
Unravelling the Adaptation Mechanisms to High Pressure in Proteins

 ,
,  , , , ,
, , , ,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
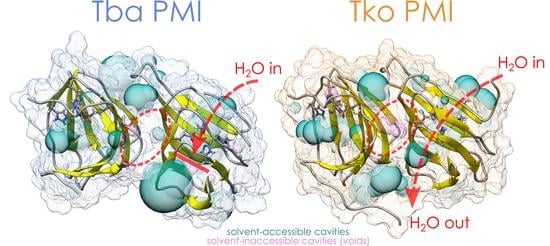
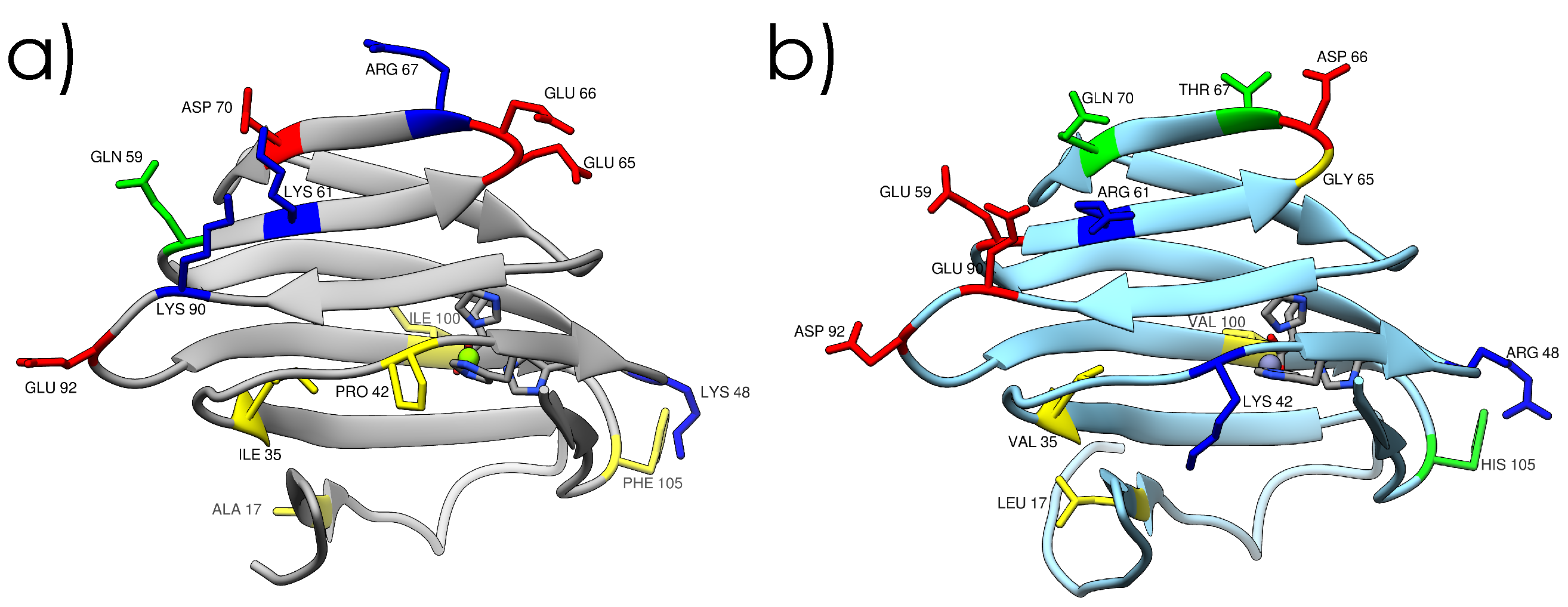
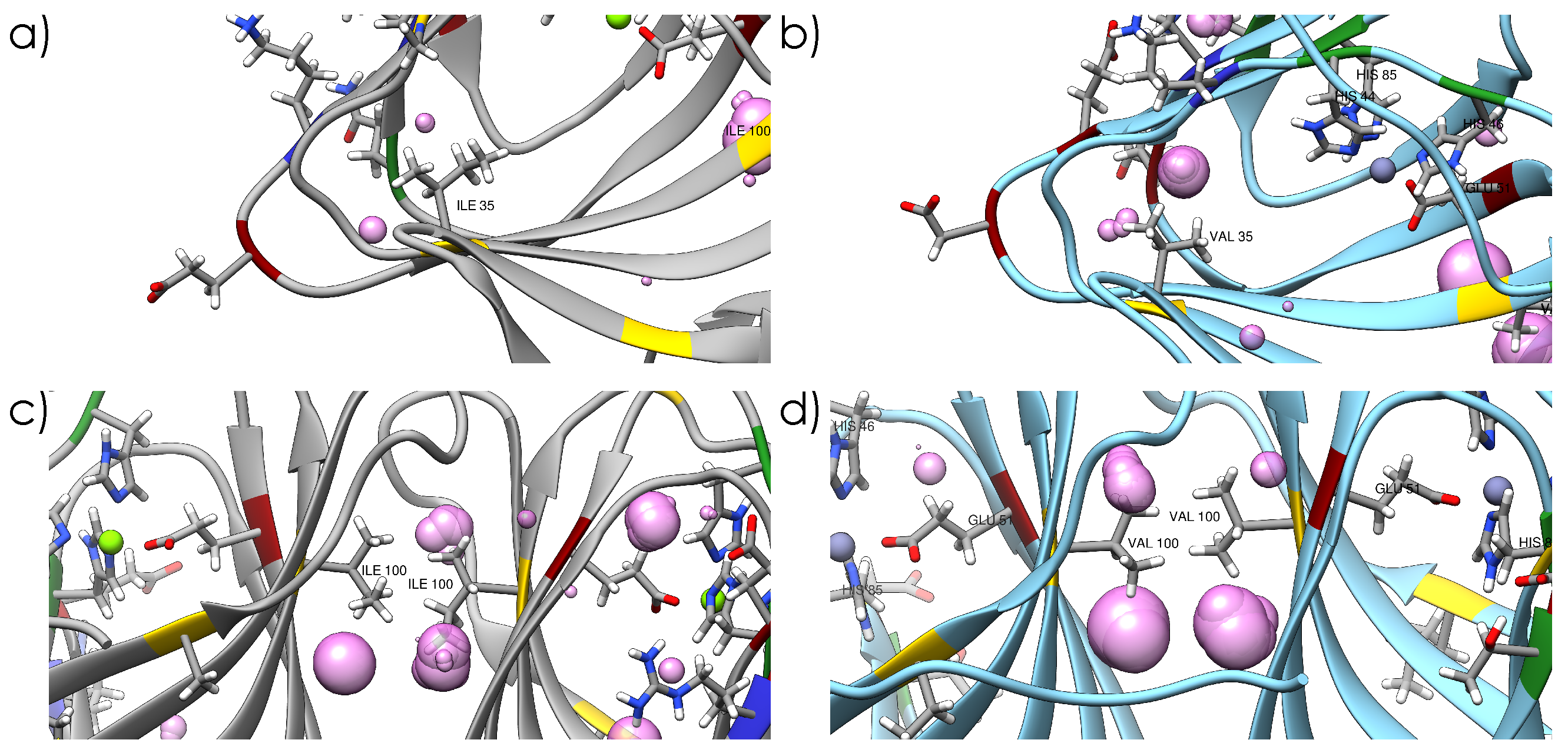
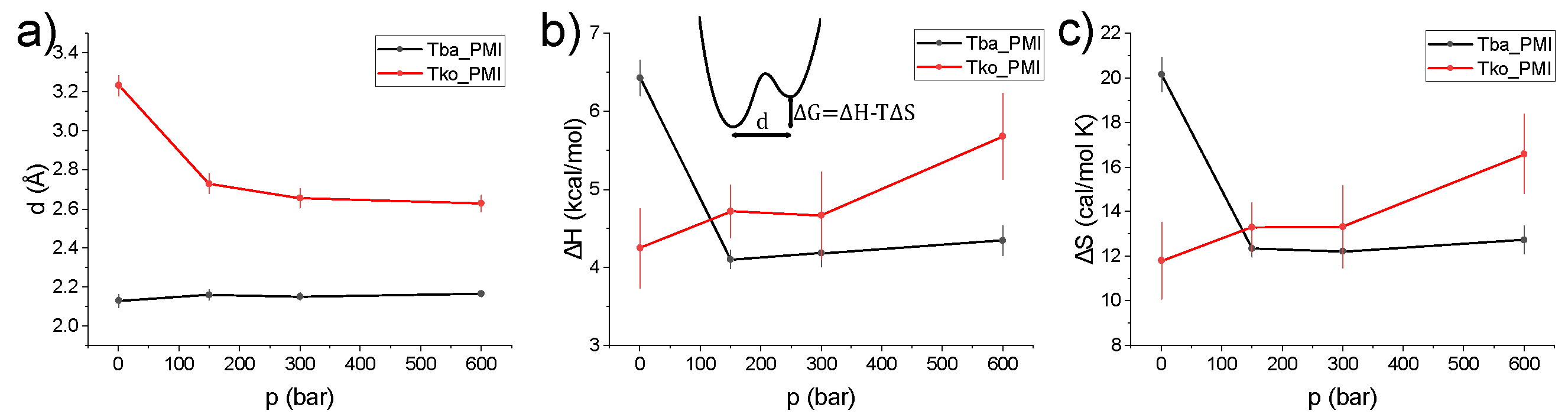
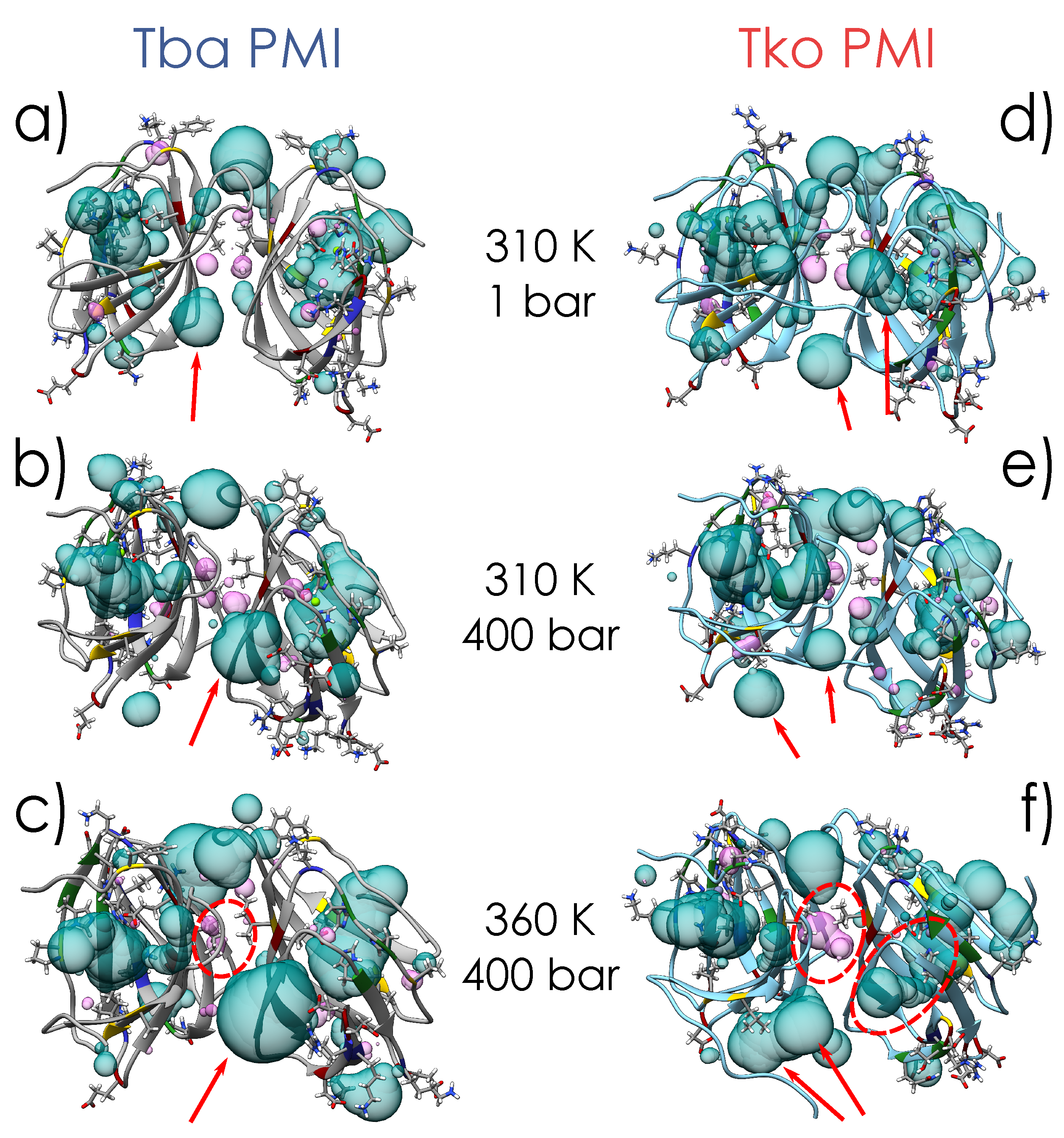
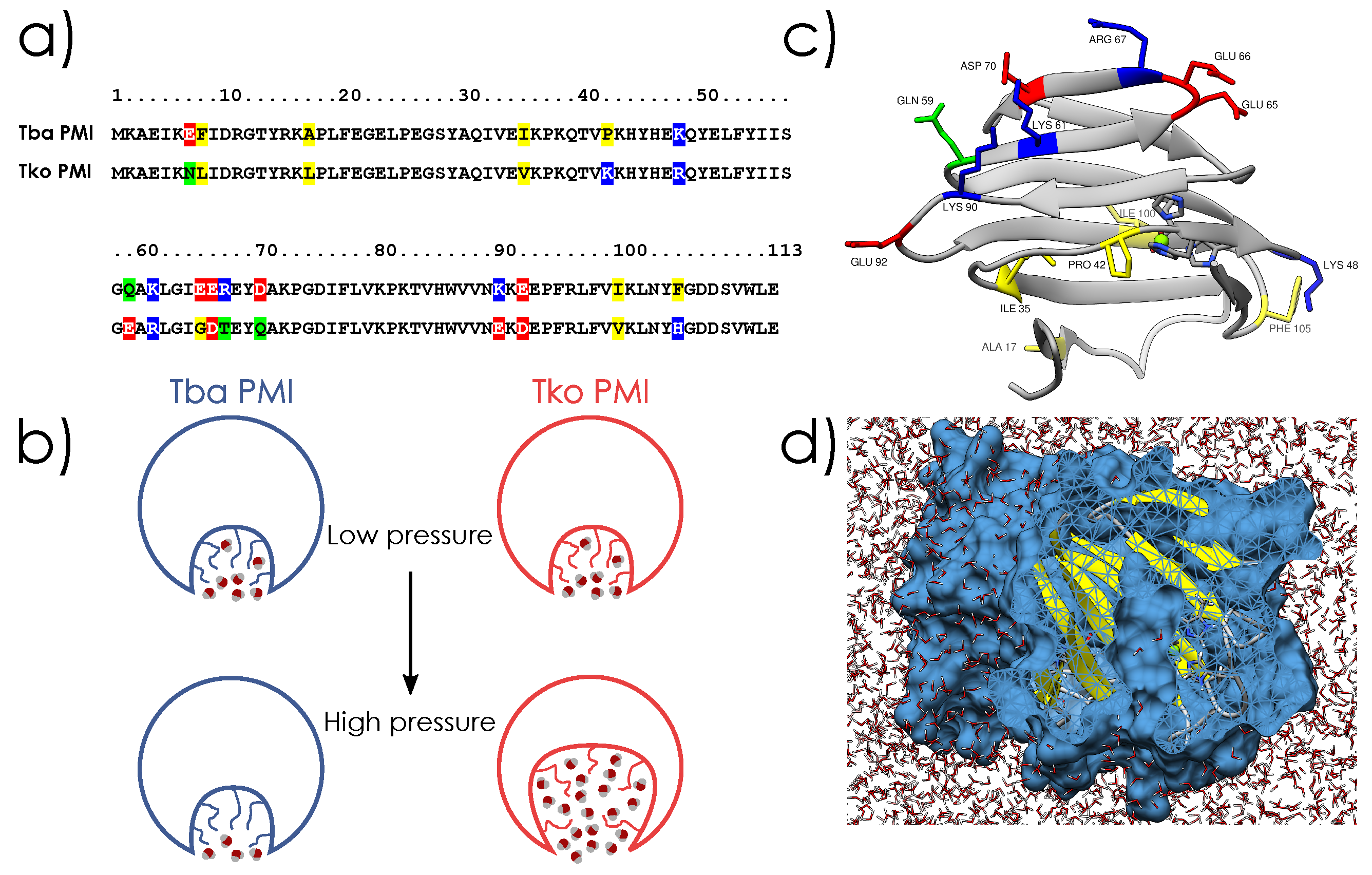
2.1. Crystal Structures and Molecular Dynamics Simulations
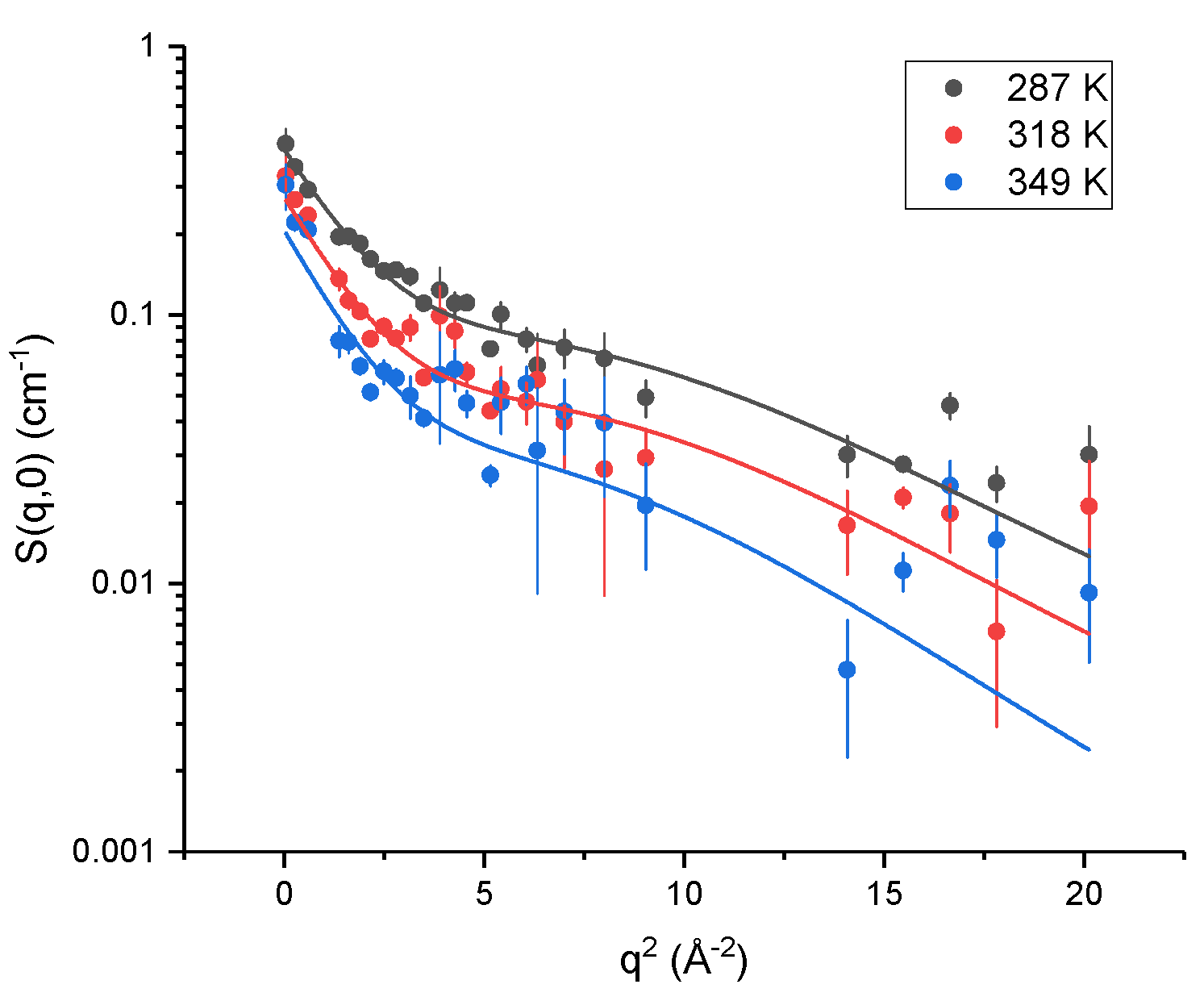
2.2. Elastic Incoherent Neutron Scattering (EINS)
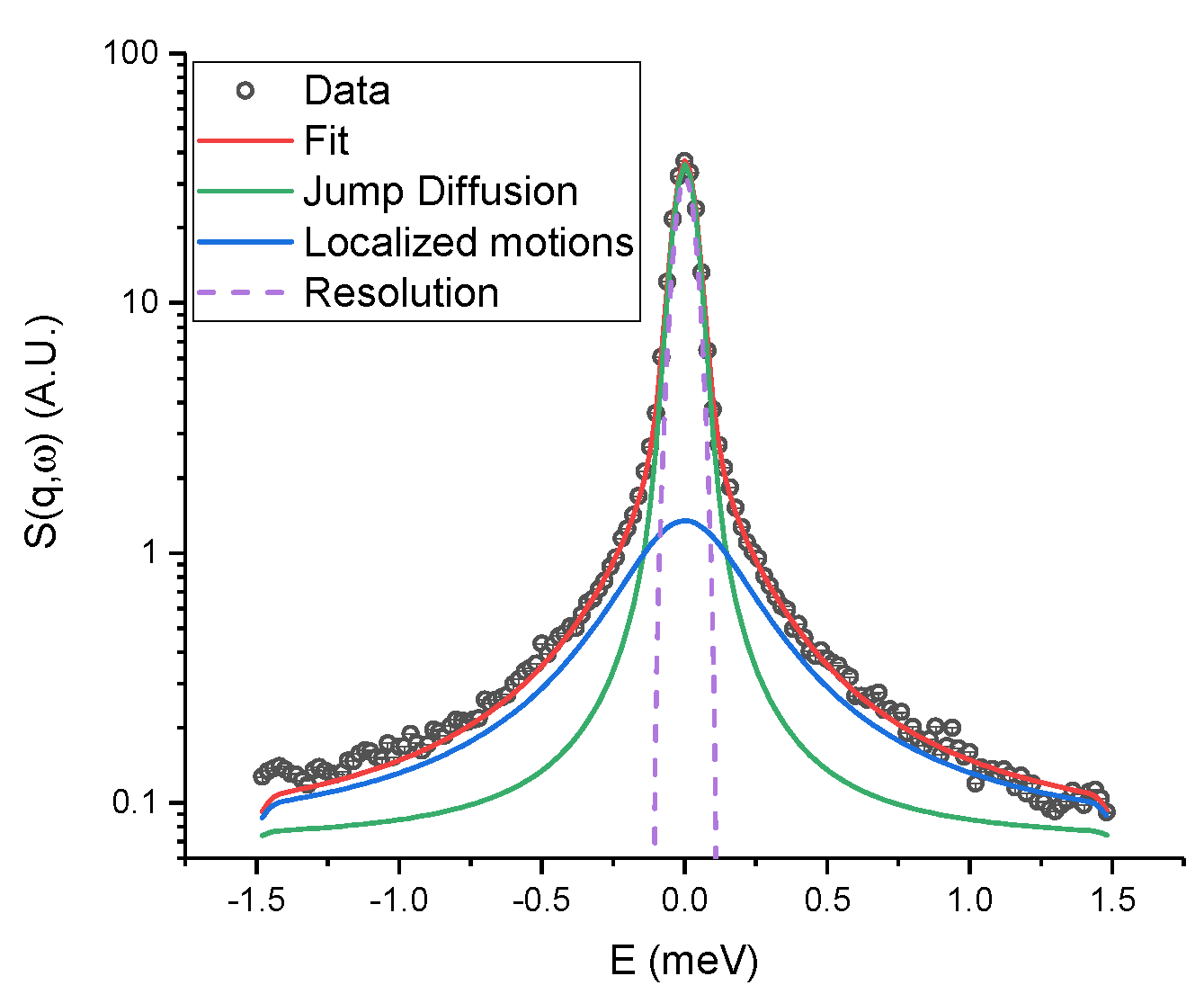
2.3. Quasi-Elastic Neutron Scattering (QENS)
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. X-ray Crystallography
4.3. Elastic Incoherent Neutron Scattering (EINS)
4.4. Quasi-Elastic Neutron Scattering (QENS)
4.5. Molecular Dynamics Simulations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, S.L.; Bada, J.L. Submarine hot springs and the origin of life. Nature 1988, 334, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Martin, W.; Baross, J.; Kelley, D.; Russell, M.J. Hydrothermal vents and the origin of life. Nat. Rev. Microbiol. 2008, 6, 805–814. [Google Scholar] [CrossRef]
- Brininger, C.; Spradlin, S.; Cobani, L.; Evilia, C. The more adaptive to change, the more likely you are to survive: Protein adaptation in extremophiles. Semin. Cell Dev. Biol. 2018, 84, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Price, P.B. A habitat for psychrophiles in deep Antarctic ice. Proc. Natl. Acad. Sci. USA 2000, 97, 1247–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auerbach, G.; Ostendorp, R.; Prade, L.; Korndörfer, I.; Dams, T.; Huber, R.; Jaenicke, R. Lactate dehydrogenase from the hyperthermophilic bacterium Thermotoga maritima: The crystal structure at 2.1 å resolution reveals strategies for intrinsic protein stabilization. Structure 1998, 6, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Martinez, N.; Michoud, G.; Cario, A.; Olliver, J.; Franzetti, B.; Jebbar, M.; Oger, P.; Peters, J. High protein flexibility and reduced hydration water dynamics are key pressure adaptive strategies in prokaryotes. Sci. Rep. 2016, 6, 