
Single-Circulating Tumor Cell Whole Genome Amplification to Unravel Cancer Heterogeneity and Actionable Biomarkers


 ,
,
Abstract
:1. Introduction
2. CTC, WGA and Single-Cell Analysis
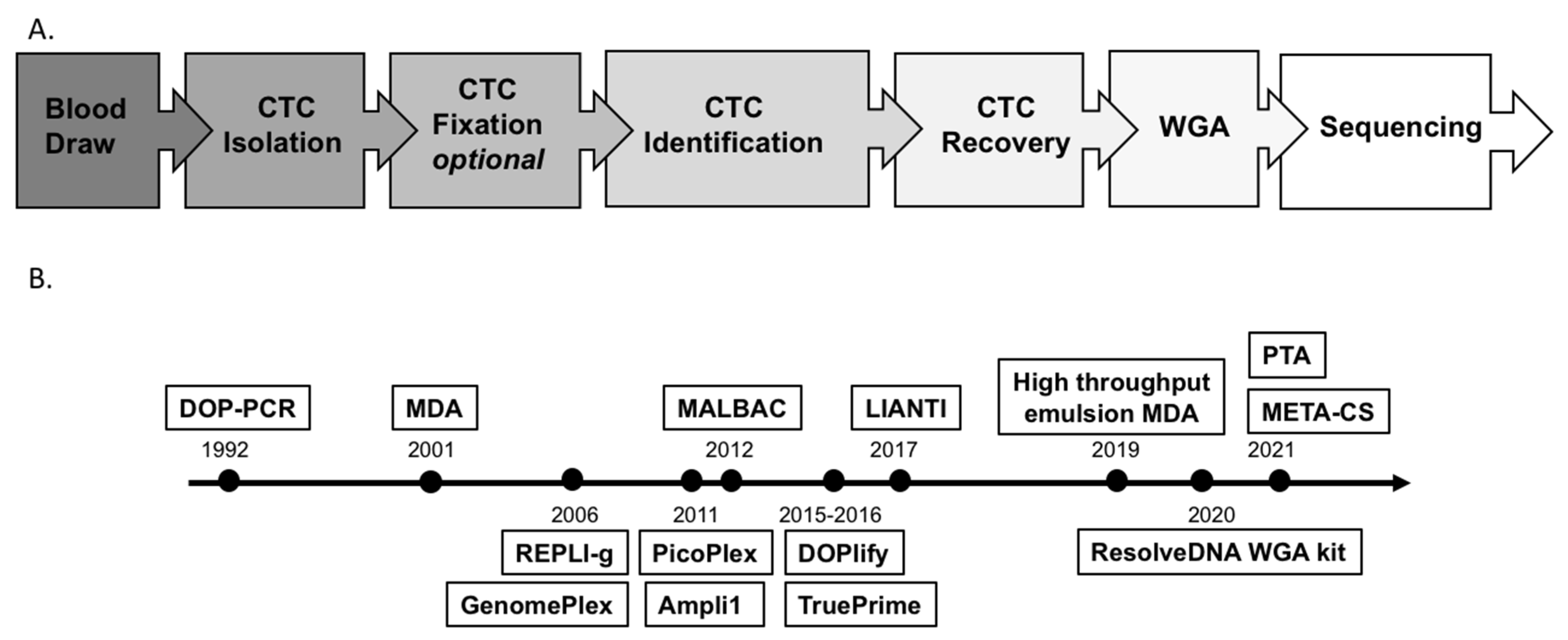
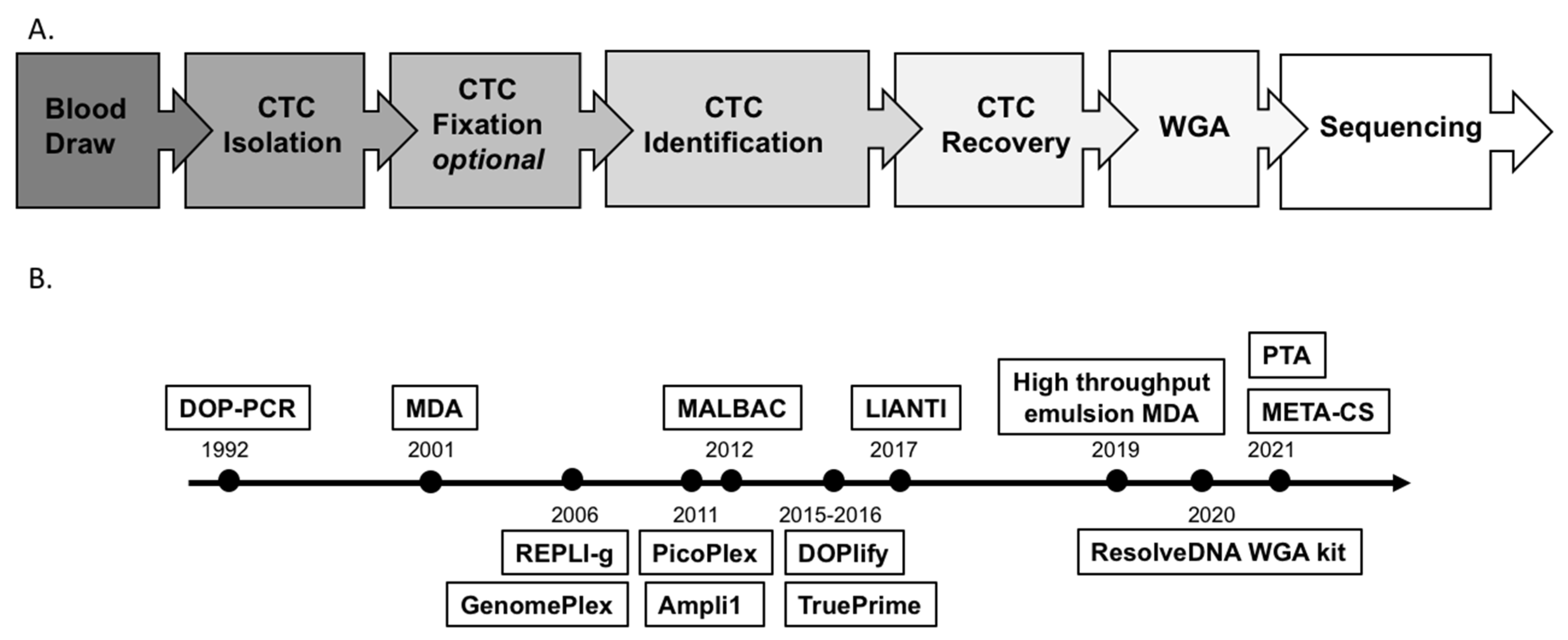
2.1. CTC Isolation, Identification and Recovery
2.2. Challenges and Technical Advances of WGA
2.3. Downstream Analysis of Single-Cell WGAs
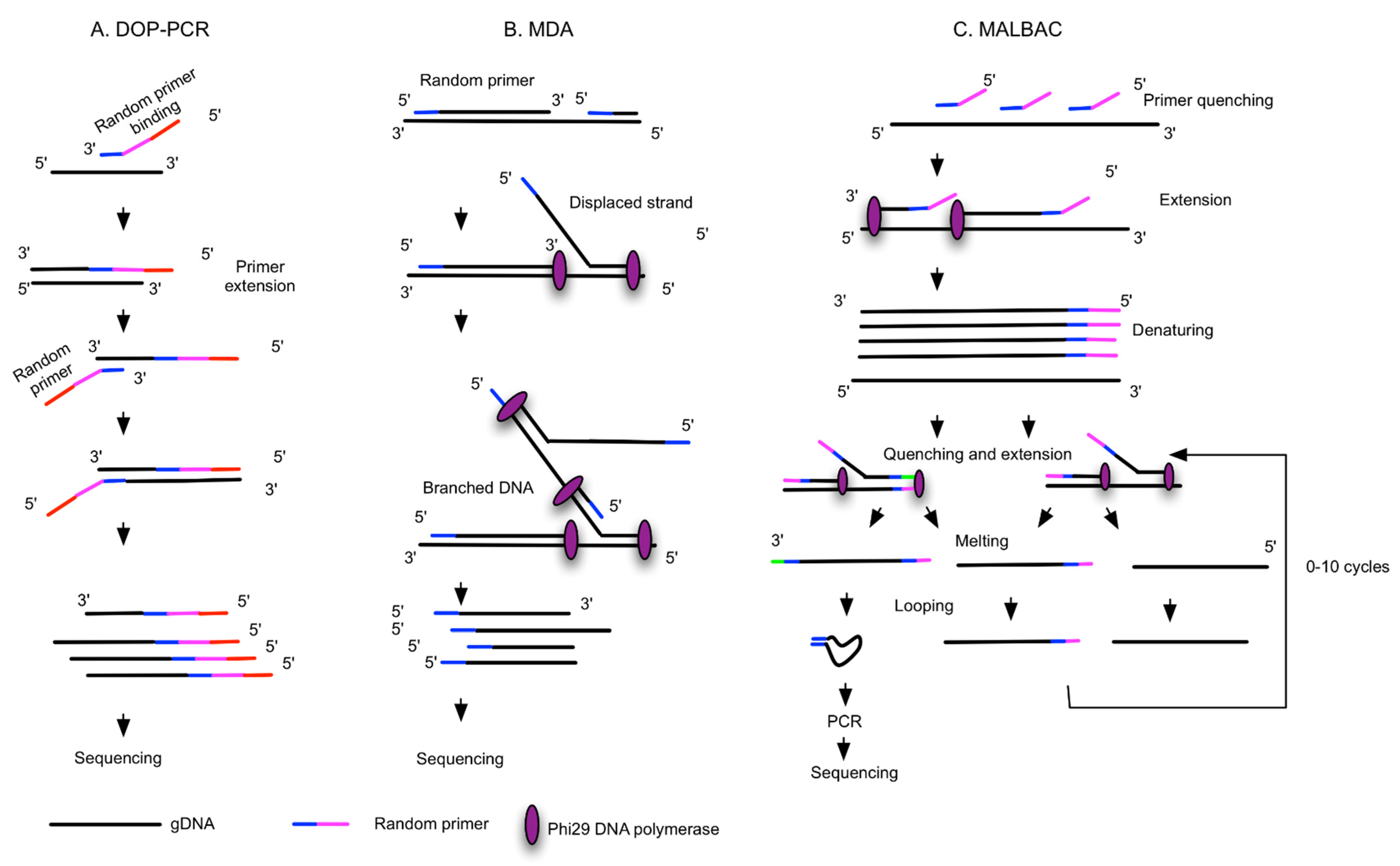
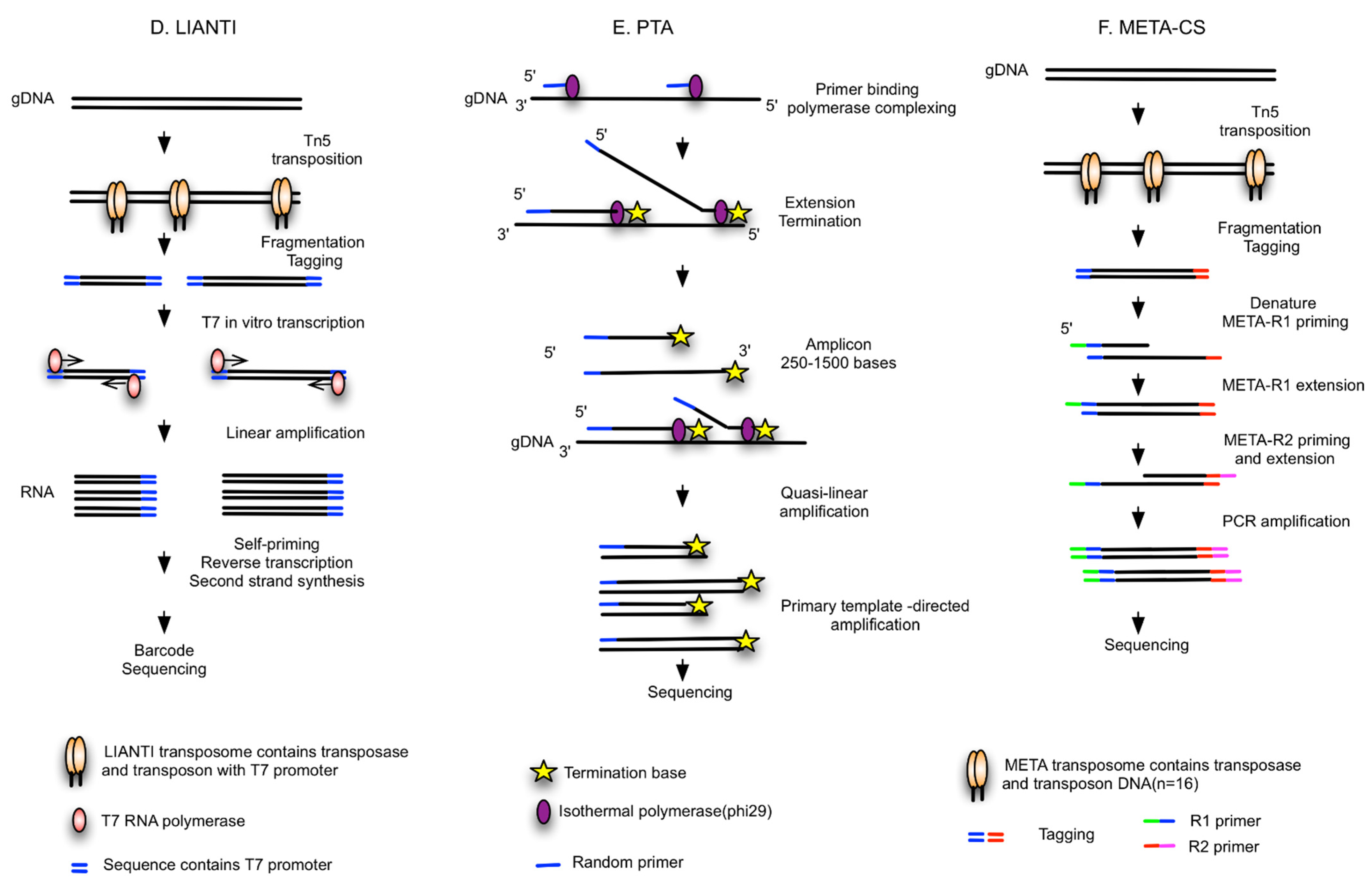

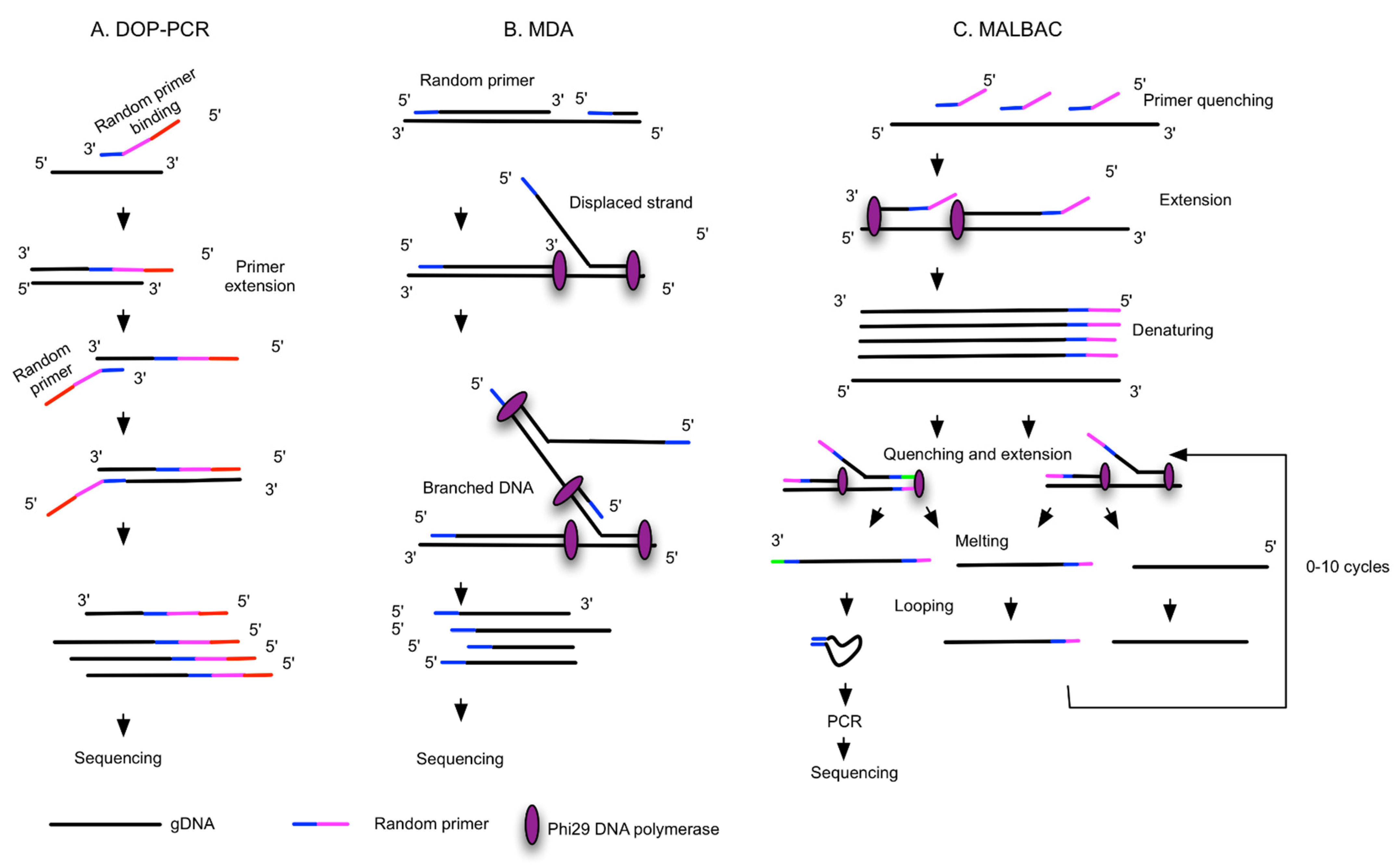
{kind=link}
{kind=link}
{kind=link}
| WGA Method | Principles and Polymerase | Commercial Kits | Technical Challenges | Advantages | Preferred Downstream Analysis |
|---|---|---|---|---|---|
| DOP-PCR [33,50] | Random priming and PCR amplification, Taq Polymerases | Sigma GenomePlex Single Cell WGA Kit, PerkinElmer DOPlify WGA kit | Low genome coverage (40–50%), better uniformity of amplification, high FP and FN, low success rate | Quick, no need of normalization | CNV, STR analysis |
| MDA and improved MDA [52,53] | Random priming and isothermal exponential amplification, Phi29 or Bst polymerases | Qiagen REPLI-g Single Cell Kit, GE GenomiPhi DNA Amplification Kit, AmpliQ Genomic Amplifier Kit, Sygnis TruePrime WGA kit | Less uniformity, artifact of C>T transitional mutation, non-reproducible from cell to cell, low chimera rate | More genome coverage (80%), low FP and FN, compatible with digital droplet MDA | Mutation detection, SNP |
| MALBAC [52,54,55,56,57] | Isothermal preamplification and PCR, Bst polymerase, deep vent (exo-) DNA Polymerase | Yikon Genomics Single Cell WGA Kit, Rubicon Genomics PicoPLEX WGA Kit, TakaRa PicoPLEX | Complicated procedure, intermediate coverage and uniformity, intermediate FP and FN | Reproducible from cell to cell, low ADO | CNV |
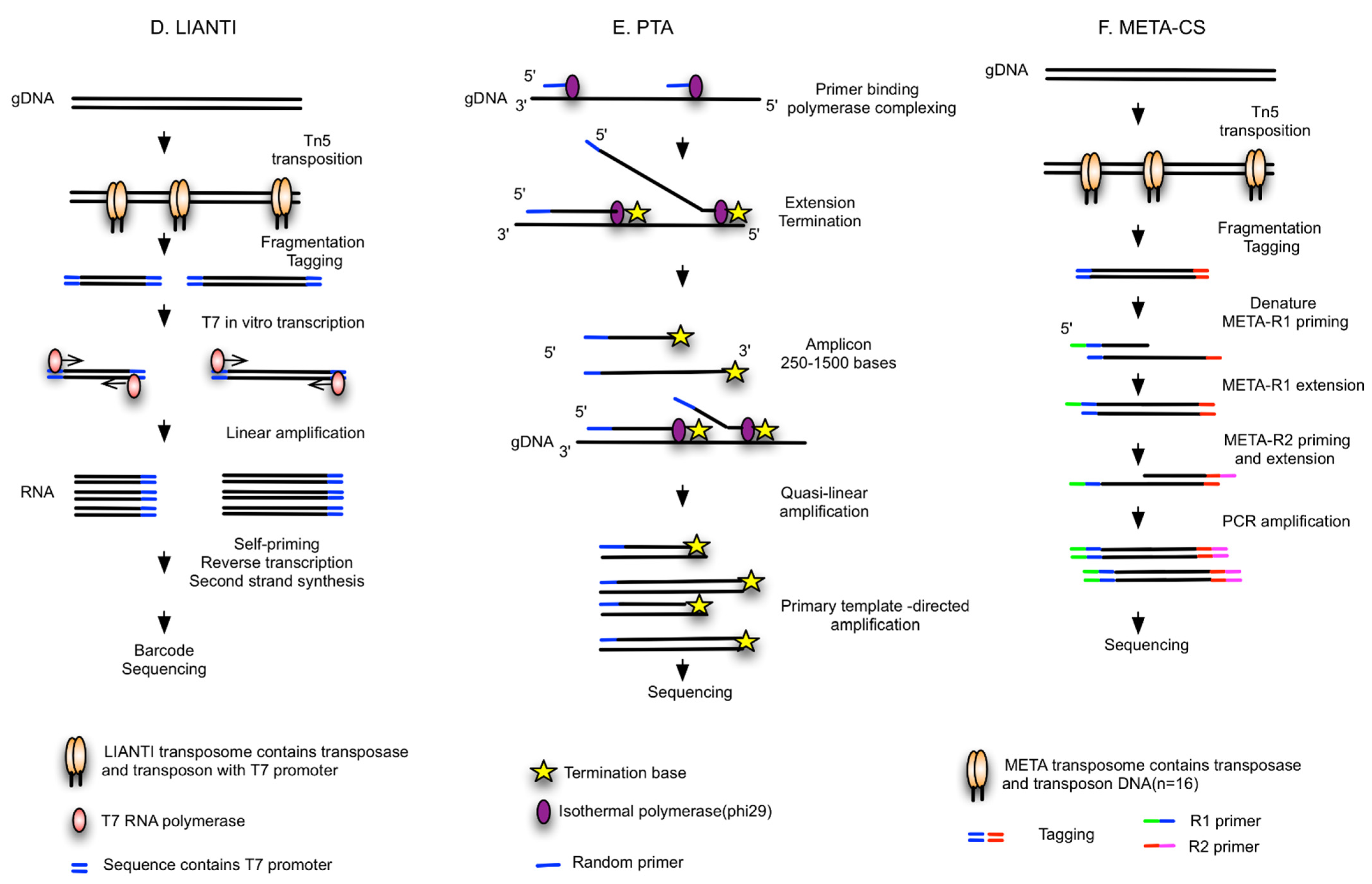
| LIANTI [39] | Random fragments tagged by T7 promoters, linear amplification of RNA, reverse transcription | NA | Needs further study | High genome coverage (97%) and low ADO (17%), low FP for SNV detection | SNV |
| PTA [40] | Isothermal WGA, quasi-linear process, Phi29 polymerase | BioSkryb ResolveDNA WGA kit | Needs further study | High coverage (95%), reproducible, high uniformity and accuracy, compatible with high-throughput reactions in microfluidic devices or emulsions | Improved capacity to call SNVs, CNVs and SVs; superior SNV sensitivity |
| META-CS [41] | Fragmented by Tn5 transposase, randomly tagged with transposon sequences, DNA pre-amplification | NA | Needs further study | High success rate (90%), single tube reaction to minimize loss, high amplification uniformity | SNVs, insertions, deletions, SVs |
3. Single-Cell Analysis of CTCs and Biomarker Detections
3.1. Breast Cancer
| Studies (Author, Year) | CTC Isolation | CTC Recovery | WGA Kits | Downstream Molecular Analysis | CTCs+ Patients Analyzed | CTC Nr Analyzed for WGA | Main Findings in Genetic Mutations and Alterations |
|---|---|---|---|---|---|---|---|
| mBC or HER2- mBC | |||||||
| Babayan, A. et al., 2013 [20] | Density gradient | Micromanipulator TransferMan NK2 | PicoPlex | Multiplex PCR | 4 | 8 single CTCs | ESR1 mutations in exons 4, 6 and 8 were not found |
| De Luca, F. et al., 2016 [68] | CellSearch | DEPArray | Ampli1 | NGS (Ion AmpliSeq Cancer Hotspot panel v2) | 4 | 3–5 single CTCs per patient | 51 sequence variants in 25 genes were found, including somatic mutations in TP53 (8 mutations) and PDGFRA (3 mutations). High intra- and inter-patient heterogeneity, discordance in mutational status between CTCs and primary tissue |
| Gasch, C. et al., 2016 [64] | CellSearch | Micromanipulator TransferMan NK2 | GenomiPhi, Ampli1 | Sanger sequencing, PCR | 33 | 114 single CTCs | PIK3CA mutations in exon 9 and 20 |
| Kaur, P. et al., 2020 [70] | Microfluidic ANGLE Parsortix | NA | REPLI-g | WES (SNVs, CNAs and SVs) | 5 | 5 CTCs and 5 WBCs | Elevated C>T mutational signature in patient samples. Low VAFs for somatic variants in CTCs compared to metastasis, complex rearrangement patterns were observed, high discordance between paired samples, marked heterogeneity of somatic landscape |
| Li, S. et al., 2020 [59] | CellCollector | CellCollector | REPLI-g | NGS (HiSeq X-Ten Illumina) | 17 | 0–15 CTCs | Different metastatic sites have their own corresponding high-frequency mutation genes |
| Neumann, M. H. et al., 2016 [65] | CellSearch | CellCelector | Ampli1 | For library preparation, the multiplex PCR-based Ion Torrent AmpliSeqTM technology with Ampli1 CHPCustom Beta panel | 2 | 7 single CTCs | Functional PIK3CA SNP (G to A, E545K) was detected in CTCs of patient 1 but not in CTCs of patient 2 |
