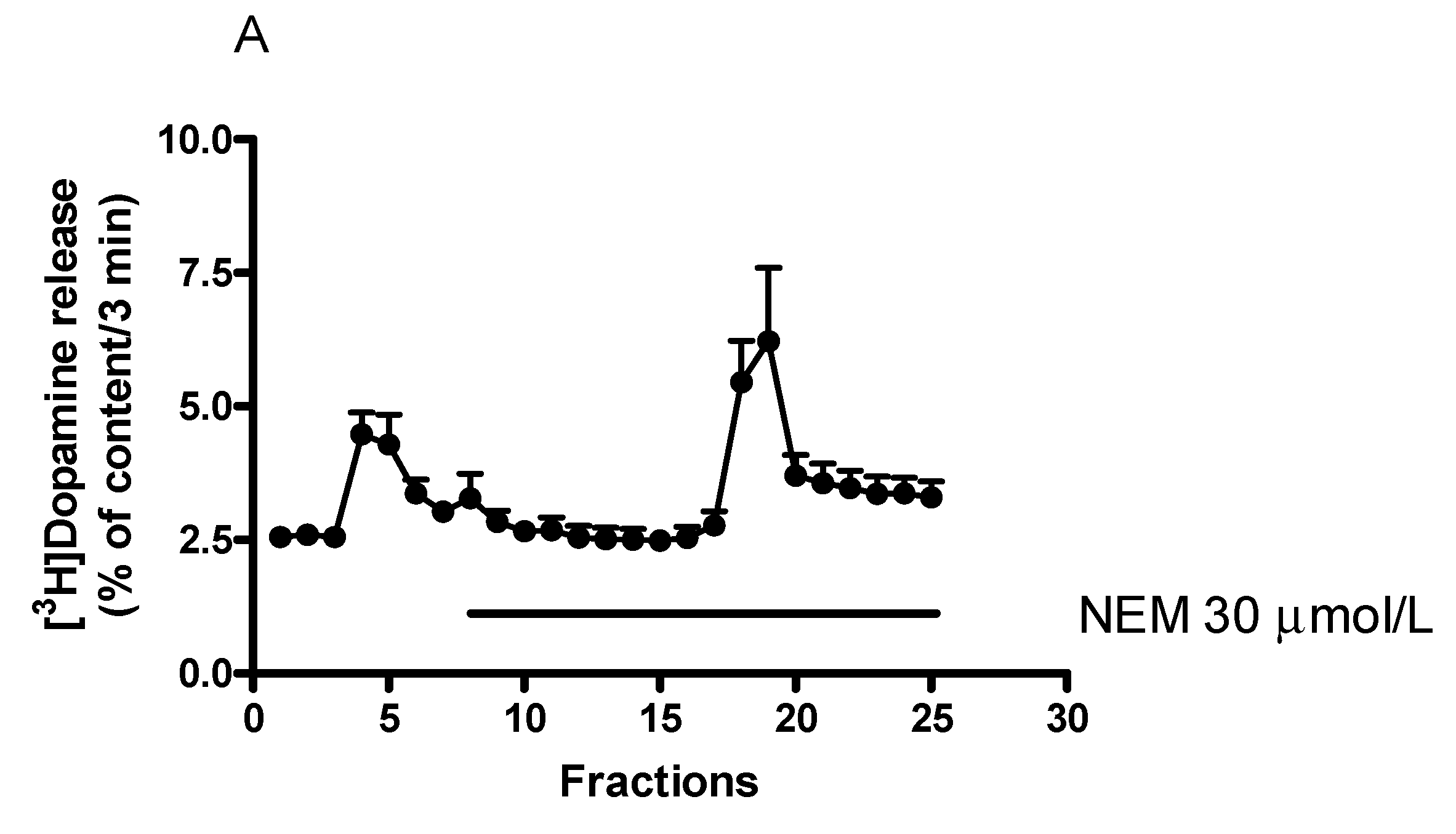
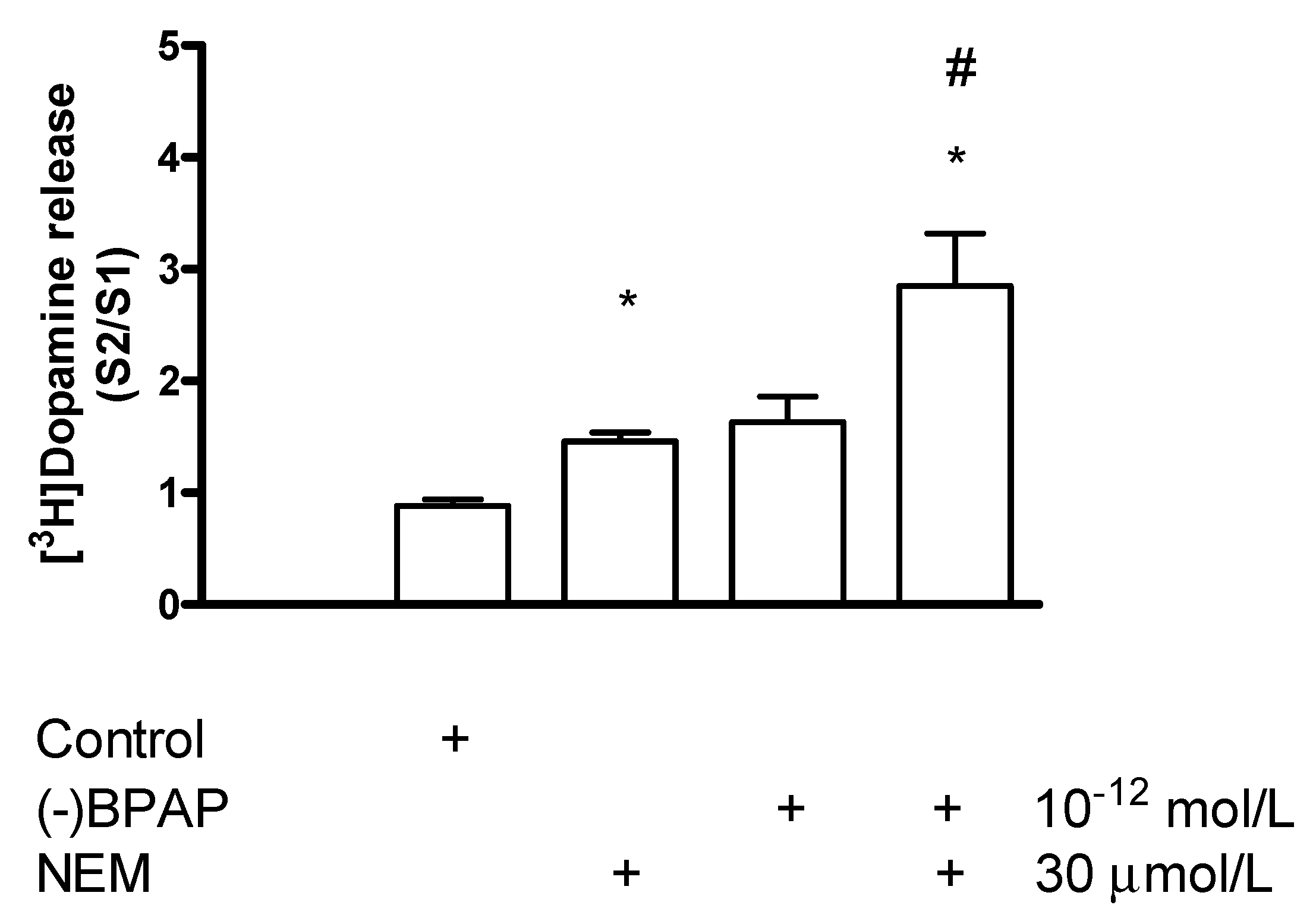
2.8. Effect of N-Ethylmaleimide-Sensitive Factor Inhibitor N-Ethylmaleimide on [3H]Dopamine Release in Rat Striatum
Effect of N-Ethylmaleimide on (−)BPAP-Stimulated [3H]Dopamine Release
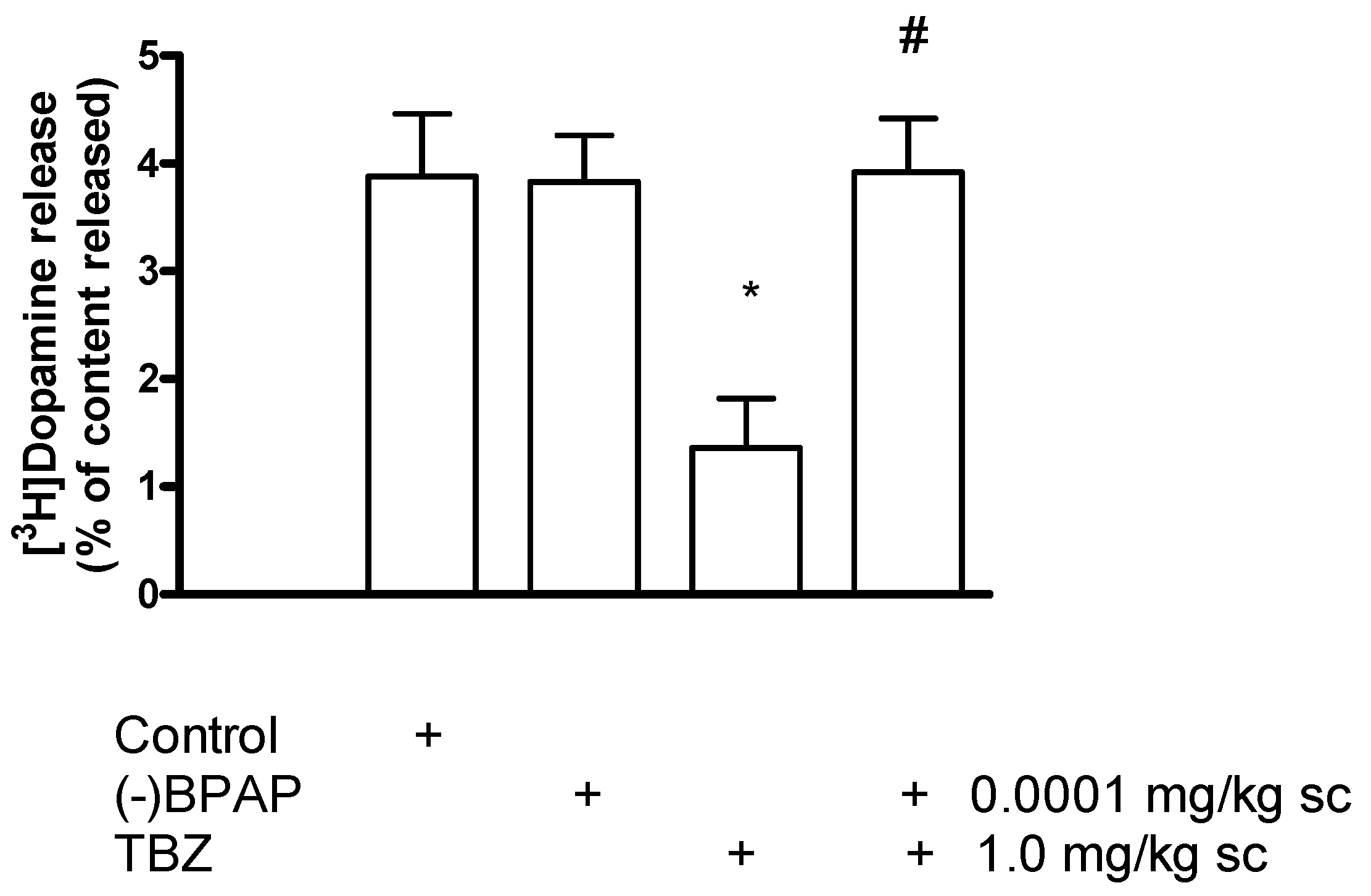
2.9. The Role of the Vesicular Monoamine Transporter 2 in [3H]Dopamine Release in Rat Striatum: Effect of Tetrabenazine
Effect of Phorbol 12-Myristate 13-Acetate on [3H]Dopamine Release in Tetrabenazine-Pretreated Rat Striatum
2.10. The Role of TAAR1 in D2 Receptor-Mediated Presynaptic Negative Feedback Inhibition
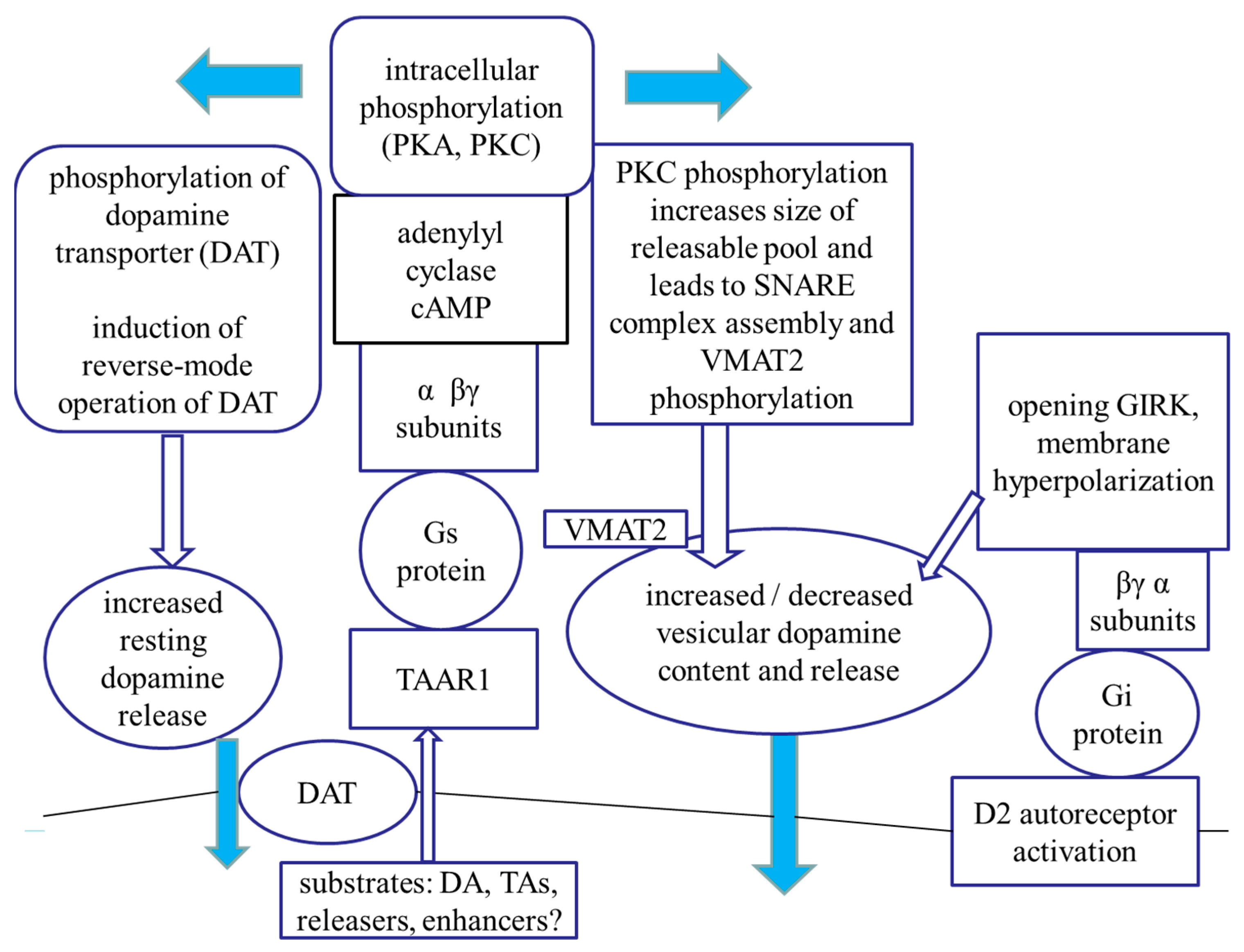
3. Discussion
3.1. Effects of the Enhancer Compound (−)BPAP on [3H]Dopamine Release from Striatal Slices of the Rat
3.1.1. Evidence That TAAR1 Is Involved in the Mechanism of Action of the Enhancer Compound (−)BPAP
3.1.2. Evidence That DAT Is Involved in Entry of the Enhancer Compound (−)BPAP into the Presynaptic Axon Terminals
3.1.3. Vesicular Dopamine Release following TAAR1 Activation: The Role of Intracellular Phosphorylation
3.1.4. The Role of SNARE Core Complex in the Enhancer Regulation of Dopamine Release
3.1.5. The Role of Vesicular Monoamine Transporter 2 in the Enhancer Regulation of Dopamine Release in Rat Striatum
3.2. Effects of the Releaser Compound (±)Methamphetamine on [3H]Dopamine Release from Striatal Slices of the Rat
3.2.1. Differential Effect of TAAR1 in (±)Methamphetamine-Induced Vesicular and Non-Vesicular Dopamine Release
3.2.2. Evidence That DAT Is Involved in Entry of (±)Methamphetamine into the Presynaptic Axon Terminals
3.2.3. The Role of Intracellular Phosphorylation in Non-Vesicular Dopamine Release following TAAR1 Activation
3.2.4. The Methamphetamine-Induced Vesicular Dopamine Release: The Role of Vesicular Monoamine Transporter 2
3.3. The TAAR Signaling: A Central Regulator of Presynaptic Dopaminergic Neurotransmission Events
4. Materials and Methods
4.1. Animals and Drug Treatments
4.2. Preparation of Rat Brain Slices
4.3. Release of [3H]Dopamine from Rat Brain Slices
4.4. Determination of [3H]Dopamine Efflux
4.5. Statistical Analyses
4.6. Materials
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| (−)BPAP | (R)-(−)-1-(benzofuran-2-yl)-2-propylaminopentane hydrochloride |
| CAE | catecholaminergic activity enhancer |
| DAT | dopamine transporter |
| DEA | N,α-diethylphenethylamine |
| EPPTB | N-(3-ethoxyphenyl)-4-pyrrolidin-1-yl-3-trifluoromethylbenzamide |
| GIRK | G-protein-coupled inwardly-rectifying K+ channel |
| IPAP | 1-(indol-3-yl)-2-propylaminopentane |
| MAO | monoamine oxidase |
| NEM | N-ethylmaleimide |
| NSF | N-ethylmaleimide-sensitive factor |
| PKA | protein kinase A |
| PKC | protein kinase C |
| PMA | phorbol 12-myristate 13-acetate |
| PPAP | 1-phenyl-2-propylaminopentane |
| TAAR1 | trace amine-associated receptor 1 |
| TBZ | tetrabenazine |