32816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, J.; Martinez, N.; Michoud, G.; Cario, A.; Franzetti, B.; Oger, P.; Jebbar, M. Deep Sea Microbes Probed by Incoherent Neutron Scattering Under High Hydrostatic Pressure. Z. Phys. Chem. 2014, 228, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Golub, M.; Martinez, N.; Michoud, G.; Ollivier, J.; Jebbar, M.; Oger, P.; Peters, J. The Effect of Crowding on Protein Stability, Rigidity, and High Pressure Sensitivity in Whole Cells. Langmuir 2018, 34, 10419–10425. [Google Scholar] [CrossRef] [PubMed]
- Salvador-Castell, M.; Golub, M.; Martinez, N.; Ollivier, J.; Peters, J.; Oger, P. The first study on the impact of osmolytes in whole cells of high temperature-adapted microorganisms. Soft Matter 2019, 15, 8381–8391. [Google Scholar] [CrossRef] [PubMed]
- Cario, A.; Jebbar, M.; Thiel, A.; Kervarec, N.; Oger, P.M. Molecular chaperone accumulation as a function of stress evidences adaptation to high hydrostatic pressure in the piezophilic archaeon Thermococcus barophilus. Sci. Rep. 2016, 6, 29483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinar, H.; Fetahaj, Z.; Cinar, S.; Vernon, R.M.; Chan, H.S.; Winter, R.H.A. Temperature, Hydrostatic Pressure, and Osmolyte Effects on Liquid–Liquid Phase Separation in Protein Condensates: Physical Chemistry and Biological Implications. Chem. Eur. J. 2019, 25, 13049–13069. [Google Scholar] [CrossRef]
- Roche, J.; Caro, J.A.; Norberto, D.R.; Barthe, P.; Roumestand, C.; Schlessman, J.L.; Garcia, A.E.; García-Moreno E., B.; Royer, C.A. Cavities determine the pressure unfolding of proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 6945–6950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, S.A. Reversible pressure-temperature denaturation of chymotrypsinogen. Biochemistry 1971, 10, 2436–2442. [Google Scholar] [CrossRef] [PubMed]
- Royer, C.A. Revisiting volume changes in pressure-induced protein unfolding. Biochim. Biophys. Acta (BBA)—Protein Struct. Mol. Enzymol. 2002, 1595, 201–209. [Google Scholar] [CrossRef]
- Frauenfelder, H.; Alberding, N.A.; Ansari, A.; Braunstein, D.; Cowen, B.R.; Hong, M.K.; Iben, I.E.T.; Johnson, J.B.; Luck, S.; Marden, M.C.; et al. Proteins and pressure. J. Phys. Chem. 1990, 94, 1024–1037. [Google Scholar] [CrossRef]
