
Metabolic Remodeling with Hepatosteatosis Induced Vascular Oxidative Stress in Hepatic ERK2 Deficiency Mice with High Fat Diets

 , ,
, ,  and
and
Abstract
:
1. Introduction
2. Results
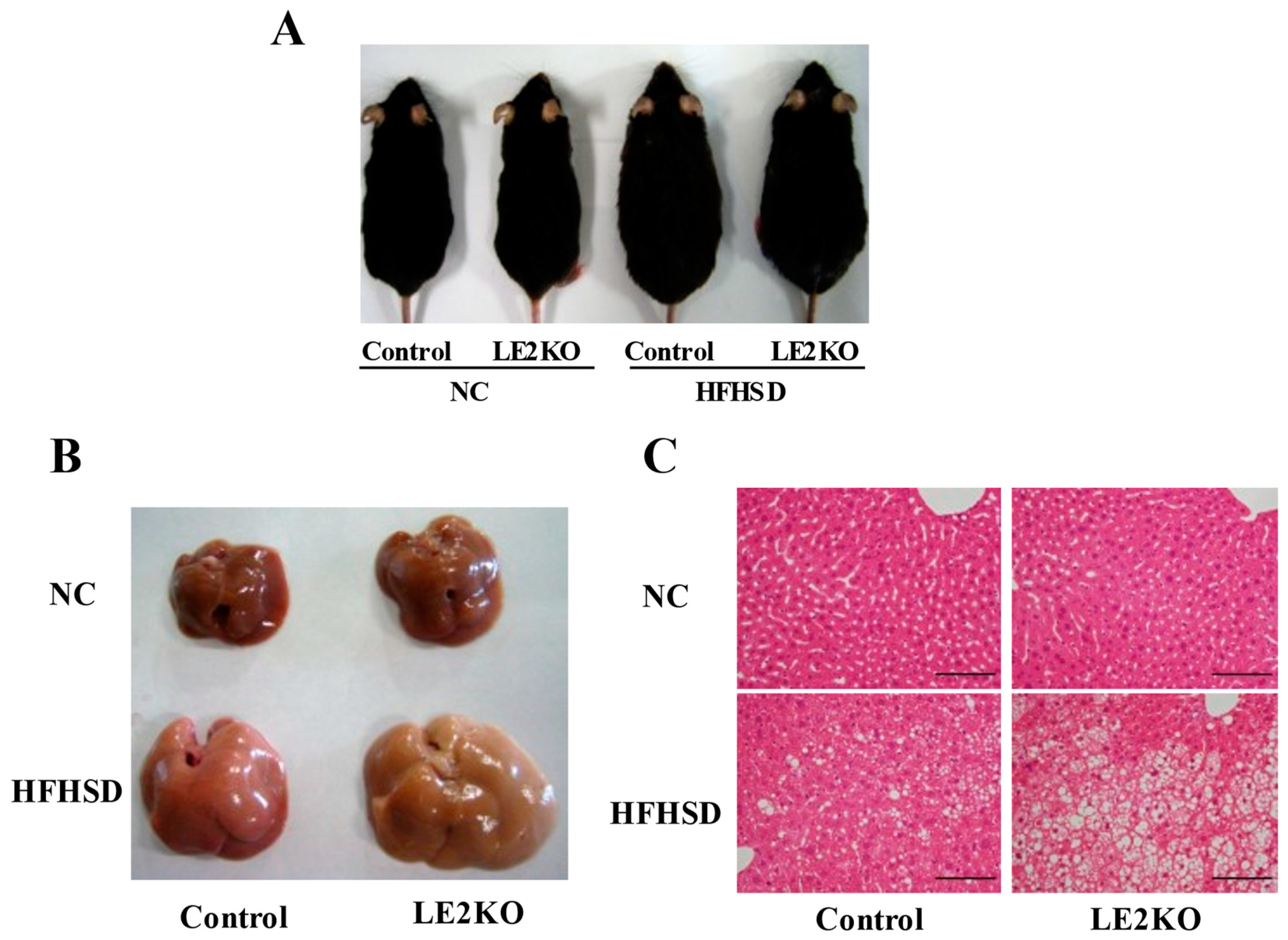
2.1. The Aggravation of HST without Changing Body Weight in HFHSD-LE2KO
2.2. Decreased Phosphorylation of AMPK/ACC and the Increased Expression of SREBP-1c in Liver from HFHSD-LE2KO
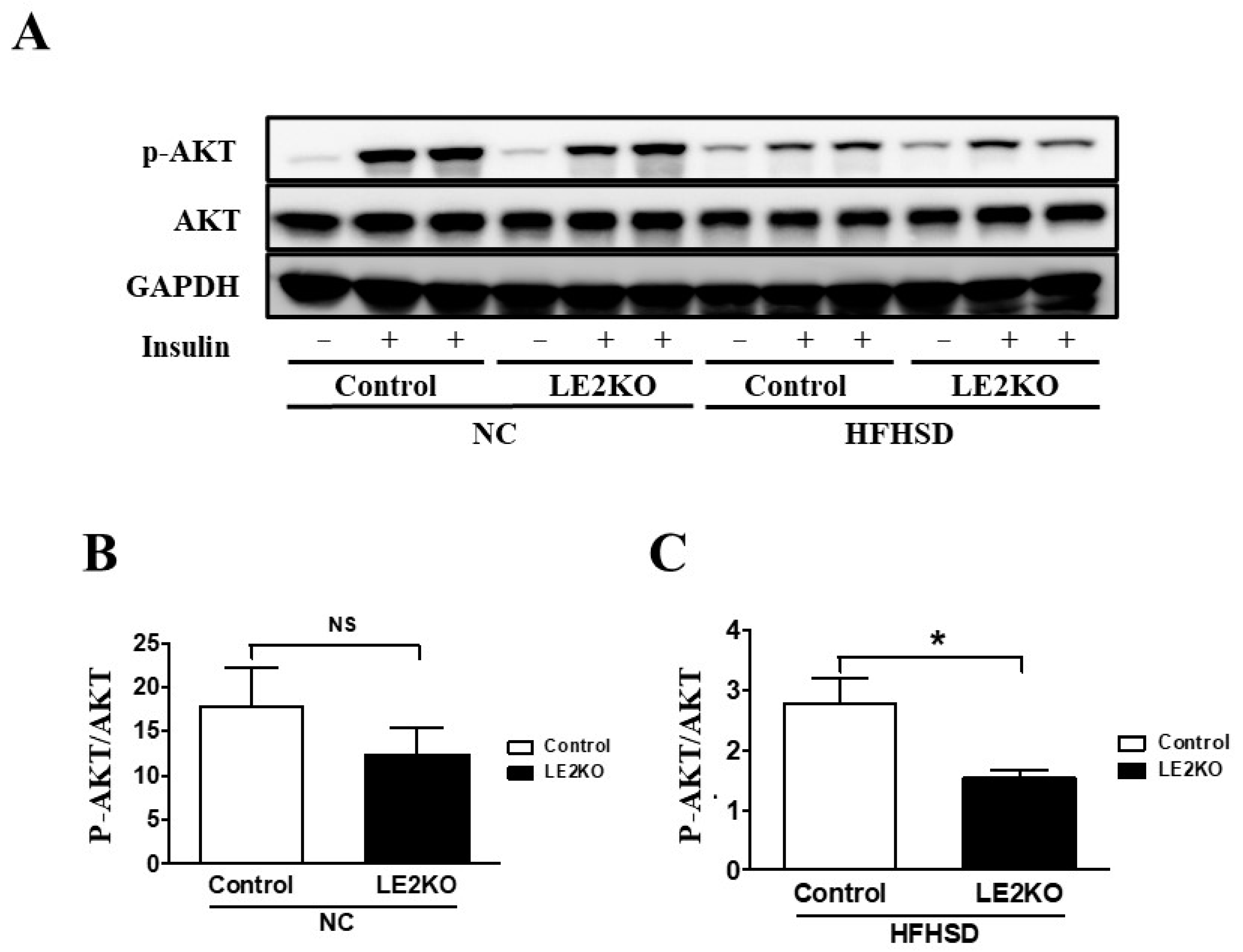
2.3. Elevation of Serum Glucose and Insulin Levels and Insulin Resistance in Skeletal Muscle from HFHSD-LE2KO
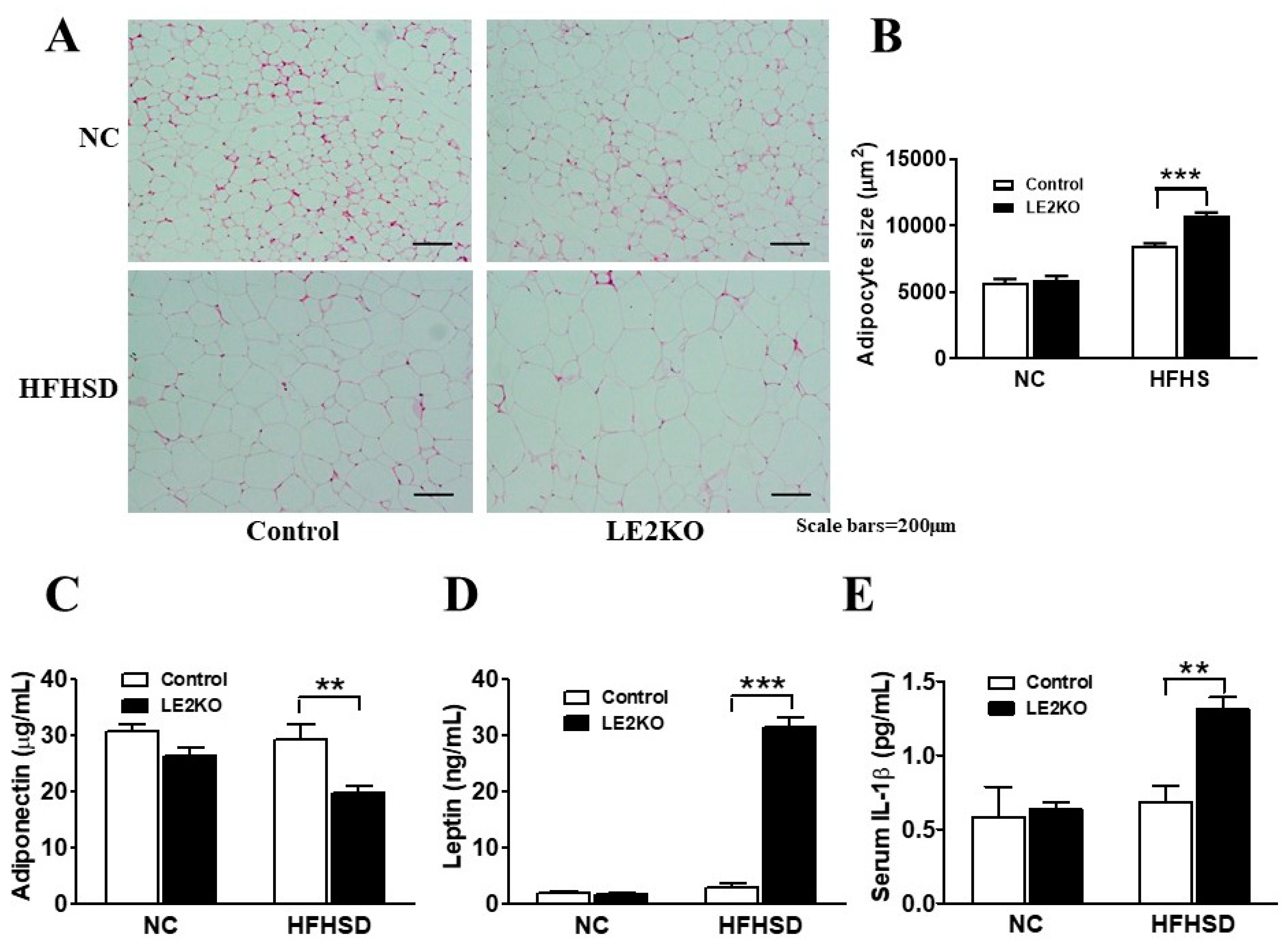
2.4. Adipocytes Enlargement and Changes in Serum Adipocytokines in Epididymal Fat
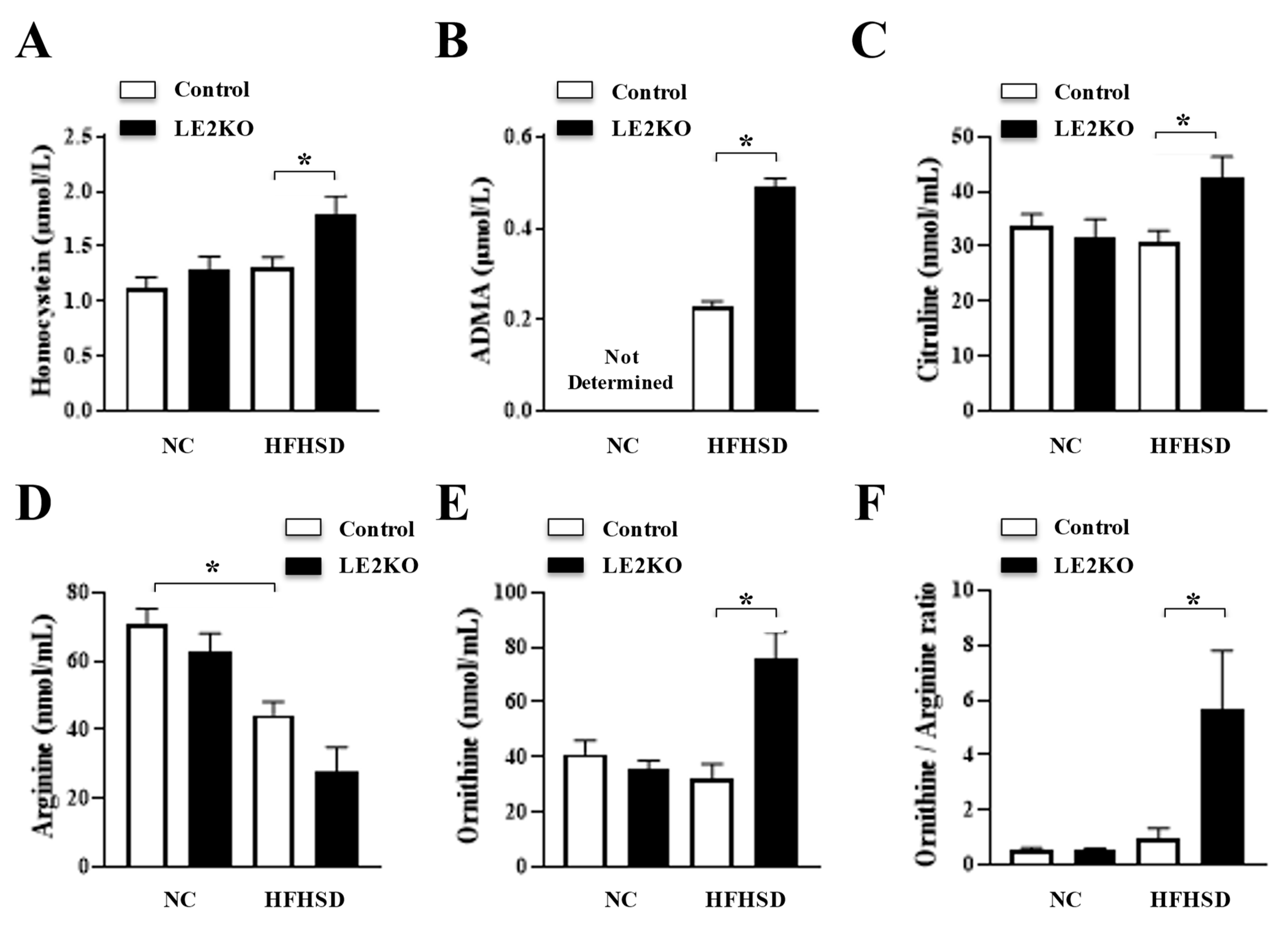
2.5. Serum FA Fraction and Amino Acid Analysis
2.6. Serum Amino Acid Analysis
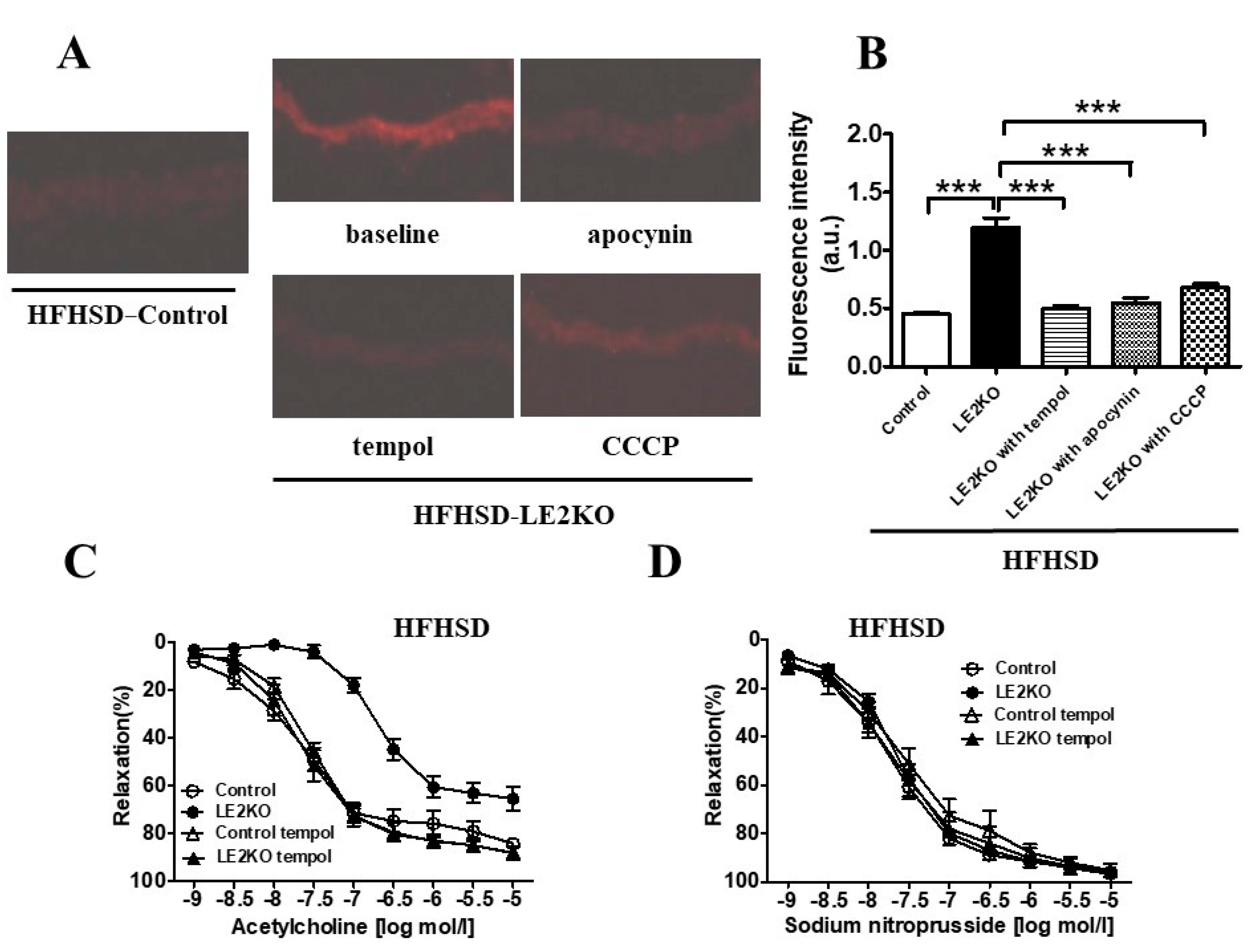
2.7. HFHSD-LE2KO Demonstrated Increased Aortic Superoxide Production and Impaired Endothelium-Dependent Relaxation
3. Discussion
4. Materials and Methods
4.1. Animals, Genotyping, and Diets
4.2. Biochemical Analysis of Tissues
4.3. Tissue Preparation and Histology
4.4. Measurements of Metabolites and Adipocytokines
4.5. Detection of Insulin-Induced p-AKT in Skeletal Muscle
4.6. Quantitative Real-Time PCR for SREBP1c
- SREBP1c (forward), 5′-TGAGAAGCGCTACCGGTCTT-3′;
- SREBP1c (reverse), 5′-AAGCGGATGTAGTCGATGGC-3′;
- 18S rRNA (forward), 5′-TTCCGATAACGAACGAGACTCT-3′;
- 18S rRNA (reverse), 5′-TGGCTGAACGCCACTTGTC-3′.
4.7. Gene Microarray Analysis
4.8. Measurement of Vascular Superoxide Production
4.9. Preparation of Aortic Rings and Organ Chamber Experiments
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations and Acronyms
| AA | Arachidonic acid |
| DHE | dihydroethidium |
| LE2KO | Liver-specific ERK2 knockout |