| VMAT2 | vesicular monoamine transporter 2 |
References
- Borowsky, B.; Adham, N.; Jones, K.A.; Raddatz, R.; Artymyshyn, R.; Ogozalek, K.L.; Durkin, M.M.; Lakhlani, P.P.; Bonini, J.A.; Pathirana, S.; et al. Trace amines: Identification of a family of mammalian G-protein-coupled receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 8966–8971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunzow, J.R.; Sonders, M.S.; Arttamangkul, S.; Harrison, L.M.; Zhang, G.; Quigley, D.I.; Darland, T.; Suchland, K.L.; Pasumamula, S.; Kennedy, J.L.; et al. Amphetamine, 3,4-methylenedioxymethamphetamine, lysergic acid diethylamide, and metabolites of the catecholamine neurotransmitters are agonists of a rat trace amine receptors. Mol. Pharmacol. 2001, 60, 1181–1188. [Google Scholar] [CrossRef]
- Grandy, D.K. Trace amine-associated receptor 1—Family archetype or iconoclast? Pharmacol. Ther. 2007, 116, 355–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gainetdinov, R.R.; Hoener, M.C.; Berry, M.D. Trace amines and their receptors. Pharmacol. Rev. 2018, 70, 549–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, Y.; Asif-Malik, A.; Canales, J.J. Trace amines and the trace amine-associated receptor1: Pharmacology, neurochemistry and clinical implications. Front. Neurosci. 2016, 10, 148. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Miller, G.M. Trace amine-associated receptor 1 as a monoaminergic modulation in brain. Biochem. Pharmacol. 2009, 78, 1095–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, G.M.; Verrico, C.D.; Jassen, A.; Konar, M.; Yang, H.; Panas, H.; Bahn, M.; Johnson, R.; Madras, B.K. Primate trace amine receptor 1 modulation by the dopamine transporter. J. Pharmacol. Exp. Ther. 2005, 313, 983–994. [Google Scholar] [CrossRef] [Green Version]
- Bradaia, A.; Trube, G.; Stalder, H.; Norcross, R.D.; Ozmen, L.; Wettstein, J.G.; Pinard, A.; Buchy, D.; Gassmann, M.; Hoener, M.C.; et al. The selective antagonist EPPTB reveals TAAR1-mediated regulatory mechanisms in dopaminergic neurons of the mesolimbic system. Proc. Natl. Acad. Sci. USA 2009, 106, 20081–20086. [Google Scholar] [CrossRef] [Green Version]
- Revel, F.G.; Moreau, J.L.; Gainetdinov, R.R.; Bradaia, A.; Sotnikova, T.D.; Mory, R.; Durkin, S.; Zbinden, K.G.; Norcross, R.; Meyer, C.A.; et al. TAAR1 activation modulates monoaminergic neurotransmission, preventing hyperdopaminergic and hypoglutamatergic activity. Proc. Natl. Acad. Sci. USA 2011, 108, 8485–8490. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Westmoreland, S.V.; Bahn, M.E.; Chen, G.L.; Yang, H.; Vallender, E.J.; Yao, W.D.; Madras, B.K.; Miller, G.M. Rhesus monkey trace amine-associated receptor 1 signaling: Enhancement by monoamine transporters and attenuation by the D2 autoreceptor in vitro. J. Pharmacol. Exp. Ther. 2007, 321, 116–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.; Miller, G.M. A receptor mechanism for methamphetamine action in dopamine transporter regulation in brain. J. Pharmacol. Exp. Ther. 2009, 330, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Miller, G.M. The emerging role of trace amine-associated receptor 1 in the functional regulation of monoamine transporters and dopaminergic activity. J. Neurochem. 2011, 116, 164–176. [Google Scholar] [CrossRef] [Green Version]