| Neves, R. P. et al., 2014 [63] | CellSearch | FACS | Ampli1 | aCGH (CNAs), qPCR | 30 | 192 single CTCs | 72.9% WGA success rate, 46.2% of WGA products show CCND1 amplification, mutations in PIK3CA exon 20 in c.3140 were found in CTCs (2/12 analyzed patients), TP53 mutations in exons 5, 7 and 8 were not found |
| Paolillo, C. et al., 2017 [25] | CellSearch | DEPArray | MALBAC | Sanger sequencing | 3 | 40 single CTCs and 12 WBCs | ESR1 mutations (Y537S and T570I) were identified |
| Pestrin, M. et al., 2014 [26] | CellSearch | DEPArray | Ampli1 | Sanger sequencing (hotspot regions in PIK3CA exon 9, 20) | 18 | 115 single CTCs | 33% of patients had an identified PI3KCA mutation. Six different mutations in the PI3KCA gene, such as c.3140A>G, c.1633G>A, c.1624G>A, c.1624G>A, etc., were identified |
| Polzer, B. et al., 2014 [27] | CellSearch | DEPArray | Ampli1 | ERBB2 qPCR (CNV), PIK3CA Sequencing, aCGH | 66 | 510 single CTCs and 189 leukocytes | PIK3CA mutations in exon 9 and 20. Analysis of ERBB2 alterations |
| Schneck, H. et al., 2013 [44] | CellSearch | NA | Ampli1 | Multiplex PCR, SNaPshot | 44 | NA | PIK3CA mutations in exon 9 and 20, such as E545K and H1047R, were detected, but E542K, E545G and E545A were not found |
| Wang, Y. et al., 2018 [22] | FACS combined with oHSV1-hTERT-GFP viral infection | FACS | MALBAC | WGS for CTC, WGS and WES for matched primary and metastatic tissue | 8 | 11 single CTCs | SNVs accumulated sporadically among CTCs and matched primary tumors, at least 2 CTCs shared 394 SNVs, SNV mutations in APC and LRP1B genes co-occurred in CTC-shared and bulk tissue, CTC behaviour-related SNVs were verified |
| Zou, L. et al., 2020 [21] | CellSearch | Micropipetting | MALBAC | WGS (CNV and gene set enrichment analysis) | 2 | Single CTCs, but number is unknown | Different frequencies of CNVs between newly diagnosed and recurrent liver metastasis; similar CNV patterns among isolated CTCs of recurrent BCLM and recurrent liver metastasis; 25 genes were identified as CNV signatures of BCLM, including β-defensins and defensins |
| PC or mCRPC | |||||||
| Faugeroux, V. et al., 2018 [6] | ISET filtration, CellSearch, Rosettesep | Self-seeding microwell chips, FACS, laser microdissection | Ampli1 | WES (10x depth coverage) | 11 | 179 WGA samples or 34 WES | Shared GRM8, TP53 and PTEN mutations in epithelial CTC samples and other CTC-exclusive variants |
| Greene, S. B. et al., 2016 [71] | Epic Sciences | Eppendorf TransferMan NK4 micromanipulator | SeqPlex Enhanced | Sequencing with Illumina NextSeq500 using a High Output kit in a Paired-End 2x150 format (PE 2x150) (CNV) | 7 | 67 single CTCs | AR amplification and PTEN loss |
| Gupta, S. et al., 2016 [72] | CellSearch, RBC lysis and CD45 depletion | IE/FACS | RepliGene, WGA4 | aCGH (CNV) | 16 | 16 CTCs and matched leukocytes | AR amplification in 50% of CTC samples, ERG genomic amplification in 40% of patients, PTEN loss, genomic alteration in chromatin reading and proliferative pathways |
| Magbanua, M. J. et al., 2012 [73] | CellSearch, IE/FACS | IE/FACS | WGA4 | aCGH | 12 | 9 patient bulk CTCs | Gains in 8q and loss in 8p; gains in the AR region of chr X of CTCs, including AR gains in 78% of cases |
| Rangel-Pozzo, A. et al., 2020 [17] | ScreenCell filtration | Laser microdissection | Ampli1 | WES | 9 | 21 single CTCs and 4 lymphocytes | Genetic variations in nine telomere maintenance pathways, including telomeric repeat-binding factor 2 (TRF2), SNVs and indels associated with telomere maintenance genes and known cancer drug response; presence of CNAs in 11 different pathways, including the DNA damage repair (DDR) pathway |
| Wu, Y. et al., 2016 [4] | Density gradient, negative and positive selection with magnetic beads | Laser microdissection | PicoPLEX (<40 cells), WGA2 kit (GenomePlex for microdissected tissues) | SNP array profiling (CytoSNP-12 and omni1-Quad bead chips, NspI 250k, SNP6.0, and CytoScanHD arrays), Nanostring (nCounter Cancer CN panel) | 8 | 8 disseminated tumor cells (bulk cells) | Gain of Ch 7 and 8q, loss in 8p, 12q23, 10q26, 13q and 16q21. AR gain, TMPRSS2/ERG alterations and MYC and other gained regions, FOXO1 gene deletion |
| Lung Cancer | |||||||
| He, Y. et al., 2017 [74] | CellCollector | CellCollector | REPLI-g | NGS (hotspot panel v2) | 5 | 6 CTCs | 44 cancer-related genes existed in mutations in the analyzed CTCs and some cancer-related mutations were identified in KIT, SMARCB1 and TP53 genes |