- Ortore, M.G.; Spinozzi, F.; Mariani, P.; Paciaroni, A.; Barbosa, L.R.S.; Amenitsch, H.; Steinhart, M.; Ollivier, J.; Russo, D. Combining structure and dynamics: Non-denaturing high-pressure effect on lysozyme in solution. J. R. Soc. Interface 2009, 6, S619–S634. [Google Scholar] [CrossRef] [Green Version]
- Erlkamp, M.; Marion, J.; Martinez, N.; Czeslik, C.; Peters, J.; Winter, R. Influence of Pressure and Crowding on the Sub-Nanosecond Dynamics of Globular Proteins. J. Phys. Chem. B 2015, 119, 4842–4848. [Google Scholar] [CrossRef] [PubMed]
- Marion, J.; Trovaslet, M.; Martinez, N.; Masson, P.; Schweins, R.; Nachon, F.; Trapp, M.; Peters, J. Pressure-induced molten globule state of human acetylcholinesterase: Structural and dynamical changes monitored by neutron scattering. Phys. Chem. Chem. Phys. 2015, 17, 3157–3163. [Google Scholar] [CrossRef] [PubMed]
- Meinhold, L.; Smith, J.C.; Kitao, A.; Zewail, A.H. Picosecond fluctuating protein energy landscape mapped by pressure–temperature molecular dynamics simulation. Proc. Natl. Acad. Sci. USA 2007, 104, 17261–17265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, U.R.; Bhowmik, D.; Copley, J.R.D.; Tyagi, M.; Leão, J.B.; Chu, X.Q. Effects of pressure on the dynamics of an oligomeric protein from deep-sea hyperthermophile. Proc. Natl. Acad. Sci. USA 2015, 112, 13886–13891. [Google Scholar] [CrossRef] [Green Version]
- Librizzi, F.; Carrotta, R.; Peters, J.; Cupane, A. The effects of pressure on the energy landscape of proteins. Sci. Rep. 2018, 8, 2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharakoz, D.P. Protein Compressibility, Dynamics, and Pressure. Biophys. J. 2000, 79, 511–525. [Google Scholar] [CrossRef] [Green Version]
- Grigera, J.R.; McCarthy, A.N. The Behavior of the Hydrophobic Effect under Pressure and Protein Denaturation. Biophys. J. 2010, 98, 1626–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studier, F.W.; Moffatt, B.A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1986, 189, 113–130. [Google Scholar] [CrossRef]