- Leo, D.; Mus, L.; Espinoza, S.; Hoener, M.C.; Sotnikova, T.D.; Gainetdinov, R.R. Taar1-mediated modulation of presynaptic dopaminergic neurotransmission: Role of D2 dopamine autoreceptors. Neuropharmacology 2014, 81, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Geracitano, R.; Federici, M.; Prisco, S.; Bernardi, G.; Mercuri, N.B. Inhibitory effects of trace amines on rat midbrain dopaminergic neurons. Neuropharmacology 2004, 46, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, L.; Ebeling, M.; Kratochwil, N.A.; Bunzow, J.R.; Grandy, D.K.; Hoener, M.C. Trace amine-associated receptors form structurally and functionally distinct subfamilies of novel G protein-coupled receptors. Genomics 2005, 85, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Liberles, S.D. Trace amine associated receptors: Ligands, neural circuits, and behaviors. Curr. Opin. Neurobiol. 2015, 34, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulton, A.A. Trace Amines and the Neurosciences. In Neurobiology of the Trace Amines; Boulton, A.A., Baker, G.B., Dewhurst, W.G., Sandler, M., Eds.; Human Press: Clifton, NJ, USA, 1984; pp. 13–24. [Google Scholar] [CrossRef]
- Berry, M.D. Mammalian central nervous system trace amines. Pharmacologic amphetamines, physiologic neuromodulators. J. Neurochem. 2004, 90, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Zucchi, R.; Chiellini, G.; Scanlan, T.S.; Grandy, D.K. Trace amine-associated receptors and their ligands. Br. J. Pharmacol. 2006, 149, 967–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.Y.T.; Neff, N.H. β-Phenethylamine: A specific substrate for type B monoamine oxidase of brain. J. Pharmacol. Exp. Ther. 1973, 187, 365–371. [Google Scholar]
- Xie, Z.; Miller, G.M. β-Phenylethylamine alters monoamine transporter function via trace amine-associated receptor 1: Implication for modulatory roles of trace amines in brain. J. Pharmacol. Exp. Ther. 2008, 325, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Westmoreland, S.V.; Miller, G.V. Modulation of monoamine transporters by common biogenic amines via trace amine-associated receptor 1 and monoamine autoreceptors in human embryonic kidney 293 cells and brain synaptosomes. J. Pharmacol. Exp. Ther. 2008, 325, 629–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zsilla, G.; Hegyi, D.E.; Baranyi, M.; Vizi, E.S. 3,4-Methylenedioxymethamphetamine, mephedrone, and β-phenylethylamine release dopamine from the cytoplasm by means of transporters and keep the concentration high and constant by blocking reuptake. Eur. J. Pharmacol. 2018, 837, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.A.; Travis, J.C.; Venhuis, B.J. A methamphetamine analog (N,α-diethyl-phenylethylamine) identified in a mainstream dietary supplement. Drug Test. Anal. 2014, 6, 805–807. [Google Scholar] [CrossRef] [PubMed]