| eNOS | endothelial nitric oxide synthase |
| EPA | Eicosapentaenoic acid |
| ERK | extracellular signal-regulated kinase |
| FA | Fatty acid |
| HST | hepatosteatosis |
| HFHSD | high-fat/high-sucrose diet |
| MetS | metabolic syndrome |
| NC | normal chow |
| NO | nitric oxide |
| PI3K | phosphatidylinositol-3 kinase |
| Alb-Cre | transgenic mice expressing Cre recombinase under the control of the albumin promoter-Cre |
References
- Muzurovic, E.; Mikhailidis, D.P.; Mantzoros, C. Non-alcoholic fatty liver disease, insulin resistance, metabolic syndrome and their association with vascular risk. Metabolism 2021, 119, 154770. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, L.S.; Curzen, N.P.; Calder, P.C.; Byrne, C.D. Non-alcoholic fatty liver disease: A new and important cardiovascular risk factor? Eur. Heart J. 2012, 33, 1190–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahfood Haddad, T.; Hamdeh, S.; Kanmanthareddy, A.; Alla, V.M. Nonalcoholic fatty liver disease and the risk of clinical cardiovascular events: A systematic review and meta-analysis. Diabetes Metab. Syndr. 2017, 11, S209–S216. [Google Scholar] [CrossRef]
- Ismaiel, A.; Dumitrascu, D.L. Cardiovascular Risk in Fatty Liver Disease: The Liver-Heart Axis-Literature Review. Front. Med. 2019, 6, 202. [Google Scholar] [CrossRef] [Green Version]
- Toh, J.Z.K.; Pan, X.H.; Tay, P.W.L.; Ng, C.H.; Yong, J.N.; Xiao, J.; Koh, J.H.; Tan, E.Y.; Tan, E.X.X.; Dan, Y.Y.; et al. A Meta-Analysis on the Global Prevalence, Risk factors and Screening of Coronary Heart Disease in Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, M.S.; Russo, V.; Nigro, G.; Durante-Mangoni, E.; Zampino, R. Interplay between Heart Disease and Metabolic Steatosis: A Contemporary Perspective. J. Clin. Med. 2021, 10, 1569. [Google Scholar] [CrossRef] [PubMed]
