
Spinal Cord Injury and Loss of Cortical Inhibition

 ,
, 
Abstract

{kind=link}
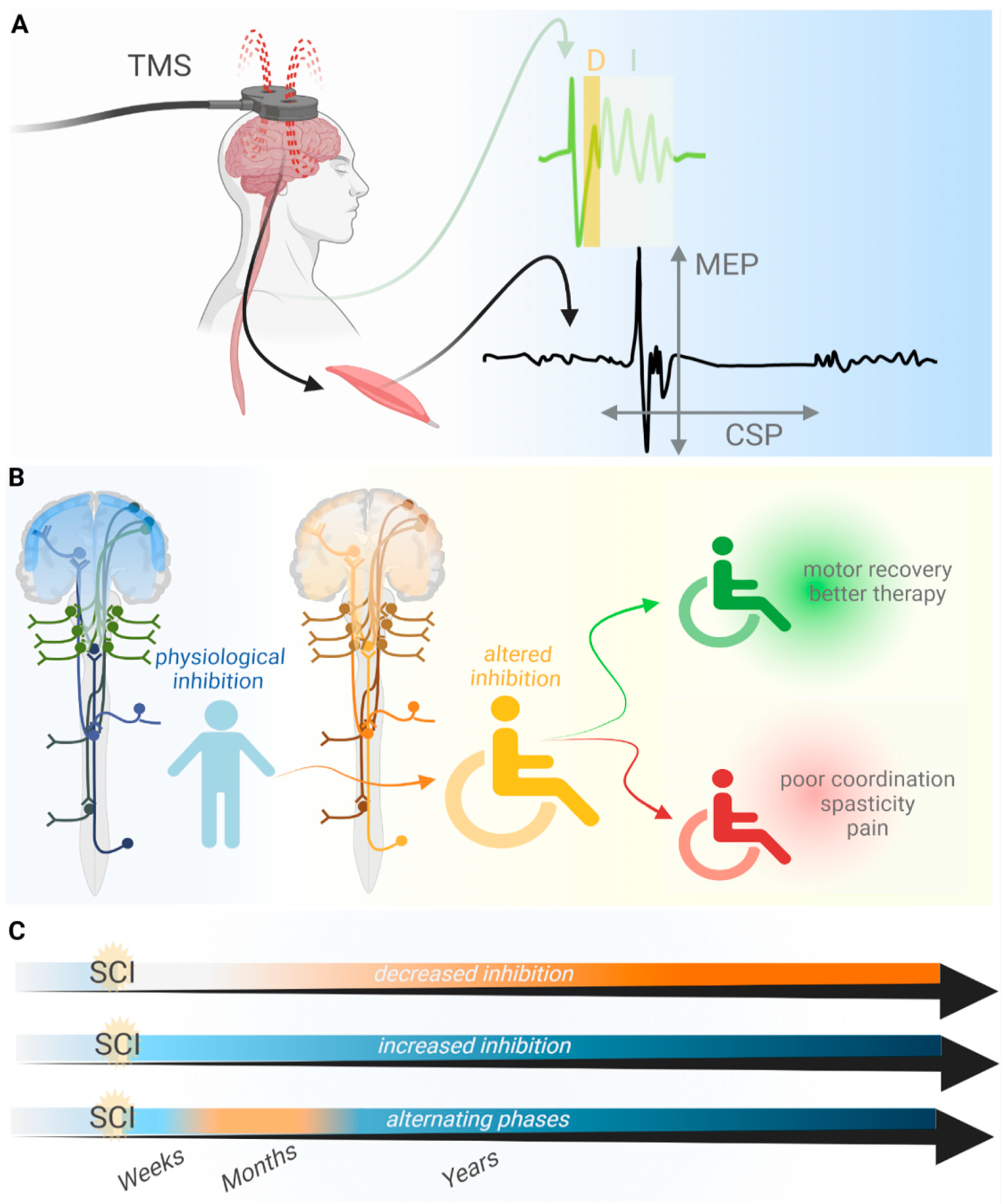{kind=link}
{kind=link}
1. Introduction: SCI Harms the Brain
2. TMS as a Method to Analyze the Loss of Inhibition after SCI
3. Loss of Inhibition Promotes Motor Recovery after SCI
4. The Loss of Inhibition Aggravates Symptoms after SCI
5. Alternative Mechanisms and Explanations for Imbalanced Cortical Excitability
6. Possible Causes for the Loss of Inhibition
6.1. Metabolic Stress
6.2. Inflammation
6.3. Remodeling of Perineuronal Nets
6.4. Altered Astrocyte Metabolism and Physiology
6.5. Rewiring of Cortical Circuits
6.6. Hyperexcitability of Excitatory Neurons
7. Can Therapy Rely on the Loss of Inhibition?
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahuja, C.S.; Wilson, J.R.; Nori, S.; Kotter, M.R.N.; Curt, A.; Fehlings, M.G. Traumatic spinal cord injury. Nat. Rev. 2017, 3, 17018. [Google Scholar] [CrossRef] [PubMed]
- Masri, R.; Keller, A. Chronic Pain Following Spinal Cord Injury. Adv. Exp. Med. Biol. 2012, 760, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Lifshutz, J.; Colohan, A. A brief history of therapy for traumatic spinal cord injury. Neurosurg. Focus 2004, 16, 1–8. [Google Scholar] [CrossRef]
- Hulsebosch, C.E. Recent advances in pathophysiology and treatment of spinal cord injury. Am. J. Physiol. Adv. Physiol. Educ. 2002, 26, 238–255. [Google Scholar] [CrossRef] [PubMed]
- Cadotte, D.W.; Fehlings, M.G. Spinal cord injury: A systematic review of current treatment options. Clin. Orthop. Relat. Res. 2011, 469, 732–741. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Venkatesh, K. Spinal cord injury: Pathophysiology, treatment strategies, associated challengeenges. Cell Tissue Res. 2019, 377, 125–151. [Google Scholar] [CrossRef]
- Clayton, K.S.; Chubon, R.A. Factors associated with the quality of life of long-term spinal cord injured persons. Arch. Phys. Med. Rehabil. 1994, 75, 633–638. [Google Scholar] [CrossRef]
- Krause, J.; Saunders, L. Risk of mortality and life expectancy after spinal cord injury: The role of health behaviors and participation. Top. Spinal Cord Inj. Rehabil. 2010, 16, 53–60. [Google Scholar] [CrossRef][Green Version]
- Anneken, V.; Hanssen-Doose, A.; Hirschfeld, S.; Scheuer, T.; Thietje, R. Influence of physical exercise on quality of life in individuals with spinal cord injury. Spinal Cord 2010, 48, 393–399. [Google Scholar] [CrossRef]
- Van Der Woude, L.H.V.; De Groot, S.; Postema, K.; Bussmann, J.B.J.; Janssen, T.W.J.; Post, M.W.M. Active LifestyLe Rehabilitation Interventions in aging Spinal Cord injury (ALLRISC): A multicentre research program. Disabil. Rehabil. 2013, 35, 1097–1103. [Google Scholar] [CrossRef]
- Van den Akker, L.E.; Holla, J.F.M.; Dadema, T.; Visser, B.; Valent, L.J.; de Groot, S.; Dallinga, J.M.; Deutekom, M. Determinants of physical activity in wheelchair users with spinal cord injury or lower limb amputation: Perspectives of rehabilitation professionals and wheelchair users. Disabil. Rehabil. 2020, 42, 1934–1941. [Google Scholar] [CrossRef] [PubMed]
- Nas, K.; Yazmalar, L.; Şah, V.; Aydin, A.; Öneş, K. Rehabilitation of spinal cord injuries. World J. Orthop. 2015, 6, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Behrman, A.L.; Harkema, S.J. Physical Rehabilitation as an Agent for Recovery After Spinal Cord Injury. Phys. Med. Rehabil. Clin. N. Am. 2007, 18, 183–202. [Google Scholar] [CrossRef] [PubMed]
- Spooren, A.I.F.; Janssen-Potten, Y.J.M.; Kerckhofs, E.; Seelen, H.A.M. Outcome of motor training programmes on arm and hand functioning in patients with cervical spinal cord injury according to different levels of the ICF: A systematic review. J. Rehabil. Med. 2009, 41, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Montesinos-Magraner, L.; Serra-Ano, P.; García-Masso, X.; Ramírez-Garcerán, L.; González, L.M.; González-Viejo, M. Comorbidity and physical activity in people with paraplegia: A descriptive cross-sectional study. Spinal Cord 2018, 56, 52–56. [Google Scholar] [CrossRef]
- Floríndez, L.I.; Carlson, M.E.; Pyatak, E.; Blanchard, J.; Cogan, A.M.; Sleight, A.G.; Hill, V.; Diaz, J.; Blanche, E.; Garber, S.L.; et al. A qualitative analysis of pressure injury development among medically underserved adults with spinal cord injury. Disabil. Rehabil. 2020, 42, 2093–2099. [Google Scholar] [CrossRef]