- Knoll, J.; Yoneda, F.; Knoll, B.; Ohde, H.; Miklya, I. (−)1-Benzofuran-2-yl)-2- propylaminopentane, [(−)BPAP], a selective enhancer of the impulse propagation mediated release of catecholamines and serotonin in the rat brain. Br. J. Pharmacol. 1999, 128, 1723–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneda, F.; Moto, T.; Sakae, M.; Ohde, H.; Knoll, B.; Miklya, I.; Knoll, J. Structure-activity studies leading to (−)1-(benzofuran-2-yl)-2-propylaminopentane ((−)BPAP, a highly potent, selective enhancer of the impulse propagation mediated release of catecholamines and serotonin in the brain. Bioorg. Med. Chem. Lett. 2001, 9, 1197–1212. [Google Scholar] [CrossRef]
- Miklya, I.; Knoll, J. Analysis of the effect of (−)-BPAP, a selective enhancer of the impulse propagation mediated release of catecholamines and serotonin in the brain. Life Sci. 2003, 72, 2915–2921. [Google Scholar] [CrossRef]
- Shimazu, S.; Miklya, I. Pharmacological studies with endogenous enhancer substances: β-phenylethylamine, tryptamine, and their synthetic derivatives. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2004, 28, 421–427. [Google Scholar] [CrossRef]
- Oka, T.; Yasusa, T.; Ando, T.; Watanabe, M.; Yoneda, F.; Ishida, T.; Knoll, J. Enantioselective synthesis and absolute configuration of (−)-1-(benzofuran-2-yl)-2-propylaminopentane, ((−)-BPAP, a highly potent and selective catecholaminergic activity enhancer. Bioorganic. Med. Chem. 2001, 9, 1213–1219. [Google Scholar] [CrossRef]
- Knoll, J. Antiaging compounds: (−)deprenyl (selegiline) and (−)1-(benzofuran-2-yl)-2-propylaminopentane, [(−)BPAP], a selective highly potent enhancer of the impulse propagation mediated release of catecholamines and serotonin in the brain. CNS Drug Rev. 2001, 7, 317–345. [Google Scholar] [CrossRef]
- Knoll, J.; Miklya, I. Longevity study with low doses of selegiline/(−)deprenyl and (2R)-1-(1-benzfuran-2-yl)-N-propylpentane-2-amine (BPAP). Life Sci. 2016, 167, 32–38. [Google Scholar] [CrossRef]
- Knoll, J.; Baghy, K.; Eckhardt, S.; Ferdinandy, P.; Garami, M.; Harsing, L.G., Jr.; Hauser, P.; Mervai, Z.; Pocza, T.; Schaff, Z.; et al. A longevity study with enhancer substances (selegiline, BPAP) detected an unknown tumor-manifestation-suppressing regulation in rat brain. Life Sci. 2017, 182, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Mervai, Z.; Reszegi, A.; Miklya, I.; Knoll, J.; Schaff, Z.; Kovalszky, I.; Baghy, K. Inhibitory effect of (2R)-1-(1-benzofuran-2-yl)-N-propylpentan-2-amine on lung adenocarcinoma. Pathol. Oncol. Res. 2020, 26, 727–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaszner, P.; Miklya, I. The use of the synthetic enhancer substances (−)deprenyl and (−)-BPAP in major depression. Neuropsychopharmacol. Hung. 2004, 6, 210–220. [Google Scholar] [PubMed]