- Tabor, S.; Richardson, C.C. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. USA 1985, 82, 1074–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frauenfelder, H.; Parak, F.; Young, R.D. Conformational Substates in Proteins. Annu. Rev. Biophys. Biophys. Chem. 1988, 17, 451–479. [Google Scholar] [CrossRef]
- Frauenfelder, H.; Young, R.D.; Fenimore, P.W. Dynamics and the Free-Energy Landscape of Proteins, Explored with the Mössbauer Effect and Quasi-Elastic Neutron Scattering. J. Phys. Chem. B 2013, 117, 13301–13307. [Google Scholar] [CrossRef] [PubMed]
- Bée, M. Quasielastic Neutron Scattering: Principles and Applications in Solid State, Chemistry, Biology and Materials Science; Adam Hilger: London, UK, 1988. [Google Scholar]
- Sears, V.F. Neutron scattering lengths and cross sections. Neutron News 1992, 3, 26–37. [Google Scholar] [CrossRef]
- Roh, J.H.; Novikov, V.N.; Gregory, R.B.; Curtis, J.E.; Chowdhuri, Z.; Sokolov, A.P. Onsets of Anharmonicity in Protein Dynamics. Phys. Rev. Lett. 2005, 95, 038101. [Google Scholar] [CrossRef]
- Doster, W.; Cusack, S.; Petry, W. Dynamical transition of myoglobin revealed by inelastic neutron scattering. Nature 1989, 337, 754–756. [Google Scholar] [CrossRef]
- Zaccai, G. How Soft Is a Protein? A Protein Dynamics Force Constant Measured by Neutron Scattering. Science 2000, 288, 1604–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bicout, D.J.; Zaccai, G. Protein Flexibility from the Dynamical Transition: A Force Constant Analysis. Biophys. D 2001, 80, 1115–1123. [Google Scholar]
- Peters, J. High Hydrostatic Pressure—A Key Element to Investigate Molecular Dynamics in Biosystems. Front. Phys. 2022, 9, 801539. [Google Scholar] [CrossRef]
- Marteinsson, V.; Birrien, J.; Reysenbach, A.; Vernet, M.; Marie, D.; Gambacorta, A.; Messner, P.; Sleytr, U.; Prieur, D. Thermococcus barophilus sp. nov., a new barophilic and hyperthermophilic archaeon isolated under high hydrostatic pressure from a deep-sea hydrothermal vent. Int. J. Syst. Evol. Microbiol. 1999, 49, 351–359. [Google Scholar] [CrossRef]
- Le Châtelier, H. Sur un Énoncé gènèral des lois des Équilibres chimiques. C. R. Acad. Sci. 1884, 99, 786–789. [Google Scholar]