- Griffin, J.M.; Bradke, F. Therapeutic repair for spinal cord injury: Combinatory approaches to address a multifaceted problem. EMBO Mol. Med. 2020, 12, e11505. [Google Scholar] [CrossRef]
- Huang, H.; Sharma, H.S.; Chen, L.; Otom, A.; Al Zoubi, Z.M.; Saberi, H.; Muresanu, D.F.; He, X. Review of clinical neurorestorative strategies for spinal cord injury: Exploring history and latest progresses. J. Neurorestoratology 2018, 1, 171–178. [Google Scholar] [CrossRef]
- Shah, M.; Peterson, C.; Yilmaz, E.; Halalmeh, D.R.; Moisi, M. Current advancements in the management of spinal cord injury: A comprehensive review of literature. Surg. Neurol. Int. 2020, 11, 2. [Google Scholar] [CrossRef]
- Hutson, T.H.; Di Giovanni, S. The translational landscape in spinal cord injury: Focus on neuroplasticity and regeneration. Nat. Rev. Neurol. 2019, 15, 732–745. [Google Scholar] [CrossRef]
- Hains, B.C.; Black, J.; Waxman, S.G. Primary cortical motor neurons undergo apoptosis after axotomizing spinal cord injury. J. Comp. Neurol. 2003, 462, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Nagendran, T.; Larsen, R.S.; Bigler, R.L.; Frost, S.B.; Philpot, B.D.; Nudo, R.J.; Taylor, A.M. Distal axotomy enhances retrograde presynaptic excitability onto injured pyramidal neurons via trans-synaptic signaling. Nat. Commun. 2017, 8, 625. [Google Scholar] [CrossRef] [PubMed]
- Beaud, M.; Schmidlin, E.; Wannier, T.; Freund, P.; Bloch, J.; Mir, A.; Schwab, M.E.; Rouiller, E.M. Anti-Nogo-A antibody treatment does not prevent cell body shrinkage in the motor cortex in adult monkeys subjected to unilateral cervical cord lesion. BMC Neurosci. 2008, 9, 5. [Google Scholar] [CrossRef]
- Onifer, S.M.; Smith, G.M.; Fouad, K. Plasticity After Spinal Cord Injury: Relevance to Recovery and Approaches to Facilitate It. Neurotherapeutics 2011, 8, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Dietz, V.; Fouad, K. Restoration of sensorimotor functions after spinal cord injury. Brain 2014, 137, 654–667. [Google Scholar] [CrossRef]
- Van Middendorp, J.J.; Goss, B.; Urquhart, S.; Atresh, S.; Williams, R.P.; Schuetz, M. Diagnosis and Prognosis of Traumatic Spinal Cord Injury. Glob. Spine J. 2011, 1, 001–007. [Google Scholar] [CrossRef] [PubMed]
- Nardone, R.; Höller, Y.; Brigo, F.; Orioli, A.; Tezzon, F.; Schwenker, K.; Christova, M.; Golaszewski, S.; Trinka, E. Descending motor pathways and cortical physiology after spinal cord injury assessed by transcranial magnetic stimulation: A systematic review. Brain Res. 2015, 1619, 139–154. [Google Scholar] [CrossRef]
- Nardone, R.; Langthaler, P.B.; Höller, Y.; Bathke, A.; Frey, V.N.; Brigo, F.; Trinka, E. Modulation of non-painful phantom sensation in subjects with spinal cord injury by means of rTMS. Brain Res. Bull. 2015, 118, 82–86. [Google Scholar] [CrossRef]
- Yuemin, D.; Abba, K.J.; Weihong, P. Neural plasticity after spinal cord injury. Neural Regen. Res. 2005, 7, 386–391. [Google Scholar] [CrossRef]
- Xerri, C. Plasticity of cortical maps: Multiple triggers for adaptive reorganization following brain damage and spinal cord injury. Neuroscientist 2012, 18, 133–148. [Google Scholar] [CrossRef]
- Nishimura, Y.; Isa, T. Compensatory changes at the cerebral cortical level after spinal cord injury. Neuroscientist 2009, 15, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Orlando, C.; Raineteau, O. Integrity of cortical perineuronal nets influences corticospinal tract plasticity after spinal cord injury. Brain Struct. Funct. 2015, 220, 1077–1091. [Google Scholar] [CrossRef] [PubMed]
- Nardone, R.; Höller, Y.; Brigo, F.; Seidl, M.; Christova, M.; Bergmann, J.; Golaszewski, S.; Trinka, E. Functional brain reorganization after spinal cord injury: Systematic review of animal and human studies. Brain Res. 2013, 1504, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Florence, S.L.; Kaas, J.H. Reorganization of somatosensory cortex after nerve and spinal cord injury. News Physiol. Sci. 1998, 13, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Hasbargen, T.; Ahmed, M.M.; Miranpuri, G.; Li, L.; Kahle, K.T.; Resnick, D.; Sun, D. Role of NKCC1 and KCC2 in the development of chronic neuropathic pain following spinal cord injury. Ann. N. Y. Acad. Sci. 2010, 1198, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.; Hamm, T.M.; Crook, S.M.; Jung, R. Modulation of inhibitory strength and kinetics facilitates regulation of persistent inward currents and motoneuron excitability following spinal cord injury. J. Neurophysiol. 2011, 106, 2167–2179. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zheng, J.; Xiong, L.; Zimmermann, M.; Yang, J. Spinal cord injury-induced attenuation of GABAergic inhibition in spinal dorsal horn circuits is associated with down-regulation of the chloride transporter KCC2 in rat. J. Physiol. 2008, 586, 5701–5715. [Google Scholar] [CrossRef]