- Tsunekawa, H.; Noda, Y.; Miyazaki, M.; Yoneda, F.; Nabeshima, T.; Wang, D. Effects of (R)-(−)-1-(benzofuran-2-yl)-2-propylaminopentane hydrochloride [(−)-BPAP] in animal models of mood disorders. Behav. Brain Res. 2008, 189, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Knoll, J.; Zelena, D.; Timar, J.; Baghy, K.; Mervai, Z.; Miklya, I. Synthetic enhancer compounds, besides acting on biogenic amine system, influence the glutamate transmission and stress response. Behav. Brain Res. 2020, 378, 112290. [Google Scholar] [CrossRef] [PubMed]
- Knoll, J. The Brain and Its Self. A Neurochemical Concept of the Innate and Acquired Drives; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2005. [Google Scholar] [CrossRef]
- Harsing, L.G., Jr. Dopamine and the dopaminergic systems of the brain. In Handbook of Neurochemistry and Molecular Neurobiology, Neurotransmitter Systems, 1st ed.; Lajtha, A., Ed.; Springer Science and Business Media: New York, NY, USA, 2008; pp. 149–170. [Google Scholar]
- Hoffmann, I. Pharmacology of nomifensine. Int. Pharm. 1982, 17, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Reith, M.E.A.; Kuhar, M.J.; Carroll, F.I.; Garris, P.A. Preferential increases in nucleus accumbens dopamine after systemic cocaine administration are caused by unique characteristics of dopamine neurotransmission. J. Neurosci. 2001, 21, 6338–6347. [Google Scholar] [CrossRef] [Green Version]
- Schacht, U.; Heptner, W. Effect of nomifensine (Hoe 984), a new antidepressant, on uptake of noradrenaline and serotonin and on release of noradrenaline in rat brain synaptosomes. Biochem. Pharmacol. 1974, 23, 3413–3422. [Google Scholar] [CrossRef]
- Hunt, P.; Raynaud, J.P.; Leven, M.; Schacht, U. Dopamine uptake inhibitors and releasing agents differentiated by the use of synaptosomes and field-stimulated brain slices in vitro. Biochem. Pharmacol. 1979, 28, 2011–2016. [Google Scholar] [CrossRef]
- Lonart, G.; Südhof, T.C. Assembly of SNARE core complexes prior to neurotransmitter release sets the readily releasable pool of synaptic vesicles. J. Biol. Chem. 2000, 275, 27703–27707. [Google Scholar] [CrossRef] [Green Version]
- Gnegy, M.E. The effect of phosphorylation on amphetamine-mediated outward transport. Eur. J. Pharmacol. 2003, 479, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.F.; Sullivan, J.M. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron 1998, 21, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Horton, D.B.; Nickell, J.B.; Zheng, G.; Crooks, P.A.; Dwoskin, L.P. GZ-793A, a lobeline analog, interacts with the vesicular monoamine transporter-2 to inhibit the effect of methamphetamine. J. Neurochem. 