- Kujiraoka, T.; Satoh, Y.; Ayaori, M.; Shiraishi, Y.; Arai-Nakaya, Y.; Hakuno, D.; Yada, H.; Kuwada, N.; Endo, S.; Isoda, K.; et al. Hepatic extracellular signal-regulated kinase 2 suppresses endoplasmic reticulum stress and protects from oxidative stress and endothelial dysfunction. J. Am. Heart. Assoc. 2013, 2, e000361. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Kagami, K.; Sato, A.; Osaki, A.; Ito, K.; Horii, S.; Toya, T.; Masaki, N.; Yasuda, R.; Nagatomo, Y.; et al. Sarco/Endoplasmic Reticulum Ca(2+) ATPase 2 Activator Ameliorates Endothelial Dysfunction; Insulin Resistance in Diabetic Mice. Cells 2022, 11, 1488. [Google Scholar] [CrossRef] [PubMed]
- Rask-Madsen, C.; Kahn, C.R. Tissue-specific insulin signaling, metabolic syndrome, and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2052–2059. [Google Scholar] [CrossRef] [Green Version]
- Michael, M.D.; Kulkarni, R.N.; Postic, C.; Previs, S.F.; Shulman, G.I.; Magnuson, M.A.; Kahn, C.R. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell 2000, 6, 87–97. [Google Scholar] [CrossRef]
- Biddinger, S.B.; Hernandez-Ono, A.; Rask-Madsen, C.; Haas, J.T.; Aleman, J.O.; Suzuki, R.; Scapa, E.F.; Agarwal, C.; Carey, M.C.; Stephanopoulos, G.; et al. Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 2008, 7, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Dufresne, S.D.; Bjørbaek, C.; El-Haschimi, K.; Zhao, Y.; Aschenbach, W.G.; Moller, D.E.; Goodyear, L.J. Altered extracellular signal-regulated kinase signaling and glycogen metabolism in skeletal muscle from p90 ribosomal S6 kinase 2 knockout mice. Mol. Cell Bio. 2001, 21, 81–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itier, R.; Guillaume, M.; Ricci, J.E.; Roubille, F.; Delarche, N.; Picard, F.; Galinier, M.; Roncalli, J. Non-alcoholic fatty liver disease and heart failure with preserved ejection fraction: From pathophysiology to practical issues. ESC Heart Fail 2021, 8, 789–798. [Google Scholar] [CrossRef]