- Bütefisch, C.M. Plasticity in the Human Cerebral Cortex: Lessons from the Normal Brain and from Stroke. Neuroscientist 2004, 10, 163–173. [Google Scholar] [CrossRef]
- Manganotti, P.; Acler, M.; Zanette, G.P.; Smania, N.; Fiaschi, A. Motor cortical disinhibition during early and late recovery after stroke. Neurorehabil. Neural Repair 2008, 22, 396–403. [Google Scholar] [CrossRef]
- Brasil-Neto, J.P.; Cohen, L.G.; Pascual-Leone, A.; Jabir, F.K.; Wall, R.T.; Hallett, M. Rapid reversible modulation of human motor outputs after transient deafferentation of the forearm: A study with transcranial magnetic stimulation. Neurology 1992, 42, 1302–1306. [Google Scholar] [CrossRef]
- Cohen, L.G.; Bandinelli, S.; Findley, T.W.; Hallett, M. Motor reorganization after upper limb amputation in man: A study with focal magnetic stimulation. Brain 1991, 114, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Candido Santos, L.; Gushken, F.; Gadotti, G.M.; de Dias, B.F.; Marinelli Pedrini, S.; Barreto, M.E.S.F.; Zippo, E.; Pinto, C.B.; de Piza, P.V.T.; Fregni, F. Intracortical Inhibition in the Affected Hemisphere in Limb Amputation. Front. Neurol. 2020, 11, 720. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Lee, L.J.H.; Jeng, J.S.; Hsieh, S.T.; Chiang, M.C.; Yeh, S.J.; Hsueh, H.W.; Chao, C.C. Pathophysiology of central poststroke pain motor cortex disinhibition and its clinical and sensory correlates. Stroke 2019, 50, 2851–2857. [Google Scholar] [CrossRef] [PubMed]
- Saturno, E.; Bonato, C.; Miniussi, C.; Lazzaro, V.; Callea, L. Motor cortex changes in spinal cord injury: A TMS study. Neurol. Res. 2008, 30, 1084–1085. [Google Scholar] [CrossRef]
- Klomjai, W.; Katz, R.; Lackmy-Vallée, A. Basic principles of transcranial magnetic stimulation (TMS) and repetitive TMS (rTMS). Ann. Phys. Rehabil. Med. 2015, 58, 208–213. [Google Scholar] [CrossRef]
- Farzan, F.; Vernet, M.; Shafi, M.M.D.; Rotenberg, A.; Daskalakis, Z.J.; Pascual-Leone, A. Characterizing and modulating brain circuitry through transcranial magnetic stimulation combined with electroencephalography. Front. Neural Circuits 2016, 10, 73. [Google Scholar] [CrossRef]
- Sack, A.T.; Linden, D.E.J. Combining transcranial magnetic stimulation and functional imaging in cognitive brain research: Possibilities and limitations. Brain Res. Rev. 2003, 43, 41–56. [Google Scholar] [CrossRef]
- Conde, V.; Tomasevic, L.; Akopian, I.; Stanek, K.; Saturnino, G.B.; Thielscher, A.; Bergmann, T.O.; Siebner, H.R. The non-transcranial TMS-evoked potential is an inherent source of ambiguity in TMS-EEG studies. Neuroimage 2019, 185, 300–312. [Google Scholar] [CrossRef]
- Potter-Baker, K.A.; Janini, D.P.; Frost, F.S.; Chabra, P.; Varnerin, N.; Cunningham, D.A.; Sankarasubramanian, V.; Plow, E.B. Reliability of TMS metrics in patients with chronic incomplete spinal cord injury. Spinal Cord 2016, 54, 980–990. [Google Scholar] [CrossRef]
- Lotze, M.; Laubis-Herrmann, U.; Topka, H. Combination of TMS and fMRI reveals a specific pattern of reorganization in M1 in patients after complete spinal cord injury. Restor. Neurol. Neurosci. 2006, 24, 97–107. [Google Scholar]
- Petersen, J.A.; Spiess, M.; Curt, A.; Dietz, V.; Schubert, M.N. Spinal cord injury: One-year evolution of motor-evoked potentials and recovery of leg motor function in 255 patients. Neurorehabil. Neural Repair 2012, 26, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Han, D.-S.; Li, C.-M.; Chang, C. Reorganization of the cortico-spinal pathway in patients with chronic complete thoracic spinal cord injury: A study of motor evoked potentials. Acta Derm. Venereol. 2008, 40, 208–212. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tazoe, T.; Perez, M.A. Effects of repetitive transcranial magnetic stimulation on recovery of function after spinal cord injury. Arch. Phys. Med. Rehabil. 2015, 96, S145–S155. [Google Scholar] [CrossRef] [PubMed]