2013, 127, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, B.; Ruoho, A.E. N-Terminus regulation of VMAT2 mediates methamphetamine-stimulated efflux. Neuroscience 2014, 259, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, M.; Harada, K.; Meller, E.; Schalling, M.; Hokfelt, T. Dopamine autoreceptors. Biochemical, pharmacological, and morphological studies. Ann. N. Y. Acad. Sci. 1990, 604, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Sara, Y.; Virmani, T.; Deak, F.; Liu, X.; Kavalali, E.T. An isolated pool of vesicles recycles at rest and drives spontaneous neurotransmission. Neuron 2005, 45, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Miller, O.H.; Moran, J.T.; Hall, B.J. Two cellular hypotheses explaining the initiation of ketamine’s antidepressant actions: Direct inhibition and disinhibition. Neuropharmacology 2016, 100, 17–26. [Google Scholar] [CrossRef]
- Miklya, I. Essential differences in the pharmacological spectrum of (−)deprenyl and rasagiline. Pharmacol. Rep. 2014, 66, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.B.; Chen, G.; Gutman, D.A.; Ewing, A.G. Dopamine levels of two classes of vesicles are differently depleted by amphetamine. Brain Res. 1998, 788, 294–301. [Google Scholar] [CrossRef]
- Partilla, J.S.; Dempsey, A.G.; Nagpal, A.S.; Blough, B.E.; Baumann, M.H.; Rothman, R.B. Interaction of amphetamines and related compounds at the vesicular monoamine transporter. J. Pharmacol. Exp. Ther. 2006, 319, 237–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stalder, H.; Hoener, M.C.; Norcross, R.D. Selective antagonists of mouse trace amine-associated receptor 1 (mTAAR1): Discovery of EPPTB (RO5212773). Bioorg. Med. Chem. Lett. 2011, 21, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Miller, G.M. Trace amine-associated receptor 1 is a modulator of the dopamine transporter. J. Pharmacol. Exp. Ther. 2007, 321, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, K.C.; Rothman, R.B.; Reith, M.A.E. Nonclassical pharmacology of the dopamine transporter: Atypical inhibitors, allosteric modulators, and partial substrates. J. Pharmacol. Exp. Ther. 2013, 346, 2–10. [Google Scholar] [CrossRef] [Green Version]
- Shimazu, S.; Tsunekawa, H.; Yoneda, F.; Katsuki, H.; Akaike, A.; Janowsky, A. Transporter-mediated actions of R-(−)-1-(benzofuran-2-yl)-2-propylaminopentane. Eur. J. Pharmacol. 2003, 482, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Sonders, M.S.; Poulsen, N.W.; Galli, A. Mechanisms of neurotransmitter release by amphetamines: A review. Prog. Neurobiol. 2005, 75, 406–433. [Google Scholar] [CrossRef] [PubMed]
- Leenders, A.G.M.; Sheng, Z.H. Modulation of neurotransmitter release by the second messenger-activated protein kinases: Implication for presynaptic plasticity. Pharmacol. Ther. 2005, 105, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Giambalvo, C.T. Protein kinase C and dopamine transport-2. Effects of amphetamine in vitro. Neuropharmacology 1992, 31, 1211–1222. [Google Scholar] [CrossRef]