- Xue, Y.; Pavlova, M.S.; Ryabov, Y.E.; Reif, B.; Skrynnikov, N.R. Methyl Rotation Barriers in Proteins from 2H Relaxation Data. Implications for Protein Structure. J. Am. Chem. Soc. 2007, 129, 6827–6838. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.L.; Ross, D. Incoherent neutron scattering functions for random jump diffusion in bounded and infinite media. Mol. Phys. 1981, 42, 673–682. [Google Scholar] [CrossRef]
- García-Colín, L.S.; del Castillo, L.F.; Goldstein, P. Theoretical basis for the Vogel-Fulcher-Tammann equation. Phys. Rev. B 1989, 40, 7040–7044. [Google Scholar] [CrossRef]
- Rault, J. Origin of the Vogel-Fulcher-Tammann law in glass-forming materials: The α-β bifurcation. J. Non-Cryst. Solids 2000, 271, 177–217. [Google Scholar] [CrossRef]
- Granato, A. A derivation of the Vogel-Fulcher-Tammann relation for supercooled liquids. J. Non-Cryst. Solids 2011, 357, 334–338. [Google Scholar] [CrossRef]
- Fomina, M.; Schirò, G.; Cupane, A. Hydration dependence of myoglobin dynamics studied with elastic neutron scattering, differential scanning calorimetry and broadband dielectric spectroscopy. Biophys. Chem. 2014, 185, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Mallamace, F.; Chen, S.H.; Broccio, M.; Corsaro, C.; Crupi, V.; Majolino, D.; Venuti, V.; Baglioni, P.; Fratini, E.; Vannucci, C.; et al. Role of the solvent in the dynamical transitions of proteins: The case of the lysozyme-water system. J. Chem. Phys. 2007, 127, 045104. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Liu, L.; Fratini, E.; Baglioni, P.; Faraone, A.; Mamontov, E. Observation of fragile-to-strong dynamic crossover in protein hydration water. Proc. Natl. Acad. Sci. USA 2006, 103, 9012–9016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagi, M.; Chu, X.; Kim, C.; Mallamace, F.; Baglioni, P.; Chen, S.H. The Low-Temperature Dynamic Crossover Phenomenon in Protein Hydration Water: Simulations vs Experiments. J. Phys. Chem. B 2008, 112, 1571–1575. [Google Scholar] [CrossRef] [PubMed]
- Vilgis, T.A. Strong and fragile glasses: A powerful classification and its consequences. Phys. Rev. B 1993, 47, 2882–2885. [Google Scholar] [CrossRef]