- Yılmaz, B.; Kesikburun, S.; Yaşar, E.; Tan, A.K. The effect of repetitive transcranial magnetic stimulation on refractory neuropathic pain in spinal cord injury. J. Spinal Cord Med. 2014, 37, 397–400. [Google Scholar] [CrossRef]
- Ellaway, P.H.; Vásquez, N.; Craggs, M. Induction of central nervous system plasticity by repetitive transcranial magnetic stimulation to promote sensorimotor recovery in incomplete spinal cord injury. Front. Integr. Neurosci. 2014, 8, 42. [Google Scholar] [CrossRef]
- Alexeeva, N.; Broton, J.G.; Calancie, B. Latency of changes in spinal motoneuron excitability evoked by transcranial magnetic brain stimulation in spinal cord injured individuals. Electroencephalogr. Clin. Neurophysiol. Electromyogr. Mot. Control 1998, 109, 297–303. [Google Scholar] [CrossRef]
- Shimizu, T.; Hino, T.; Komori, T.; Hirai, S. Loss of the muscle silent period evoked by transcranial magnetic stimulation of the motor cortex in patients with cervical cord lesions. Neurosci. Lett. 2000, 286, 199–202. [Google Scholar] [CrossRef]
- Davey, N.J. Responses of thenar muscles to transcranial magnetic stimulation of the motor cortex in patients with incomplete spinal cord injury. J. Neurol. Neurosurg. Psychiatry 1998, 65, 80–87. [Google Scholar] [CrossRef]
- Ellaway, P.H.; Catley, M.; Davey, N.J.; Kuppuswamy, A.; Strutton, P.; Frankel, H.L.; Jamous, A.; Savic, G. Review of physiological motor outcome measures in spinal cord injury using transcranial magnetic stimulation and spinal reflexes. J. Rehabil. Res. Dev. 2007, 44, 69–75. [Google Scholar] [CrossRef]
- Nardone, R.; Höller, Y.; Bathke, A.C.; Orioli, A.; Schwenker, K.; Frey, V.; Golaszewski, S.; Brigo, F.; Trinka, E. Spinal cord injury affects I-wave facilitation in human motor cortex. Brain Res. Bull. 2015, 116, 93–97. [Google Scholar] [CrossRef]
- Nardone, R.; Höller, Y.; Thomschewski, A.; Bathke, A.C.; Ellis, A.R.; Golaszewski, S.M.; Brigo, F.; Trinka, E. Assessment of corticospinal excitability after traumatic spinal cord injury using MEP recruitment curves: A preliminary TMS study. Spinal Cord 2015, 53, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Nardone, R.; De Blasi, P.; Höller, Y.; Taylor, A.C.; Brigo, F.; Trinka, E. Effects of theta burst stimulation on referred phantom sensations in patients with spinal cord injury. Neuroreport 2016, 27, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Krause, P.; Szecsi, J.; Straube, A. Concerning the article by Lotze et al., 2006: Combination of TMS and fMRI reveals a specific pattern of reorganization in M1 in patients after complete spinal cord injury. Restor. Neurol. Neurosci. 2007, 25, 611–612. [Google Scholar] [PubMed]
- Freund, P.; Rothwell, J.; Craggs, M.; Thompson, A.J.; Bestmann, S. Corticomotor representation to a human forearm muscle changes following cervical spinal cord injury. Eur. J. Neurosci. 2011, 34, 1839–1846. [Google Scholar] [CrossRef]
- Kriz, J.; Kozak, J.; Zedka, M. Primary motor cortex inhibition in spinal cord injuries. Neuroendocrinol. Lett. 2012, 33, 431–441. [Google Scholar] [PubMed]
- Ziemann, U. Pharmaco-Transcranial Magnetic Stimulation Studies of Motor Excitability, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2013; Volume 116. [Google Scholar]
- Di Lazzaro, V.; Oliviero, A.; Meglio, M.; Cioni, B.; Tamburrini, G.; Tonali, P.; Rothwell, J.C. Direct demonstration of the effect of lorazepam on the excitability of the human motor cortex. Clin. Neurophysiol. 2000, 111, 794–799. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Pilato, F.; Dileone, M.; Profice, P.; Oliviero, A.; Mazzone, P.; Insola, A.; Ranieri, F.; Meglio, M.; Tonali, P.A.; et al. The physiological basis of the effects of intermittent theta burst stimulation of the human motor cortex. J. Physiol. 2008, 586, 3871–3879. [Google Scholar] [CrossRef]
- Cirillo, J.; Calabro, F.J.; Perez, M.A. Impaired Organization of Paired-Pulse TMS-Induced I-Waves After Human Spinal Cord Injury. Cereb. Cortex 2016, 26, 2167–2177. [Google Scholar] [CrossRef]
- Di Lazzaro, V.; Profice, P.; Ranieri, F.; Capone, F.; Dileone, M.; Oliviero, A.; Pilato, F. I-wave origin and modulation. Brain Stimul. 2012, 5, 512–525. [Google Scholar] [CrossRef]