| Lu, S. et al., 2020 [28] | CellSearch | DEPArray | MALBAC, REPLI-g, WGA4, Ampli1 | Targeted sequencing, WES, WGS | 4 | 80 single CTCs and 11 WBCs | Comparative study, MALBAC WGA coupled with LP-WGS is a robust workflow for CNV profiling, but none of the WGA methods achieve sufficient sensitivity and specificity by WES |
| Mariscal, J. et al., 2016 [75] | CELLection Epithelial Enrich Dynabeads | NA | WTA2 | Gene expression profiling (Agilent 4x44k gene expression arrays), qPCR | 42 NSCLC patients and 16 controls | NA | CTC-specific expression profile associates with the PI3K/AKT, ERK1/2 and NF-kB pathways. NOTCH1, PTP4A3, LGALS3 and ITGB3 were further validated by RT-qPCR in an independent cohort of NSCLC patients |
| Nakamura, I. T. et al., 2021 [13] | AutoMACS | DEPArray | SMARTer PicoPLEX | NGS (Todai OncoPanel, AmpliSeq for Illumina comprehensive cancer panel, WGS) and Sanger sequencing | 2 | 40 single floating tumor cells in pleural effusion | EGFR exon 19 deletion was confirmed in 63.2% of samples from case 1, detection of 85% EML4-ALK fusion in case 2, alectinib- resistant mutation of ALK (p.G1202R) in case 2. A BRCA1 truncating mutation and an RAF1 oncogenic mutation were identified |
| Ni, X. et al., 2013 [5] | CellSearch | Micropipetting | MALBAC | WGS at ∼0.1× sequencing depth and WES for SNV/indel | 11 | 72 single CTCs (including 4 leucocytes) | EGFR mutations (such as one INDEL p.K746_A750del), PIK3CA (such as p.E545K), RB1 (p.R320*) and TP53 mutations (such as p.T155I) were only shared between the liver metastatic tumor and CTCs; gain region in chromosome 8q contains the c-Myc gene; gain in chromosome 5p, which contains the telomerase reverse transcriptase (TERT) gene; chromosomal regions, including 3q29, 17q22, 17q25.3 and 20p13, had significant gain in all 19 CTCs of patients |
| Colorectal Cancer | |||||||
| Fabbri, F. et al., 2013 [76] | OncoQuick | DEPArray | Ampli1 | Sequencing and pyrosequencing | 21 | 16 samples or cases | KRAS gene mutations in 50% of cases. G12C, G12D and G13D-KRAS mutations in one patient in three different groups of CTCs |
| Gasch, C. et al., 2013 [19] | CellSearch | Micromanipulator TransferMan NK2 | GenomePlex, GenomiPhi | Targeted sequencing for KRAS, BRAF and PIK3CA gene, qPCR for EGFR | 5 | 69 single CTCs | EGFR amplification in 7/26 CTCs, KRAS mutations (G12V) in 33% of CTCs, PIK3CA mutations (E545A and E542K) in 39% of CTCs, no BRAF locus change detected |
| Li, R. et al., 2019 [42] | Microfluidic chip (SCIGA-chip) | Microfluidic chip (SCIGA-chip) | MDA | Illumina sequencing (SNPs/SVs) | 1 | 2 single CTCs and 1 WBC | A novel method involving all processing steps from blood collection to WGA preparation, 11 shared somatic mutations (e.g., C18orf25, GFM2, DDX60L, etc.) and 153 structure variations were identified |
| Pancreatic Cancer | |||||||
| Court, C.M et al., 2016 [77] | Density gradient and NanoVelcro/LCM microchip | Laser microdissection | REPLI-g | Sanger sequencing | 12 | 119 single CTCs and 103 WBCs | KRAS mutations in 92% of patients and 33 out of 119 single CTCs sequenced (resulting in a 27.7% detection rate in single CTCs). No KRAS mutants were found in any WBCs |
| Melanoma | |||||||
| Reid, A. L. et al., 2014 [78] | RBC lysis, immune-magnetic beads | NA | REPLI-g | ddPCR and castPCR | 15 | 30 CTCs | Comparative study of ddPCR and castPCR. BRAF-V600E/K mutations were detected |
| Ruiz, C. et al., 2016 [79] | RBC lysis | Micromanipulator | GenomePlex | CNV analysis | 40 | Single CTCs and WBCs | Deletions of CDKN2A and PTEN; amplifications of BRAF, TERT, MDM2 and KRAS; chromosomal amplifications in chr12, 17 and 19 |
| Mixed patient cohort | |||||||
| Aljohani, H.M. et al., 2018 [23] | RBC lysis, CD45 depletion and EpCam positive selection | FACS | REPLI-g | Sanger sequencing, ddPCR | 10 | NA | Mutations (R34G, E79Q, E82G) in Nrf2 in isolated CTCs, some mutations in the Keap/Nrf2/ARE pathway |
| Ferrarini, A. et al., 2018 [80] | CellSearch | DEPArray | Ampli1 | WGS (CNAs), aCGH | 3 | 15 single CTCs and 7 WBCs | A large amplification (100 Mbp) on chr 8, including the c-MYC gene, copy number loss was detected in the BRCA2 locus |
| Gao, Y. et al., 2017 [81] | CellSearch | Micropipetting | MALBAC | WGS and WES for SNV/indels, SVs, CNs | 23 | 97 single CTCs | Homozygous deletion of PTEN; amplification of the MYC gene; 11 focal regions were identified, including well-known tumor suppressor genes or oncogenes, which were deleted or amplified |