- Zou, M.-H.; Chaomei Shi, R.A.C. High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H(2) receptor-mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial cells. Diabetes 2002, 51, 198–203. [Google Scholar]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carling, D.; Zammit, V.A.; Hardie, D.G. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987, 2, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randle, P.J.; Garland, P.B.; Hales, C.N.; Newsholme, E.A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963, 1, 785–789. [Google Scholar] [CrossRef]
- Wu, H.; Ballantyne, C.M. Skeletal muscle inflammation and insulin resistance in obesity. J. Clin Investig. 2017, 127, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kadowaki, T. Adiponectin receptor as a key player in healthy longevity and obesity-related diseases. Cell Metab. 2013, 17, 185–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef] [PubMed]
- Sztolsztener, K.; Chabowski, A.; Harasim-Symbor, E.; Bielawiec, P.; Konstantynowicz-Nowicka, K. Arachidonic Acid as an Early Indicator of Inflammation during Non-Alcoholic Fatty Liver Disease Development. Biomolecules 2020, 10, 1133. [Google Scholar] [CrossRef]
- Zuccollo, A.; Shi, C.; Mastroianni, R.; Maitland-Toolan, K.A.; Weisbrod, R.M.; Zang, M.; Xu, S.; Jiang, B.; Oliver-Krasinski, J.M.; Cayatte, A.J.; et al. The thromboxane A2 receptor antagonist S18886 prevents enhanced atherogenesis caused by diabetes mellitus. Circulation 2005, 112, 3001–3008. [Google Scholar] [CrossRef] [Green Version]