- Thickbroom, G.W. A model of the contribution of late I-waves to α-motoneuronal activation: Implications for paired-pulse TMS. Brain Stimul. 2011, 4, 77–83. [Google Scholar] [CrossRef]
- Belci, M.; Catley, M.; Husain, M.; Frankel, H.L.; Davey, N.J. Magnetic brain stimulation can improve clinical outcome in incomplete spinal cord injured patients. Spinal Cord 2004, 42, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Schnitzler, A.; Benecke, R. The silent period after transcranial magnetic stimulation is of exclusive cortical origin: Evidence from isolated cortical ischemic lesions in man. Neurosci. Lett. 1994, 180, 41–45. [Google Scholar] [CrossRef]
- Hallett, M. Transcranial magnetic stimulation and the human brain. Nature 2000, 406, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Pascual-Leone, A. Basic principles of magnetic stimulation. Lancet 2003, 2, 145–156. [Google Scholar] [CrossRef]
- Burke, D.; Hicks, R.; Gandevia, S.C.; Stephen, J.; Woodforth, I.; Crawford, M. Direct comparison of corticospinal volleys in human subjects to transcranial magnetic and electrical stimulation. J. Physiol. 1993, 470, 383–393. [Google Scholar] [CrossRef]
- Nielsen, J.; Petersen, N. Changes in the effect of magnetic brain stimulation accompanying voluntary dynamic contraction in man. J. Physiol. 1995, 484, 777–789. [Google Scholar] [CrossRef]
- Schneider, C.; Lavoie, B.A.; Barbeau, H.; Capaday, C. Timing of cortical excitability changes during the reaction time of movements superimposed on tonic motor activity. J. Appl. Physiol. 2004, 97, 2220–2227. [Google Scholar] [CrossRef]
- Geertsen, S.S.; Zuur, A.T.; Nielsen, J.B. Voluntary activation of ankle muscles is accompanied by subcortical facilitation of their antagonists. J. Physiol. 2010, 588, 2391–2402. [Google Scholar] [CrossRef]
- Hou, J.; Nelson, R.; Nissim, N.; Parmer, R.; Thompson, F.J.; Bose, P. Effect of combined treadmill training and magnetic stimulation on spasticity and gait impairments after cervical spinal cord injury. J. Neurotrauma 2014, 31, 1088–1106. [Google Scholar] [CrossRef]
- Moxon, K.A.; Oliviero, A.; Aguilar, J.; Foffani, G. Cortical reorganization after spinal cord injury: Always for good? Neuroscience 2014, 283, 78–94. [Google Scholar] [CrossRef]
- Roy, F.D.; Zewdie, E.T.; Gorassini, M.A. Short-interval intracortical inhibition with incomplete spinal cord injury. Clin. Neurophysiol. 2011, 122, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Federico, P.; Perez, M.A. Altered corticospinal function during movement preparation in humans with spinal cord injury. J. Physiol. 2017, 595, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Bunday, K.L.; Urbin, M.A.; Perez, M.A. Potentiating paired corticospinal-motoneuronal plasticity after spinal cord injury. Brain Stimul. 2018, 11, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Urbin, M.A.; Ozdemir, R.A.; Tazoe, T.; Perez, M.A. Spike-timing-dependent plasticity in lower-limb motoneurons after human spinal cord injury. J. Neurophysiol. 2017, 118, 2171–2180. [Google Scholar] [CrossRef]
- Tolmacheva, A.; Savolainen, S.; Kirveskari, E.; Lioumis, P.; Kuusela, L.; Brandstack, N.; Ylinen, A.; Mäkelä, J.P.; Shulga, A. Long-Term Paired Associative Stimulation Enhances Motor Output of the Tetraplegic Hand. J. Neurotrauma 2017, 34, 2668–2674. [Google Scholar] [CrossRef]
- Weise, D.; Mann, J.; Ridding, M.; Eskandar, K.; Huss, M.; Rumpf, J.J.; Di Lazzaro, V.; Mazzone, P.; Ranieri, F.; Classen, J. Microcircuit mechanisms involved in paired associative stimulation-induced depression of corticospinal excitability. J. Physiol. 2013, 591, 4903–4920. [Google Scholar] [CrossRef]
- Petrosyan, H.A.; Alessi, V.; Sisto, S.A.; Kaufman, M.; Arvanian, V.L. Transcranial magnetic stimulation (TMS) responses elicited in hindlimb muscles as an assessment of synaptic plasticity in spino-muscular circuitry after chronic spinal cord injury. Neurosci. Lett. 2017, 642, 37–42. [Google Scholar] [CrossRef]
- Kokotilo, K.J.; Eng, J.J.; Curt, A. Reorganization and preservation of motor control of the brain in spinal cord injury: A systematic review. J. Neurotrauma 2009, 26, 2113–2126. [Google Scholar] [CrossRef]
- Wagle-Shukla, A.; Ni, Z.; Gunraj, C.A.; Bahl, N.; Chen, R. Effects of short interval intracortical inhibition and intracortical facilitation on short interval intracortical facilitation in human primary motor cortex. J. Physiol. 2009, 587, 5665–5678. [Google Scholar] [CrossRef]