3.2. Prostate Cancer
3.3. Lung Cancer
3.4. Colorectal Cancer
3.5. Other Cancer Types
4. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gerlinger, M.; McGranahan, N.; Dewhurst, S.M.; Burrell, R.A.; Tomlinson, I.; Swanton, C. Cancer: Evolution within a Lifetime. Annu. Rev. Genet. 2014, 48, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wei, S.; Deng, Y.; Wang, Z.; Liu, L. Clinical significance of tumour mutation burden in immunotherapy across multiple cancer types: An individual meta-analysis. Jpn. J. Clin. Oncol. 2020, 50, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Wu, Y.; Schoenborn, J.R.; Morrissey, C.; Xia, J.; Larson, S.; Brown, L.G.; Qu, X.; Lange, P.H.; Nelson, P.S.; Vessella, R.L.; et al. High-Resolution Genomic Profiling of Disseminated Tumor Cells in Prostate Cancer. J. Mol. Diagn. 2016, 18, 131–143. [Google Scholar] [CrossRef]
- Ni, X.; Zhuo, M.; Su, Z.; Duan, J.; Gao, Y.; Wang, Z.; Zong, C.; Bai, H.; Chapman, A.R.; Zhao, J.; et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc. Natl. Acad. Sci. USA 2013, 110, 21083–21088. [Google Scholar] [CrossRef] [Green Version]
- Faugeroux, V.; Lefebvre, C.; Pailler, E.; Pierron, V.; Marcaillou, C.; Tourlet, S.; Billiot, F.; Dogan, S.; Oulhen, M.; Vielh, P.; et al. An Accessible and Unique Insight into Metastasis Mutational Content through Whole-Exome Sequencing of Circulating Tumor Cells in Metastatic Prostate Cancer. Eur. Urol. Oncol. 2020, 3, 498–508. [Google Scholar] [CrossRef] [Green Version]
- Gillooly, J.F.; Hein, A.; Damiani, R. Nuclear DNA content varies with cell size across human cell types. Cold Spring Harb. Perspect. Biol. 2015, 7, a019091. [Google Scholar] [CrossRef] [Green Version]
- Van Loo, P.; Voet, T. Single cell analysis of cancer genomes. Curr. Opin. Genet. Dev. 2014, 24, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Rushton, A.J.; Nteliopoulos, G.; Shaw, J.A.; Coombes, R.C. A Review of Circulating Tumour Cell Enrichment Technologies. Cancers 2021, 13, 970. [Google Scholar] [CrossRef]
- Williams, A.L.; Fitzgerald, J.E.; Ivich, F.; Sontag, E.D.; Niedre, M. Short-term circulating tumor cell dynamics in mouse xenograft models and implications for liquid biopsy. Front. Oncol. 2020, 10, 601085. [Google Scholar] [CrossRef] [PubMed]
- Swennenhuis, J.F.; van Dalum, G.; Zeune, L.L.; Terstappen, L.W. Improving the CellSearch® system. Expert Rev. Mol. Diagn. 2016, 16, 1291–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozar, T.; Jesenko, T.; Kloboves Prevodnik, V.; Cemazar, M.; Hosta, V.; Jericevic, A.; Nolde, N.; Grasic Kuhar, C. Preclinical and Clinical Evaluation of Magnetic-Activated Cell Separation Technology for CTC Isolation in Breast Cancer. Front. Oncol. 2020, 10, 554554. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.T.; Ikegami, M.; Hasegawa, N.; Hayashi, T.; Ueno, T.; Kawazu, M.; Yagishita, S.; Goto, Y.; Shinno, Y.; Kojima, Y.; et al. Development of an optimal protocol for molecular profiling of tumor cells in pleural effusions at single-cell level. Cancer Sci. 2021, 112, 2006–2019. [Google Scholar] [CrossRef] [PubMed]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Mao, X.; Imrali, A.; Syed, F.; Mutsvangwa, K.; Berney, D.; Cathcart, P.; Hines, J.; Shamash, J.; Lu, Y.J. Optimization and Evaluation of a Novel Size Based Circulating Tumor Cell Isolation System. PLoS ONE 2015, 10, e0138032. [Google Scholar] [CrossRef] [Green Version]
- Po, J.W.; Ma, Y.; Balakrishna, B.; Brungs, D.; Azimi, F.; de Souza, P.; Becker, T.M. Immunomagnetic isolation of circulating melanoma cells and detection of PD-L1 status. PLoS ONE 2019, 14, e0211866. [Google Scholar] [CrossRef] [PubMed]
- Rangel-Pozzo, A.; Liu, S.; Wajnberg, G.; Wang, X.; Ouellette, R.J.; Hicks, G.G.; Drachenberg, D.; Mai, S. Genomic Analysis of Localized High-Risk Prostate Cancer Circulating Tumor Cells at the Single-Cell Level. Cells 2020, 9, 1863. [Google Scholar] [CrossRef]
- Po, J.W.; Ma, Y.; Balakrishnar, B.; Brungs, D.; Azimi, F.; Cooper, A.; Saricilar, E.; Murthy, V.; Souza, P.d.; Becker, T.M. PD-L1 Detection on Circulating Melanoma Cells. In Melanoma; Humana: New York, NY, USA, 2021; Volume 2265, pp. 223–233. [Google Scholar]