- Wada, M.; DeLong, C.J.; Hong, Y.H.; Rieke, C.J.; Song, I.; Sidhu, R.S.; Yuan, C.; Warnock, M.; Schmaier, A.H.; Yokoyama, C.; et al. Enzymes and receptors of prostaglandin pathways with arachidonic acid-derived versus eicosapentaenoic acid-derived substrates and products. J. Biol. Chem. 2007, 282, 22254–22266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.-H.; Miyahara, H.; Hatanaka, A. Chronic administration of palmitoleic acid reduces insulin resistance and hepatic lipid accumulation in KK-Ay Mice with genetic type 2 diabetes. Lipids Health Dis. 2011, 10, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boger, R.H.; Sullivan, L.M.; Schwedhelm, E.; Wang, T.J.; Maas, R.; Benjamin, E.J.; Schulze, F.; Xanthakis, V.; Benndorf, R.A.; Vasan, R.S. Plasma asymmetric dimethylarginine and incidence of cardiovascular disease and death in the community. Circulation 2009, 119, 1592–1600. [Google Scholar] [CrossRef] [PubMed]
- Boushey, C.J.; Beresford, S.A.; Omenn, G.S.; Motulsky, A.G. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. JAMA 1995, 274, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Cho, L.; Brennan, D.M.; Hazen, S.L. Diminished global arginine bioavailability and increased arginine catabolism as metabolic profile of increased cardiovascular risk. J. Am. Coll. Cardiol. 2009, 53, 2061–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, K.; Masaki, N.; Adachi, T. Arginine deficiency measured by global arginine bioavailability ratio in patients with acute coronary syndrome.pdf. Vasc. Fail 2018, 2, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Masaki, N.; Hakuno, D.; Toya, T.; Shiraishi, Y.; Kujiraoka, T.; Namba, T.; Yada, H.; Kimura, K.; Miyazaki, K.; Adachi, T. Association between brachial-ankle pulse wave velocity and the ratio of l-arginine to asymmetric dimethylarginine in patients undergoing coronary angiography. J. Cardiol. 2015, 65, 311–317. [Google Scholar] [CrossRef]