- Gupta, S.; Jaiswal, A.; Norman, K.; DePaul, V. Progressive neuromuscular scoliosis secondary to spinal cord injury in a young patient treated with nonfusion anterior scoliosis correction. Top. Spinal Cord Inj. Rehabil. 2019, 25, 164–185. [Google Scholar] [CrossRef]
- Curt, A.; Van Hedel, H.J.A.; Klaus, D.; Dietz, V. Recovery from a spinal cord injury: Significance of compensation, neural plasticity, and repair. J. Neurotrauma 2008, 25, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Rowley, N.M.; Madsen, K.K.; Schousboe, A.; Steve White, H. Glutamate and GABA synthesis, release, transport and metabolism as targets for seizure control. Neurochem. Int. 2012, 61, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Kann, O. The interneuron energy hypothesis: Implications for brain disease. Neurobiol. Dis. 2016, 90, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Azbill, R.D.; Mu, X.; Bruce-Keller, A.J.; Mattson, M.P.; Springer, J.E. Impaired mitochondrial function, oxidative stress and altered antioxidant enzyme activities following traumatic spinal cord injury. Brain Res. 1997, 765, 283–290. [Google Scholar] [CrossRef]
- Phillips, A.A.; Ainslie, P.N.; Krassioukov, A.V.; Warburton, D.E.R. Regulation of cerebral blood flow after spinal cord injury. J. Neurotrauma 2013, 30, 1551–1563. [Google Scholar] [CrossRef]
- Melloni, L.; Molina, C.; Pena, M.; Torres, D.; Singer, W.; Rodriguez, E. Synchronization of neural activity across cortical areas correlates with conscious perception. J. Neurosci. 2007, 27, 2858–2865. [Google Scholar] [CrossRef]
- Uhlhaas, P.J.; Singer, W. Abnormal neural oscillations and synchrony in schizophrenia. Nat. Rev. Neurosci. 2010, 11, 100–113. [Google Scholar] [CrossRef]
- Craig, A.; Guest, R.; Tran, Y.; Middleton, J. Cognitive Impairment and Mood States after Spinal Cord Injury. J. Neurotrauma 2017, 34, 1156–1163. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, Z.; Sabirzhanov, B.; Stoica, B.A.; Kumar, A.; Luo, T.; Skovira, J.; Faden, A.I. Spinal cord injury causes brain inflammation associated with cognitive and affective changes: Role of cell cycle pathways. J. Neurosci. 2014, 34, 10989–11006. [Google Scholar] [CrossRef]
- Gensel, J.C.; Zhang, B. Macrophage activation and its role in repair and pathology after spinal cord injury. Brain Res. 2015, 1619, 1–11. [Google Scholar] [CrossRef]
- Ren, Y.; Young, W. Managing inflammation after spinal cord injury through manipulation of macrophage function. Neural Plast. 2013, 2013, 945034. [Google Scholar] [CrossRef] [PubMed]
- Pineau, I.; Steve, L. Proinflammatory Cytokine Synthesis in the Injured Mouse Spinal Cord: Multiphasic Expression Pattern and Identification of the Cell Types Involved. J. Comp. Neurol. 2007, 346, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Zeilhofer, H.U. Loss of glycinergic and GABAergic inhibition in chronic pain-contributions of inflammation and microglia. Int. Immunopharmacol. 2008, 8, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Ferrini, F.; De Koninck, Y. Microglia control neuronal network excitability via BDNF signalling. Neural Plast. 2013, 2013, 429815. [Google Scholar] [CrossRef]
- Pozzi, D.; Rasile, M.; Corradini, I.; Matteoli, M. Environmental regulation of the chloride transporter KCC2: Switching inflammation off to switch the GABA on? Transl. Psychiatry 2020, 10, 349. [Google Scholar] [CrossRef]
- Reichelt, A.C.; Hare, D.J.; Bussey, T.J.; Saksida, L.M. Perineuronal Nets: Plasticity, Protection, and Therapeutic Potential. Trends Neurosci. 2019, 42, 458–470. [Google Scholar] [CrossRef]
- Carstens, K.E.; Phillips, M.L.; Pozzo-Miller, L.; Weinberg, R.J.; Dudek, S.M. Perineuronal nets suppress plasticity of excitatory synapses on CA2 pyramidal neurons. J. Neurosci. 2016, 36, 6312–6320. [Google Scholar] [CrossRef]
- Kwok, J.C.F.; Dick, G.; Wang, D.; Fawcett, J.W. Extracellular matrix and perineuronal nets in CNS repair. Dev. Neurobiol. 2011, 71, 1073–1089. [Google Scholar] [CrossRef]
- Casha, S.; Zygun, D.; McGowan, M.D.; Bains, I.; Yong, V.W.; John Hurlbert, R. Results of a phase II placebo-controlled randomized trial of minocycline in acute spinal cord injury. Brain 2012, 135, 1224–1236. [Google Scholar] [CrossRef]