- Gasch, C.; Bauernhofer, T.; Pichler, M.; Langer-Freitag, S.; Reeh, M.; Seifert, A.M.; Mauermann, O.; Izbicki, J.R.; Pantel, K.; Riethdorf, S. Heterogeneity of epidermal growth factor receptor status and mutations of KRAS/PIK3CA in circulating tumor cells of patients with colorectal cancer. Clin. Chem. 2013, 59, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Babayan, A.; Hannemann, J.; Spötter, J.; Müller, V.; Pantel, K.; Joosse, S.A. Heterogeneity of estrogen receptor expression in circulating tumor cells from metastatic breast cancer patients. PLoS ONE 2013, 8, e75038. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Imani, S.; Maghsoudloo, M.; Shasaltaneh, M.D.; Gao, L.; Zhou, J.; Wen, Q.; Liu, S.; Zhang, L.; Chen, G. Genome-wide copy number analysis of circulating tumor cells in breast cancer patients with liver metastasis. Oncol. Rep. 2020, 44, 1075–1093. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guo, L.; Feng, L.; Zhang, W.; Xiao, T.; Di, X.; Chen, G.; Zhang, K. Single nucleotide variant profiles of viable single circulating tumour cells reveal CTC behaviours in breast cancer. Oncol. Rep. 2018, 39, 2147–2159. [Google Scholar] [CrossRef] [Green Version]
- Aljohani, H.M.; Aittaleb, M.; Furgason, J.M.; Amaya, P.; Deeb, A.; Chalmers, J.J.; Bahassi, E.M. Genetic mutations associated with lung cancer metastasis to the brain. Mutagenesis 2018, 33, 137–145. [Google Scholar] [CrossRef]
- Di Trapani, M.; Manaresi, N.; Medoro, G. DEPArray™ system: An automatic image-based sorter for isolation of pure circulating tumor cells. Cytometry A. 2018, 93, 1260–1266. [Google Scholar] [CrossRef] [Green Version]
- Paolillo, C.; Mu, Z.; Rossi, G.; Schiewer, M.J.; Nguyen, T.; Austin, L.; Capoluongo, E.; Knudsen, K.; Cristofanilli, M.; Fortina, P. Detection of Activating Estrogen Receptor Gene (ESR1) Mutations in Single Circulating Tumor Cells. Clin. Cancer Res. 2017, 23, 6086–6093. [Google Scholar] [CrossRef] [Green Version]
- Pestrin, M.; Salvianti, F.; Galardi, F.; De Luca, F.; Turner, N.; Malorni, L.; Pazzagli, M.; Di Leo, A.; Pinzani, P. Heterogeneity of PIK3CA mutational status at the single cell level in circulating tumor cells from metastatic breast cancer patients. Mol. Oncol. 2015, 9, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Polzer, B.; Medoro, G.; Pasch, S.; Fontana, F.; Zorzino, L.; Pestka, A.; Andergassen, U.; Meier-Stiegen, F.; Czyz, Z.T.; Alberter, B.; et al. Molecular profiling of single circulating tumor cells with diagnostic intention. EMBO Mol. Med. 2014, 6, 1371–1386. [Google Scholar] [CrossRef]
- Lu, S.; Chang, C.J.; Guan, Y.; Szafer-Glusman, E.; Punnoose, E.; Do, A.; Suttmann, B.; Gagnon, R.; Rodriguez, A.; Ers, M.; et al. Genomic Analysis of Circulating Tumor Cells at the Single-Cell Level. J. Mol. Diagn. 2020, 22, 770–781. [Google Scholar] [CrossRef]
- Chen, D.; Zhen, H.; Qiu, Y.; Liu, P.; Zeng, P.; Xia, J.; Shi, Q.; Xie, L.; Zhu, Z.; Gao, Y.; et al. Comparison of single cell sequencing data between two whole genome amplification methods on two sequencing platforms. Sci. Rep. 2018, 8, 4963. [Google Scholar] [CrossRef]
- Deleye, L.; Gansemans, Y.; de Coninck, D.; van Nieuwerburgh, F.; Deforce, D. Massively parallel sequencing of micro-manipulated cells targeting a comprehensive panel of disease-causing genes: A comparative evaluation of upstream whole-genome amplification methods. PLoS ONE 2018, 13, e0196334. [Google Scholar] [CrossRef] [Green Version]
- Sho, S.; Court, C.M.; Winograd, P.; Lee, S.; Hou, S.; Graeber, T.G.; Tseng, H.R.; Tomlinson, J.S. Precision oncology using a limited number of cells: Optimization of whole genome amplification products for sequencing applications. BMC Cancer 2017, 17, 457. [Google Scholar] [CrossRef] [Green Version]
- Vander Plaetsen, A.-S.; Deleye, L.; Cornelis, S.; Tilleman, L.; van Nieuwerburgh, F.; Deforce, D. STR profiling and Copy Number Variation analysis on single, preserved cells using current Whole Genome Amplification methods. Sci. Rep. 2017, 7, 17189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Ma, F.; Chapman, A.; Lu, S.; Xie, X.S. Single-Cell Whole-Genome Amplification and Sequencing: Methodology and Applications. Ann. Rev. Genom. Hum. Genet. 2015, 16, 79–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Lu, N.; Shi, X.; Qiao, Y.; Chen, L.; Duan, M.; Hou, Y.; Ge, Q.; Tao, Y.; Tu, J.; et al. 1D-Reactor Decentralized MDA for Uniform and Accurate Whole Genome Amplification. Anal. Chem. 2017, 89, 10147–10152. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, M.; Nishikawa, Y.; Kogawa, M.; Takeyama, H. Massively parallel whole genome amplification for single-cell sequencing using droplet microfluidics. Sci. Rep. 2017, 7, 5199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, K.; Klaus, A.; Lin, B.K.; Laks, E.; Biele, J.; Lai, D.; Bashashati, A.; Huang, Y.F.; Aniba, R.; Moksa, M.; et al. Robust high-performance nanoliter-volume single-cell multiple displacement amplification on planar substrates. Proc. Natl. Acad. Sci. USA 2016, 113, 8484–8489. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, D.; Ruan, W.; Liu, W.; Yin, K.; Tian, T.; Bi, Y.; Ruan, Q.; Zhao, Y.; Zhu, Z.; et al. Centrifugal-Driven Droplet Generation Method with Minimal Waste for Single-Cell Whole Genome Amplification. Anal. Chem. 2019, 91, 13611–13619. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhang, F.; Zhang, X.; Yin, J.; Du, M.; Jiang, M.; Liu, L.; Li, J.; Huang, Y.; Wang, J. High-throughput single-cell whole-genome amplification through centrifugal emulsification and eMDA. Commun. Biol. 2019, 2, 147. [Google Scholar] [CrossRef]
- Chen, C.; Xing, D.; Tan, L.; Li, H.; Zhou, G.; Huang, L.; Xie, X.S. Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science 2017, 356, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Pena, V.; Natarajan, S.; Xia, Y.; Klein, D.; Carter, R.; Pang, Y.; Shaner, B.; Annu, K.; Putnam, D.; Chen, W.; et al. Accurate genomic variant detection in single cells with primary template-directed amplification. Proc. Natl. Acad. Sci. USA 2021, 118, e2024176118. [Google Scholar] [CrossRef]
- Xing, D.; Tan, L.; Chang, C.-H.; Li, H.; Xie, X.S. Accurate SNV detection in single cells by transposon-based whole-genome amplification of complementary strands. Proc. Natl. Acad. Sci. USA 2021, 118, e2013106118. [Google Scholar] [CrossRef]
- Li, R.; Jia, F.; Zhang, W.; Shi, F.; Fang, Z.; Zhao, H.; Hu, Z.; Wei, Z. Device for whole genome sequencing single circulating tumor cells from whole blood. Lab Chip 2019, 19, 3168–3178. [Google Scholar] [CrossRef] [PubMed]
- Carretta, C.; Mallia, S.; Genovese, E.; Parenti, S.; Rontauroli, S.; Bianchi, E.; Fantini, S.; Sartini, S.; Tavernari, L.; Tagliafico, E.; et al. Genomic Analysis of Hematopoietic Stem Cell at the Single-Cell Level: Optimization of Cell Fixation and Whole Genome Amplification (WGA) Protocol. Int. J. Mol. Sci. 2020, 21, 7366. [Google Scholar] [CrossRef] [PubMed]
- Schneck, H.; Blassl, C.; Meier-Stiegen, F.; Neves, R.P.; Janni, W.; Fehm, T.; Neubauer, H. Analysing the mutational status of PIK3CA in circulating tumor cells from metastatic breast cancer patients. Mol. Oncol. 2013, 7, 976–986. [Google Scholar] [CrossRef]
- Estrada-Rivadeneyra, D. Sanger sequencing. FEBS J. 2017, 284, 4174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Möhlendick, B.; Stoecklein, N.H. Analysis of Copy-Number Alterations in Single Cells Using Microarray-Based Comparative Genomic Hybridization (aCGH). Curr. Protoc. Cell Biol. 2014, 65, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Zhang, X.; Marjani, S.L.; Hu, Z.; Weissman, S.M.; Pan, X.; Wu, S. Single-Cell Sequencing for Precise Cancer Research: Progress and Prospects. Cancer Res. 2016, 76, 1305. [Google Scholar] [CrossRef] [Green Version]
- Gawad, C.; Koh, W.; Quake, S.R. Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef]
- Grün, D.; van Oudenaarden, A. Design and Analysis of Single-Cell Sequencing Experiments. Cell 2015, 163, 799–810. [Google Scholar] [CrossRef] [Green Version]
- Zong, C.; Lu, S.; Chapman, A.R.; Xie, X.S. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science 2012, 338, 1622–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, F.B.; Hosono, S.; Fang, L.; Wu, X.; Faruqi, A.F.; Bray-Ward, P.; Sun, Z.; Zong, Q.; Du, Y.; Du, J.; et al. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. USA 2002, 99, 5261–5266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.; Li, C.; Lu, S.; Zhou, W.; Tang, F.; Xie, X.S.; Huang, Y. Uniform and accurate single-cell sequencing based on emulsion whole-genome amplification. Proc. Natl. Acad. Sci. USA 2015, 112, 11923–11928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, W.K.; Edge, P.; Lee, H.S.; Bansal, V.; Bafna, V.; Huang, X.; Zhang, K. Ultraaccurate genome sequencing and haplotyping of single human cells. Proc. Natl. Acad. Sci. USA 2017, 114, 12512–12517. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Xu, Y.; Zhu, L.; Su, Z.; Han, X.; Zhang, Z.; Huang, Y.; Liu, Q. Comparison of multiple displacement amplification (MDA) and multiple annealing and looping-based amplification cycles (MALBAC) in limited DNA sequencing based on tube and droplet. Micromachines 2020, 11, 645. [Google Scholar] [CrossRef]
- Chapman, A.R.; He, Z.; Lu, S.; Yong, J.; Tan, L.; Tang, F.; Xie, X.S. Single cell transcriptome amplification with MALBAC. PLoS ONE 2015, 10, e0120889. [Google Scholar] [CrossRef]