- Bowler, M.W.; Nurizzo, D.; Barrett, R.; Beteva, A.; Bodin, M.; Caserotto, H.; Delagenière, S.; Dobias, F.; Flot, D.; Giraud, T.; et al. MASSIF-1: A beamline dedicated to the fully automatic characterization and data collection from crystals of biological macromolecules. J. Synchrotron Radiat. 2015, 22, 1540–1547. [Google Scholar] [CrossRef]
- Svensson, O.; Gilski, M.; Nurizzo, D.; Bowler, M.W. Multi-position data collection and dynamic beam sizing: Recent improvements to the automatic data-collection algorithms on MASSIF-1. Acta Crystallogr. Sect. D 2018, 74, 433–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D 2019, 75, 861–877. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D 2012, 68, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Vagin, A.A.; Steiner, R.A.; Lebedev, A.A.; Potterton, L.; McNicholas, S.; Long, F.; Murshudov, G.N. REFMAC5 dictionary: Organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. Sect. D 2004, 60, 2184–2195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natali, F.; Peters, J.; Russo, D.; Barbieri, S.; Chiapponi, C.; Cupane, A.; Deriu, A.; Bari, M.T.D.; Farhi, E.; Gerelli, Y.; et al. IN13 Backscattering Spectrometer at ILL: Looking for Motions in Biological Macromolecules and Organisms. Neutron News 2008, 19, 14–18. [Google Scholar]
- Lelièvre-Berna, E.; Demé, B.; Gonthier, J.; Gonzales, J.P.; Maurice, J.; Memphis, Y.; Payre, C.; Oger, P.; Peters, J.; Vial, S. 700 MPa sample stick for studying liquid samples or solid-gas reactions down to 1.8 K and up to 550 K. J. Neutron Res. 2017, 19, 77–84. [Google Scholar] [CrossRef]
- Peters, J.; Trapp, M.; Hughes, D.; Rowe, S.; Demé, B.; Laborier, J.L.; Payre, C.; Gonzales, J.P.; Baudoin, S.; Belkhier, N.; et al. High hydrostatic pressure equipment for neutron scattering studies of samples in solutions. High Press. Res. 2012, 32, 97–102. [Google Scholar] [CrossRef]
- Sidorov, V.A.; Sadykov, R.A. Hydrostatic limits of Fluorinert liquids used for neutron and transport studies at high pressure. J. Phys. Condens. Matter 2005, 17, S3005–S3008. [Google Scholar] [CrossRef]
- Richard, D.; Ferrand, M.; Kearley, G.J. Analysis and visualisation of neutron-scattering data. J. Neutron Res. 1996, 4, 33–39. [Google Scholar] [CrossRef]