- Südhof, T.C.; Rizo, J. Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol. 2011, 3, a005637. [Google Scholar] [CrossRef]
- Fon, E.A.; Pothos, E.N.; Sun, B.C.; Killeen, N.; Sulzer, D.; Edwards, R.H. Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron 1997, 19, 1271–1283. [Google Scholar] [CrossRef] [Green Version]
- German, C.L.; Baladi, M.G.; McFadden, L.M.; Hanson, G.R.; Fleckenstein, A.E. Regulation of the dopamine and vesicular monoamine transporters: Pharmacological targets and implications for disease. Pharmacol. Rev. 2015, 67, 1005–1024. [Google Scholar] [CrossRef] [Green Version]
- Scherman, D.; Henry, J.P. Reserpine binding to bovine chromaffin granule membranes. Characterization and comparison with dihydrotetrabenazine binding. Mol. Pharmacol. 1984, 25, 113–122. [Google Scholar] [PubMed]
- Leviel, V. The reverse transport of DA, what physiological significance? Neurochem. Int. 2001, 38, 83–106. [Google Scholar] [CrossRef]
- Schmitz, Y.; Lee, C.J.; Schmauss, C.; Gonon, F.; Schulzer, D. Amphetamine distorts stimulation-dependent dopamine overflow: Effects on D2 autoreceptors, transporters, and synaptic vesicles stores. J. Neurosci. 2001, 21, 5916–5924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raiteri, M.; Cerrito, F.; Cervoni, A.M.; Levi, G. Dopamine can be released by two mechanisms differentially affected by the dopamine transport inhibitor nomifensine. J. Pharmacol. Exp. Ther. 1979, 208, 195–202. [Google Scholar]
- Butcher, S.P.; Liptrot, J.; Aburthnott, G.W. Characterization of methylphenidate and nomifensine induced dopamine release in rat striatum using in vivo brain microdialysis. Neurosci. Lett. 1991, 122, 245–248. [Google Scholar] [CrossRef]
- Kantor, L.; Keikilani, H.H.G.; Park, Y.H.; Richardson-Burns, S.M.; Mellon, M.J.; Gnegy, M.E. Protein kinase C and intracellular calcium are required for amphetamine-mediated dopamine release via the norepinephrine transporter in undifferentiated PC12 cells. J. Pharmacol. Exp. Ther. 2001, 297, 1016–1024. [Google Scholar]
- Khoshbouei, H.; Sen, N.; Guptaroy, B.; Johnson, L.; Lund, D.; Gnegy, M.E.; Galli, A.; Javitch, J.A. N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS Biol. 2004, 2, E78. [Google Scholar] [CrossRef]
- Jones, S.R.; Gainetdinov, R.R.; Wightman, R.M.; Caron, M.G. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J. Neurosci. 1998, 18, 1979–1986. [Google Scholar] [CrossRef]
- Daberkow, D.P.; Brown, H.D.; Bunner, K.D.; Kraniotis, S.A.; Doellman, M.A.; Ragozzino, M.E.; Garris, P.A.; Roitman, M.F. Amphetamine paradoxically augments exocytotic dopamine release and phasic dopamine signals. J. Neurosci. 2012, 13, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Avelar, A.J.; Juliano, S.A.; Garris, P.A. Amphetamine augments vesicular dopamine release in the dorsal and ventral striatum through different mechanisms. J. Neurochem. 2013, 125, 373–385. [Google Scholar] [CrossRef] [Green Version]