- Rossier, J.; Bernard, A.; Cabungcal, J.H.; Perrenoud, Q.; Savoye, A.; Gallopin, T.; Hawrylycz, M.; Cuénod, M.; Do, K.; Urban, A.; et al. Cortical fast-spiking parvalbumin interneurons enwrapped in the perineuronal net express the metallopeptidases Adamts8, Adamts15 and Neprilysin. Mol. Psychiatry 2015, 20, 154–161. [Google Scholar] [CrossRef]
- Härtig, W.; Derouiche, A.; Welt, K.; Brauer, K.; Grosche, J.; Mäder, M.; Reichenbach, A.; Brückner, G. Cortical neurons immunoreactive for the potassium channel Kv3.1b subunit are predominantly surrounded by perineuronal nets presumed as a buffering system for cations. Brain Res. 1999, 842, 15–29. [Google Scholar] [CrossRef]
- Lorenzo Bozzelli, P.; Alaiyed, S.; Kim, E.; Villapol, S.; Conant, K. Proteolytic Remodeling of Perineuronal Nets: Effects on Synaptic Plasticity and Neuronal Population Dynamics. Neural Plast. 2018, 2018, 5735789. [Google Scholar] [CrossRef] [PubMed]
- Cabungcal, J.H.; Steullet, P.; Morishita, H.; Kraftsik, R.; Cuenod, M.; Hensch, T.K.; Do, K.Q. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 9130–9135. [Google Scholar] [CrossRef] [PubMed]
- Morishita, H.; Cabungcal, J.H.; Chen, Y.; Do, K.Q.; Hensch, T.K. Prolonged Period of Cortical Plasticity upon Redox Dysregulation in Fast-Spiking Interneurons. Biol. Psychiatry 2015, 78, 396–402. [Google Scholar] [CrossRef]
- Agarwala, S.; Kalil, R.E. Axotomy-induced neuronal death and reactive astrogliosis in the lateral geniculate nucleus following a lesion of the visual cortex in the rat. J. Comp. Neurol. 1998, 392, 252–263. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. The tripartite synapse: Roles for gliotransmission in health and disease. Trends Mol. Med. 2007, 13, 54–63. [Google Scholar] [CrossRef]
- Le Meur, K.; Mendizabal-Zubiaga, J.; Grandes, P.; Audinat, E. GABA release by hippocampal astrocytes. Front. Comput. Neurosci. 2012, 6, 59. [Google Scholar] [CrossRef]
- Benedetti, B.; Matyash, V.; Kettenmann, H. Astrocytes control GABAergic inhibition of neurons in the mouse barrel cortex. J. Physiol. 2011, 589, 1159–1172. [Google Scholar] [CrossRef]
- Waagepetersen, H.S.; Sonnewald, U.; Schousboe, A. Compartmentation of glutamine, glutamate, and GABA metabolism in neurons and astrocytes: Functional implications. Neuroscientist 2003, 9, 398–403. [Google Scholar] [CrossRef]
- Walls, A.B.; Waagepetersen, H.S.; Bak, L.K.; Schousboe, A.; Sonnewald, U. The Glutamine–Glutamate/GABA Cycle: Function, Regional Differences in Glutamate and GABA Production and Effects of Interference with GABA Metabolism. Neurochem. Res. 2015, 40, 402–409. [Google Scholar] [CrossRef]
- MacVicar, B.A.; Choi, H.B. Astrocytes Provide Metabolic Support for Neuronal Synaptic Function in Response to Extracellular K+. Neurochem. Res. 2017, 42, 2588–2594. [Google Scholar] [CrossRef] [PubMed]
- Hassanzadeh, S.; Jalessi, M.; Jameie, S.B.; Khanmohammadi, M.; Bagher, Z.; Namjoo, Z.; Davachi, S.M. More attention on glial cells to have better recovery after spinal cord injury. Biochem. Biophys. Rep. 2021, 25, 100905. [Google Scholar] [CrossRef] [PubMed]
- Leemhuis, E.; Giuffrida, V.; De Martino, M.L.; Forte, G.; Pecchinenda, A.; De Gennaro, L.; Giannini, A.M.; Pazzaglia, M. Rethinking the Body in the Brain after Spinal Cord Injury. J. Clin. Med. 2022, 11, 388. [Google Scholar] [CrossRef] [PubMed]
- Spolidoro, M.; Sale, A.; Berardi, N.; Maffei, L. Plasticity in the adult brain: Lessons from the visual system. Exp. Brain Res. 2009, 192, 335–341. [Google Scholar] [CrossRef]
- Bachtiar, V.; Stagg, C.J. The role of inhibition in human motor cortical plasticity. Neuroscience 2014, 278, 93–104. [Google Scholar] [CrossRef]
- Kolasinski, J.; Hinson, E.L.; Divanbeighi Zand, A.P.; Rizov, A.; Emir, U.E.; Stagg, C.J. The dynamics of cortical GABA in human motor learning. J. Physiol. 2019, 597, 271–282. [Google Scholar] [CrossRef]
- Castro-Alamancos, M.; Donoghue, J.P.; Connors, B.W. Different forms of synaptic plasticity in somatosensory and motor areas of the neocortex. J. Neurosci. 1995, 15, 5324–5333. [Google Scholar] [CrossRef]
- Trepel, C.; Racine, R.J. GABAergic Modulation of Neocortical Long-Term Potentiation in the Freely Moving Rat. Synapse 2000, 35, 120–128. [Google Scholar] [CrossRef]
- Clarkson, A.N.; Huang, B.S.; Macisaac, S.E.; Mody, I.; Carmichael, S.T. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 2010, 468, 305–309. [Google Scholar] [CrossRef]