- Ollivier, J.; Mutka, H.; Didier, L. The New Cold Neutron Time-of-Flight Spectrometer IN5. Neutron News 2010, 21, 22–25. [Google Scholar] [CrossRef]
- Van Hove, L. Correlations in Space and Time and Born Approximation Scattering in Systems of Interacting Particles. Phys. Rev. 1954, 95, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Grimaldo, M.; Roosen-Runge, F.; Hennig, M.; Zanini, F.; Zhang, F.; Jalarvo, N.; Zamponi, M.; Schreiber, F.; Seydel, T. Hierarchical molecular dynamics of bovine serum albumin in concentrated aqueous solution below and above thermal denaturation. Phys. Chem. Chem. Phys. 2015, 17, 4645–4655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singwi, K.S.; Sjölander, A. Diffusive Motions in Water and Cold Neutron Scattering. Phys. Rev. 1960, 119, 863–871. [Google Scholar] [CrossRef]
- Chudley, C.T.; Elliott, R.J. Neutron Scattering from a Liquid on a Jump Diffusion Model. Proc. Phys. Soc. 1961, 77, 353–361. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannier, P.; Marteinsson, V.T.; Fridjonsson, O.H.; Oger, P.; Jebbar, M. Complete genome sequence of the hyperthermophilic, piezophilic, heterotrophic, and carboxydotrophic archaeon thermococcus barophilus mp. J. Bacteriol. 2011, 193, 1481–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, T.; Atomi, H.; Kanai, T.; Matsumi, R.; Fujiwara, S.; Imanaka, T. Complete genome sequence of the hyperthermophilic archaeon thermococcus kodakaraensis kod1 and comparison with pyrococcus genomes. Genome Res. 2005, 15, 352–363. [Google Scholar] [CrossRef] [Green Version]
- Svensson, O.; Malbet-Monaco, S.; Popov, A.; Nurizzo, D.; Bowler, M.W. Fully automatic characterization and data collection from crystals of biological macromolecules. Acta Crystallogr. Sect. D 2015, 71, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Svensson, O.; Gilski, M.; Nurizzo, D.; Bowler, M.W. A comparative anatomy of protein crystals: Lessons from the automatic processing of 56000 samples. IUCrJ 2019, 6, 822–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowler, M.W.; Svensson, O.; Nurizzo, D. Fully automatic macromolecular crystallography: The impact of massif1 on the optimum acquisition and quality of data. Crystallogr. Rev. 2016, 22, 233–249. [Google Scholar] [CrossRef] [Green Version]