- Rupp, H.; Wagner, D.; Rupp, T.; Schulte, L.M.; Maisch, B. Risk stratification by the "EPA+DHA level" and the "EPA/AA ratio" focus on anti-inflammatory and antiarrhythmogenic effects of long-chain omega-3 fatty acids. Herz 2004, 29, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Satoh, Y.; Kobayashi, Y.; Takeuchi, A.; Pages, G.; Pouyssegur, J.; Kazama, T. Deletion of ERK1 and ERK2 in the CNS causes cortical abnormalities and neonatal lethality: Erk1 deficiency enhances the impairment of neurogenesis in Erk2-deficient mice. J. Neurosci. 2011, 31, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Satoh, Y.; Endo, S.; Nakata, T.; Kobayashi, Y.; Yamada, K.; Ikeda, T.; Takeuchi, A.; Hiramoto, T.; Watanabe, Y.; Kazama, T. ERK2 contributes to the control of social behaviors in mice. J. Neurosci. 2011, 31, 11953–11967. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Hakuno, D.; Hamba, Y.; Toya, T.; Adachi, T. Plasma amino acid profiling identifies specific amino acid associations with cardiovascular function in patients with systolic heart failure. PLoS ONE 2015, 10, e0117325. [Google Scholar] [CrossRef] [PubMed]
- Jr, F.J.M.; Gutterman, D.D.; Rios, C.D.; Heistad, D.D.; Davidson, B.L. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ. Res. 1998, 1998, 1298–1305. [Google Scholar]
- Dong, Y.F.; Liu, L.; Kataoka, K.; Nakamura, T.; Fukuda, M.; Tokutomi, Y.; Nako, H.; Ogawa, H.; Kim-Mitsuyama, S. Aliskiren prevents cardiovascular complications and pancreatic injury in a mouse model of obesity and type 2 diabetes. Diabetologia 2010, 53, 180–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NC | HFHSD | p Value | |||
|---|---|---|---|---|---|