- Sulzer, D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron 2011, 69, 628–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freyberg, Z.; Sonders, M.S.; Aguilar, J.I.; Hiranita, T.; Karam, C.S.; Flores, J.; Pizzo, A.B.; Zhang, Y.; Farino, Z.J.; Chen, A.; et al. Mechanisms of amphetamine action illuminated through optical monitoring of dopamine synaptic vesicles in Drosophila brain. Nat. Commun. 2016, 7, 10652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feschenko, M.S.; Stevenson, E.; Sweadner, K.J. Interaction of protein kinase C and cAMP-dependent pathways in the phosphorylation of the Na,K-ATP-ase. J. Biol. Chem. 2000, 275, 34693–34700. [Google Scholar] [CrossRef] [Green Version]
- Hucho, T.B.; Dina, O.A.; Levone, J.D. Epac mediates cAMP-to-PKC signaling in inflammatory pain: An isolectin B4(+) neuron-sspecific mechanism. J. Neurosci. 2005, 25, 6119–6126. [Google Scholar] [CrossRef] [Green Version]
- Lindemann, L.; Meyer, C.A.; Jeanneau, K.; Bradaia, A.; Ozmen, L.; Bluethmann, H.; Bettler, B.; Wettstein, J.G.; Borroni, E.; Moreau, J.L.; et al. Trace amine-associated receptor 1 modulates dopaminergic activity. J. Pharmacol. Exp. Ther. 2008, 324, 948–956. [Google Scholar] [CrossRef] [Green Version]
- Starke, K.; Gothert, M.; Kilbinger, H. Modulation of neurotransmitter release by presynaptic autoreceptors. Physiol. Rev. 1989, 69, 864–989. [Google Scholar] [CrossRef]
- Glowinski, J.; Iversen, L.L. Regional studies of catecholamines in the rat brain. I. The disposition of 3H-norepinephrine, 3H-dopamine, and 3H-dopa in various regions of the brain. J. Neurochem. 1966, 13, 655–669. [Google Scholar] [CrossRef]
- Harsing, L.G., Jr.; Sershen, H.; Lajtha, A. Dopamine efflux from striatum after chronic nicotine: Evidence for autoreceptor desensitization. J. Neurochem. 1992, 59, 48–54. [Google Scholar] [CrossRef]




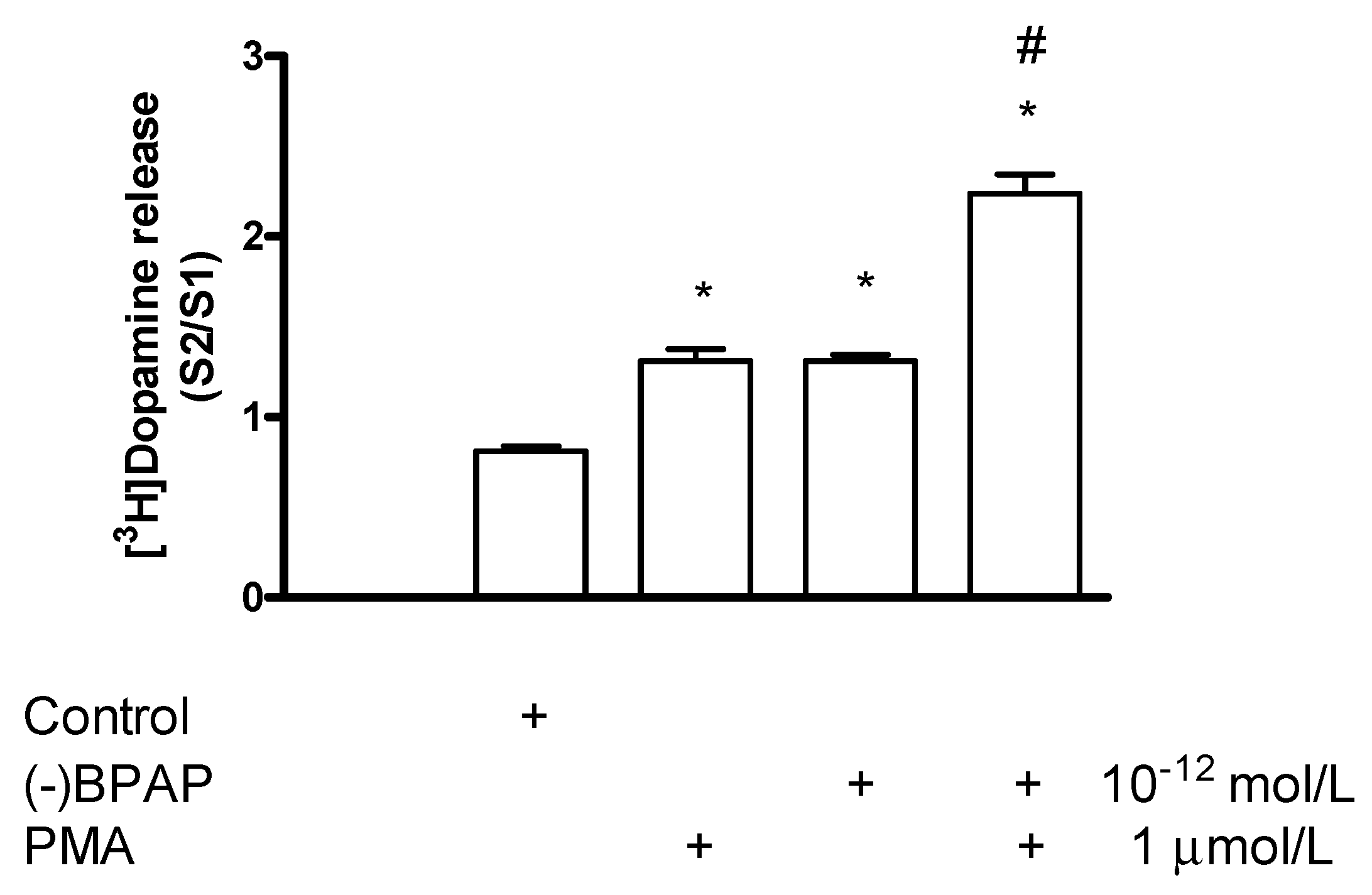



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harsing, L.G.; Knoll, J.; Miklya, I. Enhancer Regulation of Dopaminergic Neurochemical Transmission in the Striatum. Int. J. Mol. Sci. 2022, 23, 8543. https://doi.org/10.3390/ijms23158543
Harsing LG, Knoll J, Miklya I. Enhancer Regulation of Dopaminergic Neurochemical Transmission in the Striatum. International Journal of Molecular Sciences. 2022; 23(15):8543. https://doi.org/10.3390/ijms23158543
Chicago/Turabian StyleHarsing, Laszlo G., Joseph Knoll, and Ildiko Miklya. 2022. "Enhancer Regulation of Dopaminergic Neurochemical Transmission in the Striatum" International Journal of Molecular Sciences 23, no. 15: 8543. https://doi.org/10.3390/ijms23158543
APA StyleHarsing, L. G., Knoll, J., & Miklya, I. (2022). Enhancer Regulation of Dopaminergic Neurochemical Transmission in the Striatum. International Journal of Molecular Sciences, 23(15), 8543. https://doi.org/10.3390/ijms23158543