- Murphy, S.C.; Palmer, L.M.; Nyffeler, T.; Müri, R.M.; Larkum, M.E. Transcranial magnetic stimulation (TMS) inhibits cortical dendrites. eLife 2016, 5, e13598. [Google Scholar] [CrossRef]
- Nardone, R.; Höller, Y.; Thomschewski, A.; Höller, P.; Lochner, P.; Golaszewski, S.; Brigo, F.; Trinka, E. Serotonergic transmission after spinal cord injury. J. Neural Transm. 2014, 122, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Araneda, R.; Andrade, R. 5-Hydroxytryptamine2 and 5-hydroxytryptamine1A receptors mediate opposing responses on membrane excitability in rat association cortex. Neuroscience 1991, 40, 399–412. [Google Scholar] [CrossRef]
- Azmitia, E.C.; Gannon, P.J.; Kheck, N.M.; Whitaker-Azmitia, P.M. Cellular localization of the 5-HT(1A) receptor in primate brain neurons and glial cells. Neuropsychopharmacology 1996, 14, 35–46. [Google Scholar] [CrossRef]
- Sheldon, P.W.; Aghajanian, G.K. Excitatory responses to serotonin (5-HT) in neurons of the rat piriform cortex: Evidence for mediation by 5-HT1C receptors in pyramidal cells and 5-HT2 receptors in interneurons. Synapse 1991, 9, 208–218. [Google Scholar] [CrossRef]
- Sheldon, P.W.; Aghajanian, G.K. Serotonin (5-HT) induces IPSPs in pyramidal layer cells of rat piriform cortex: Evidence for the involvement of a 5-HT2 -activated interneuron. Brain Res. 1990, 506, 62–69. [Google Scholar] [CrossRef]
- Fouad, K.; Rank, M.M.; Vavrek, R.; Murray, K.C.; Sanelli, L.; Bennett, D.J. Locomotion after spinal cord injury depends on constitutive activity in serotonin receptors. J. Neurophysiol. 2010, 104, 2975–2984. [Google Scholar] [CrossRef]
- Giménez Y Ribotta, M.; Provencher, J.; Feraboli-Lohnherr, D.; Rossignol, S.; Privát, A.; Orsal, D. Activation of locomotion in adult chronic spinal rats is achieved by transplantation of embryonic raphe cells reinnervating a precise lumbar level. J. Neurosci. 2000, 20, 5144–5152. [Google Scholar] [CrossRef]
- Guertin, P.A. Role of NMDA receptor activation in serotonin agonist-induced air-stepping in paraplegic mice. Spinal Cord 2004, 42, 185–190. [Google Scholar] [CrossRef]
- Long, J.; Federico, P.; Perez, M.A. A novel cortical target to enhance hand motor output in humans with spinal cord injury. Brain 2017, 140, 1619–1632. [Google Scholar] [CrossRef]
- Versace, V.; Langthaler, P.B.; Höller, Y.; Frey, V.N.; Brigo, F.; Sebastianelli, L.; Saltuari, L.; Nardone, R. Abnormal cortical neuroplasticity induced by paired associative stimulation after traumatic spinal cord injury: A preliminary study. Neurosci. Lett. 2018, 664, 167–171. [Google Scholar] [CrossRef]
- Kuppuswamy, A.; Balasubramaniam, A.V.; Maksimovic, R.; Mathias, C.J.; Gall, A.; Craggs, M.D.; Ellaway, P.H. Action of 5Hz repetitive transcranial magnetic stimulation on sensory, motor and autonomic function in human spinal cord injury. Clin. Neurophysiol. 2011, 122, 2452–2461. [Google Scholar] [CrossRef] [PubMed]
- Cortes, M.; Elder, J.; Rykman, A.; Murray, L.; Avedissian, M.; Stampa, A.; Thickbroom, G.W.; Pascual-Leone, A.; Krebs, H.I.; Valls-Sole, J.; et al. Improved motor performance in chronic spinal cord injury following upper-limb robotic training. NeuroRehabilitation 2013, 33, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.G. Gabaergic neurons and their role in cortical plasticity in primates. Cereb. Cortex 1993, 3, 361–372. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benedetti, B.; Weidenhammer, A.; Reisinger, M.; Couillard-Despres, S. Spinal Cord Injury and Loss of Cortical Inhibition. Int. J. Mol. Sci. 2022, 23, 5622. https://doi.org/10.3390/ijms23105622
Benedetti B, Weidenhammer A, Reisinger M, Couillard-Despres S. Spinal Cord Injury and Loss of Cortical Inhibition. International Journal of Molecular Sciences. 2022; 23(10):5622. https://doi.org/10.3390/ijms23105622
Chicago/Turabian StyleBenedetti, Bruno, Annika Weidenhammer, Maximilian Reisinger, and Sebastien Couillard-Despres. 2022. "Spinal Cord Injury and Loss of Cortical Inhibition" International Journal of Molecular Sciences 23, no. 10: 5622. https://doi.org/10.3390/ijms23105622
APA StyleBenedetti, B., Weidenhammer, A., Reisinger, M., & Couillard-Despres, S. (2022). Spinal Cord Injury and Loss of Cortical Inhibition. International Journal of Molecular Sciences, 23(10), 5622. https://doi.org/10.3390/ijms23105622