| Control | LE2KO | Control | LE2KO | ||
| Body weight | 31.4 ± 0.7 | 31.0 ± 0.8 | 41.5 ± 0.9 * | 41.6 ± 0.9 | <0.0001 |
| Myristic acid | 11.7 ± 3.8 | 16.2 ± 8.8 | 9.7 ± 1.1 | 7.4 ± 1.0 # | 0.03 |
| Myristoleic acid | 0.7 ± 0.4 | 1.1 ± 0.9 | 1.3 ± 0.3 | 1.2 ± 0.8 | 0.28 |
| Palmitic acid | 677.1 ± 64.1 | 840.0 ± 180.0 | 694.7 ± 88.8 | 756.1 ± 122.2 | 0.05 |
| Palmitoleic acid | 63.6 ± 11.8 | 86.3 ± 22.8 | 76.5 ± 12.2 | 57.2 ± 6.9 # | 0.005 |
| ThankStearic acid | 183.4 ± 18.7 | 221.0 ± 37.8 | 215.2 ± 17.0 | 353.3 ± 73.1# | <0.0001 |
| Oleic acid | 371.7 ± 50.2 | 519.4 ± 194.0 | 556.6 ± 96.4 * | 676.2 ± 85.6 | 0.0003 |
| Linoleic acid | 792.8 ± 72.6 | 972.8 ± 284.1 | 633.7 ± 71.0 | 458.1 ± 83.4 | <0.0001 |
| γ-Linolenic acid | 18.0 ± 2.9 | 22.9 ± 8.6 | 14.3 ± 1.5 | 9.7 ± 1.5 # | 0.002 |
| α-Linolenic acid | 9.9 ± 2.5 | 14.3 ± 7.8 | 6.7 ± 1.2 * | 2.6 ± 0.6 # | 0.002 |
| Arachidic acid | 2.6 ± 0.4 | 3.3 ± 1.0 | 3.8 ± 0.6 | 5.3 ± 1.4 # | <0.0001 |
| Eicosenoic acid | 7.4 ± 2.0 | 10.3 ± 5.8 | 8.3 ± 1.2 | 9.4 ± 2.3 | 0.35 |
| Eicosadienoic acid | 3.2 ± 1.0 | 4.2 ± 1.9 | 3.0 ± 0.8 | 3.7 ± 1.1 | 0.31 |
| 5-8-11 Eicosatetraenoic acid | 2.2 ± 0.4 | 2.9 ± 0.8 | 5.2 ± 0.9 * | 21.3 ± 9.7 # | <0.0001 |
| Dihomo-γ-linolenic acid | 19.7 ± 2.4 | 25.9 ± 5.5 | 27.7 ± 5.7 * | 73.4 ± 28.5 # | 0.0003 |
| Arachidonic acid | 310.3 ± 40.0 | 346.3 ± 50.5 | 488.5 ± 53.4 * | 992.2 ± 282.1 # | <0.0001 |
| Eicosapentaenoic acid | 30.6 ± 2.8 | 39.2 ± 10.4 | 19.4 ± 2.9 * | 8.7 ± 2.9 # | <0.0001 |
| Behenic acid | 6.7 ± 0.7 | 7.0 ± 0.8 | 9.7 ± 1.4 * | 12.8 ± 3.5 | 0.0002 |
| Erucic acid | 1.9 ± 0.1 | 1.2 ± 0.4 | 1.6 ± 0.3 | 2.2 ± 0.6 # | <0.0001 |
| Docosatetraenoic acid | 2.9 ± 0.5 | 1.9 ± 1.5 | 4.7 ± 0.9 | 9.4 ± 2.4 # | <0.0001 |
| Docosapentaenoic acid | 11.9 ± 1.6 | 15.2 ± 3.2 | 9.8 ± 1.8 | 7.6 ± 1.2 | <0.0001 |