- Nurizzo, D.; Bowler, M.W.; Caserotto, H.; Dobias, F.; Giraud, T.; Surr, J.; Guichard, N.; Papp, G.; Guijarro, M.; Mueller-Dieckmann, C.; et al. RoboDiff: Combining a sample changer and goniometer for highly automated macromolecular crystallography experiments. Acta Crystallogr. Sect. D 2016, 72, 966–975. [Google Scholar] [CrossRef]
- Hutin, S.; Van Laer, B.; Mueller-Dieckmann, C.; Leonard, G.; Nurizzo, D.; Bowler, M.W. Fully autonomous characterization and data collection from crystals of biological macromolecules. JoVE 2019, 145, e59032. [Google Scholar] [CrossRef] [PubMed]
- Ortega, A.; Amorós, D.; García de la Torre, J. Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 2011, 101, 892–898. [Google Scholar] [CrossRef] [Green Version]
- Millero, F.J.; Dexter, R.; Hoff, E. Density and viscosity of deuterium oxide solutions from 5–70.deg. J. Chem. Eng. Data 1971, 16, 85–87. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caliò, A.; Dubois, C.; Fontanay, S.; Koza, M.M.; Hoh, F.; Roumestand, C.; Oger, P.; Peters, J. Unravelling the Adaptation Mechanisms to High Pressure in Proteins. Int. J. Mol. Sci. 2022, 23, 8469. https://doi.org/10.3390/ijms23158469
Caliò A, Dubois C, Fontanay S, Koza MM, Hoh F, Roumestand C, Oger P, Peters J. Unravelling the Adaptation Mechanisms to High Pressure in Proteins. International Journal of Molecular Sciences. 2022; 23(15):8469. https://doi.org/10.3390/ijms23158469
Chicago/Turabian StyleCaliò, Antonino, Cécile Dubois, Stéphane Fontanay, Michael Marek Koza, François Hoh, Christian Roumestand, Philippe Oger, and Judith Peters. 2022. "Unravelling the Adaptation Mechanisms to High Pressure in Proteins" International Journal of Molecular Sciences 23, no. 15: 8469. https://doi.org/10.3390/ijms23158469
APA StyleCaliò, A., Dubois, C., Fontanay, S., Koza, M. M., Hoh, F., Roumestand, C., Oger, P., & Peters, J. (2022). Unravelling the Adaptation Mechanisms to High Pressure in Proteins. International Journal of Molecular Sciences, 23(15), 8469. https://doi.org/10.3390/ijms23158469