- Bidard, F.-C.; Proudhon, C.; Pierga, J.-Y. Circulating tumor cells in breast cancer. Mol. Oncol. 2016, 10, 418–430. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Yang, S.; Shi, J.; Ding, Y.; Gao, W.; Cheng, M.; Sun, Y.; Xie, Y.; Sang, M.; Yang, H.; et al. Recognition of the organ-specific mutations in metastatic breast cancer by circulating tumor cells isolated in vivo. Neoplasma 2021, 68, 31–39. [Google Scholar] [CrossRef]
- Allison, K.H.; Hammond, M.E.H.; Dowsett, M.; McKernin, S.E.; Carey, L.A.; Fitzgibbons, P.L.; Hayes, D.F.; Lakhani, S.R.; Chavez-MacGregor, M.; Perlmutter, J.; et al. Estrogen and Progesterone Receptor Testing in Breast Cancer: ASCO/CAP Guideline Update. J. Clin. Oncol. 2020, 38, 1346–1366. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C.J.F.i.O. PI3K/Akt/mTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front. Oncol. 2022, 12, 819128. [Google Scholar] [CrossRef]
- Fusco, N.; Malapelle, U.; Fassan, M.; Marchiò, C.; Buglioni, S.; Zupo, S.; Criscitiello, C.; Vigneri, P.; Dei Tos, A.P.; Maiorano, E.; et al. PIK3CA Mutations as a Molecular Target for Hormone Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Front. Oncol. 2021, 11, 644737. [Google Scholar] [CrossRef] [PubMed]
- Neves, R.P.; Raba, K.; Schmidt, O.; Honisch, E.; Meier-Stiegen, F.; Behrens, B.; Möhlendick, B.; Fehm, T.; Neubauer, H.; Klein, C.A.; et al. Genomic high-resolution profiling of single CKpos/CD45neg flow-sorting purified circulating tumor cells from patients with metastatic breast cancer. Clin. Chem. 2014, 60, 1290–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasch, C.; Oldopp, T.; Mauermann, O.; Gorges, T.M.; Andreas, A.; Coith, C.; Müller, V.; Fehm, T.; Janni, W.; Pantel, K.; et al. Frequent detection of PIK3CA mutations in single circulating tumor cells of patients suffering from HER2-negative metastatic breast cancer. Mol. Oncol. 2016, 10, 1330–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, M.H.; Schneck, H.; Decker, Y.; Schömer, S.; Franken, A.; Endris, V.; Pfarr, N.; Weichert, W.; Niederacher, D.; Fehm, T.; et al. Isolation and characterization of circulating tumor cells using a novel workflow combining the CellSearch(®) system and the CellCelector(™). Biotechnol. Prog. 2016, 33, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, E.M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. J. Cancer Cell 2007, 12, 395–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandarlapaty, S.; Sakr, R.A.; Giri, D.; Patil, S.; Heguy, A.; Morrow, M.; Modi, S.; Norton, L.; Rosen, N.; Hudis, C. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. J. Clin. Cancer Res. 2012, 18, 6784–6791. [Google Scholar] [CrossRef] [Green Version]
- De Luca, F.; Rotunno, G.; Salvianti, F.; Galardi, F.; Pestrin, M.; Gabellini, S.; Simi, L.; Mancini, I.; Vannucchi, A.M.; Pazzagli, M.; et al. Mutational analysis of single circulating tumor cells by next generation sequencing in metastatic breast cancer. Oncotarget 2016, 7, 26107–26119. [Google Scholar] [CrossRef]
- Jeffreys, S.A.; Becker, T.M.; Khan, S.; Soon, P.; Neubauer, H.; de Souza, P.; Powter, B. Prognostic and Predictive Value of CCND1/Cyclin D1 Amplification in Breast Cancer With a Focus on Postmenopausal Patients: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2022, 13, 895729. [Google Scholar] [CrossRef]
- Kaur, P.; Campo, D.; Porras, T.B.; Ring, A.; Lu, J.; Chairez, Y.; Su, Y.; Kang, I.; Lang, J.E. A Pilot Study for the Feasibility of Exome-Sequencing in Circulating Tumor Cells Versus Single Metastatic Biopsies in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 4826. [Google Scholar] [CrossRef] [PubMed]
- Greene, S.B.; Dago, A.E.; Leitz, L.J.; Wang, Y.; Lee, J.; Werner, S.L.; Gendreau, S.; Patel, P.; Jia, S.; Zhang, L.; et al. Chromosomal Instability Estimation Based on Next Generation Sequencing and Single Cell Genome Wide Copy Number Variation Analysis. PLoS ONE 2016, 11, e0165089. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Li, J.; Kemeny, G.; Bitting, R.L.; Beaver, J.; Somarelli, J.A.; Ware, K.E.; Gregory, S.; Armstrong, A.J. Whole Genomic Copy Number Alterations in Circulating Tumor Cells from Men with Abiraterone or Enzalutamide-Resistant Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2017, 23, 1346–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magbanua, M.J.; Sosa, E.V.; Scott, J.H.; Simko, J.; Collins, C.; Pinkel, D.; Ryan, C.J.; Park, J.W. Isolation and genomic analysis of circulating tumor cells from castration resistant metastatic prostate cancer. BMC Cancer 2012, 12, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Shi, J.; Shi, G.; Xu, X.; Liu, Q.; Liu, C.; Gao, Z.; Bai, J.; Shan, B. Using the New CellCollector to Capture Circulating Tumor Cells from Blood in Different Groups of Pulmonary Disease: A Cohort Study. Sci. Rep. 2017, 7, 9542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariscal, J.; Alonso-Nocelo, M.; Muinelo-Romay, L.; Barbazan, J.; Vieito, M.; Abalo, A.; Gomez-Tato, A.; Maria de Los Angeles, C.C.; Garcia-Caballero, T.; Rodriguez, C.; et al. Molecular Profiling of Circulating Tumour Cells Identifies Notch1 as a Principal Regulator in Advanced Non-Small Cell Lung Cancer. Sci. Rep. 2016, 6, 37820. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, F.; Carloni, S.; Zoli, W.; Ulivi, P.; Gallerani, G.; Fici, P.; Chiadini, E.; Passardi, A.; Frassineti, G.L.; Ragazzini, A.; et al. Detection and recovery of circulating colon cancer cells using a dielectrophoresis-based device: KRAS mutation status in pure CTCs. Cancer Lett. 2013, 335, 225–231. [Google Scholar] [CrossRef]
- Court, C.M.; Ankeny, J.S.; Sho, S.; Hou, S.; Li, Q.; Hsieh, C.; Song, M.; Liao, X.; Rochefort, M.M.; Wainberg, Z.A.; et al. Reality of Single Circulating Tumor Cell Sequencing for Molecular Diagnostics in Pancreatic Cancer. J. Mol. Diagn. 2016, 18, 688–696. [Google Scholar] [CrossRef] [Green Version]
- Reid, A.L.; Freeman, J.B.; Millward, M.; Ziman, M.; Gray, E.S. Detection of BRAF-V600E and V600K in melanoma circulating tumour cells by droplet digital PCR. Clin. Biochem. 2014, 48, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, C.; Li, J.; Luttgen, M.S.; Kolatkar, A.; Kendall, J.T.; Flores, E.; Topp, Z.; Samlowski, W.E.; McClay, E.; Bethel, K.; et al. Limited genomic heterogeneity of circulating melanoma cells in advanced stage patients. Phys. Biol. 2016, 12, 016008. [Google Scholar] [CrossRef] [Green Version]
- Ferrarini, A.; Forcato, C.; Buson, G.; Tononi, P.; Del Monaco, V.; Terracciano, M.; Bolognesi, C.; Fontana, F.; Medoro, G.; Neves, R.; et al. A streamlined workflow for single-cells genome-wide copy-number profiling by low-pass sequencing of LM-PCR whole-genome amplification products. PLoS ONE 2018, 13, e0193689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Ni, X.; Guo, H.; Su, Z.; Ba, Y.; Tong, Z.; Guo, Z.; Yao, X.; Chen, X.; Yin, J.; et al. Single-cell sequencing deciphers a convergent evolution of copy number alterations from primary to circulating tumor cells. Genome Res. 2017, 27, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Khan, T.; Becker, T.M.; Scott, K.F.; Descallar, J.; de Souza, P.; Chua, W.; Ma, Y. Prognostic and Predictive Value of Liquid Biopsy-Derived Androgen Receptor Variant 7 (AR-V7) in Prostate Cancer: A Systematic Review and Meta-Analysis. Front. Oncol. 2022, 12, 868031. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.; Strickler, J.; Bekaii-Saab, T.; Yaeger, R. BRAF-Mutated Advanced Colorectal Cancer: A Rapidly Changing Therapeutic Landscape. J. Clin. Oncol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Gemenetzis, G.; Kinny-Köster, B.; Habib, J.R.; Groot, V.P.; Teinor, J.; Yin, L.; Pu, N.; Hasanain, A.; van Oosten, F.; et al. Pancreatic circulating tumor cell detection by targeted single-cell next-generation sequencing. Cancer Lett. 2020, 493, 245–253. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, T.; Becker, T.M.; Po, J.W.; Chua, W.; Ma, Y. Single-Circulating Tumor Cell Whole Genome Amplification to Unravel Cancer Heterogeneity and Actionable Biomarkers. Int. J. Mol. Sci. 2022, 23, 8386. https://doi.org/10.3390/ijms23158386
Khan T, Becker TM, Po JW, Chua W, Ma Y. Single-Circulating Tumor Cell Whole Genome Amplification to Unravel Cancer Heterogeneity and Actionable Biomarkers. International Journal of Molecular Sciences. 2022; 23(15):8386. https://doi.org/10.3390/ijms23158386
Chicago/Turabian StyleKhan, Tanzila, Therese M. Becker, Joseph W. Po, Wei Chua, and Yafeng Ma. 2022. "Single-Circulating Tumor Cell Whole Genome Amplification to Unravel Cancer Heterogeneity and Actionable Biomarkers" International Journal of Molecular Sciences 23, no. 15: 8386. https://doi.org/10.3390/ijms23158386
APA StyleKhan, T., Becker, T. M., Po, J. W., Chua, W., & Ma, Y. (2022). Single-Circulating Tumor Cell Whole Genome Amplification to Unravel Cancer Heterogeneity and Actionable Biomarkers. International Journal of Molecular Sciences, 23(15), 8386. https://doi.org/10.3390/ijms23158386