| Lignoceric acid | 5.5 ± 0.5 | 5.8 ± 0.6 | 5.8 ± 0.5 | 6.6 ± 1.6 | 0.18 |
| Docosahexaenoic acid | 225.4 ± 25.1 | 266.9 ± 32.1 | 230.1 ± 21.5 | 271.9 ± 40.0 # | 0.006 |
| Nervonic acid | 15.9 ± 1.1 | 17.4 ± 2.0 | 21.4 ± 1.4 * | 32.8 ± 4.9 # | <0.0001 |
| EPA/AA ratio | 30.6 ± 2.8 | 39.2 ± 10.4 | 19.4 ± 2.9 * | 8.7 ± 1.1 # | <0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kujiraoka, T.; Kagami, K.; Kimura, T.; Ishinoda, Y.; Shiraishi, Y.; Ido, Y.; Endo, S.; Satoh, Y.; Adachi, T. Metabolic Remodeling with Hepatosteatosis Induced Vascular Oxidative Stress in Hepatic ERK2 Deficiency Mice with High Fat Diets. Int. J. Mol. Sci. 2022, 23, 8521. https://doi.org/10.3390/ijms23158521
Kujiraoka T, Kagami K, Kimura T, Ishinoda Y, Shiraishi Y, Ido Y, Endo S, Satoh Y, Adachi T. Metabolic Remodeling with Hepatosteatosis Induced Vascular Oxidative Stress in Hepatic ERK2 Deficiency Mice with High Fat Diets. International Journal of Molecular Sciences. 2022; 23(15):8521. https://doi.org/10.3390/ijms23158521
Chicago/Turabian StyleKujiraoka, Takehiko, Kazuki Kagami, Toyokazu Kimura, Yuki Ishinoda, Yasunaga Shiraishi, Yasuo Ido, Shogo Endo, Yasushi Satoh, and Takeshi Adachi. 2022. "Metabolic Remodeling with Hepatosteatosis Induced Vascular Oxidative Stress in Hepatic ERK2 Deficiency Mice with High Fat Diets" International Journal of Molecular Sciences 23, no. 15: 8521. https://doi.org/10.3390/ijms23158521
APA StyleKujiraoka, T., Kagami, K., Kimura, T., Ishinoda, Y., Shiraishi, Y., Ido, Y., Endo, S., Satoh, Y., & Adachi, T. (2022). Metabolic Remodeling with Hepatosteatosis Induced Vascular Oxidative Stress in Hepatic ERK2 Deficiency Mice with High Fat Diets. International Journal of Molecular Sciences, 23(15), 8521. https://doi.org/10.3390/ijms23158521