
Exogenous ANP Treatment Ameliorates Myocardial Insulin Resistance and Protects against Ischemia–Reperfusion Injury in Diet-Induced Obesity

 , ,
, ,
Abstract
1. Introduction
2. Results
2.1. Heart Weight and Cardiac Function in HFD Mice
2.2. ANP Treatment Ameliorated Cardiac Insulin Signaling in HFD Mice
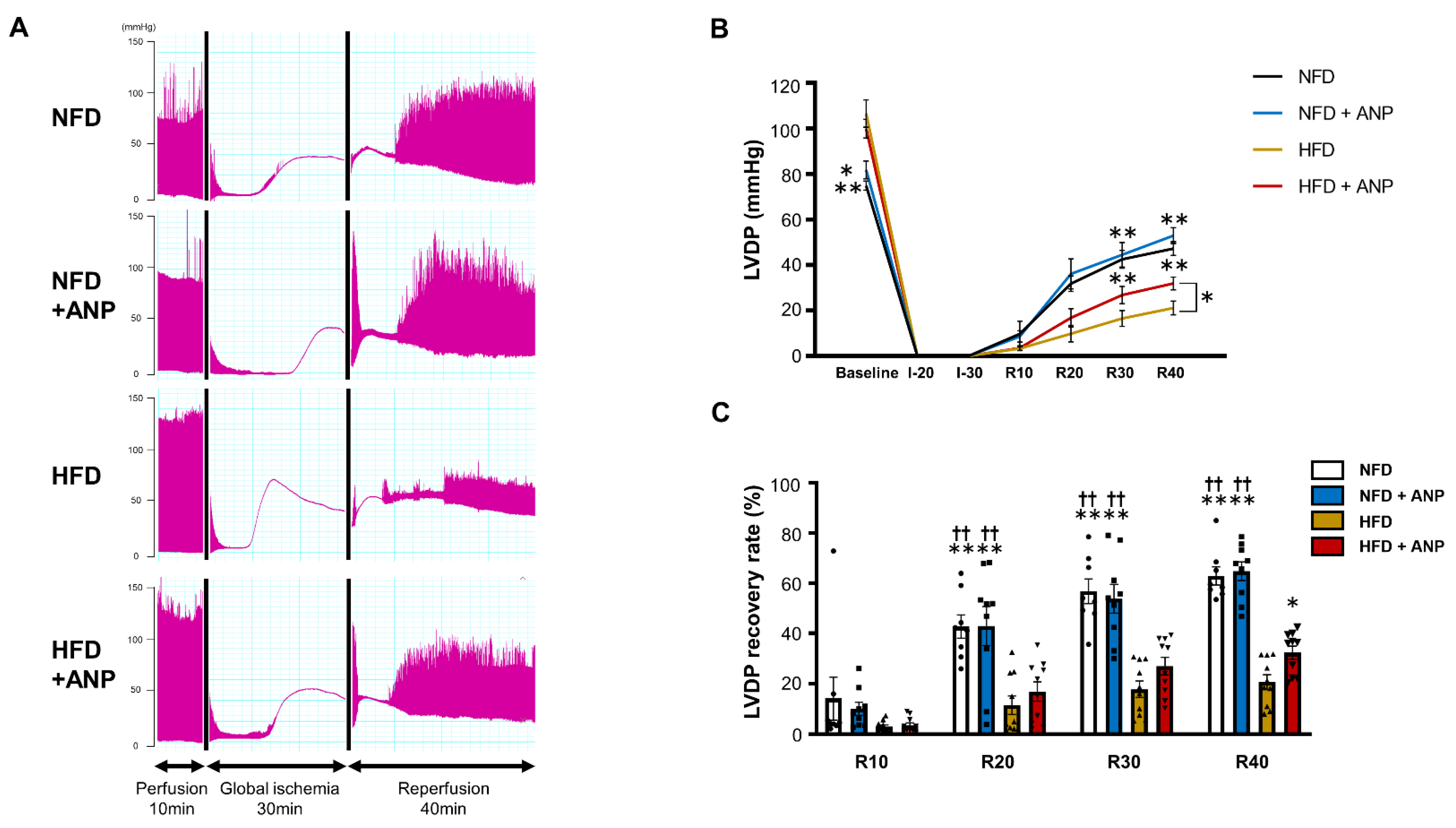
2.3. Effects of ANP Treatment on the Cardiac Function during IRI in HFD Mice
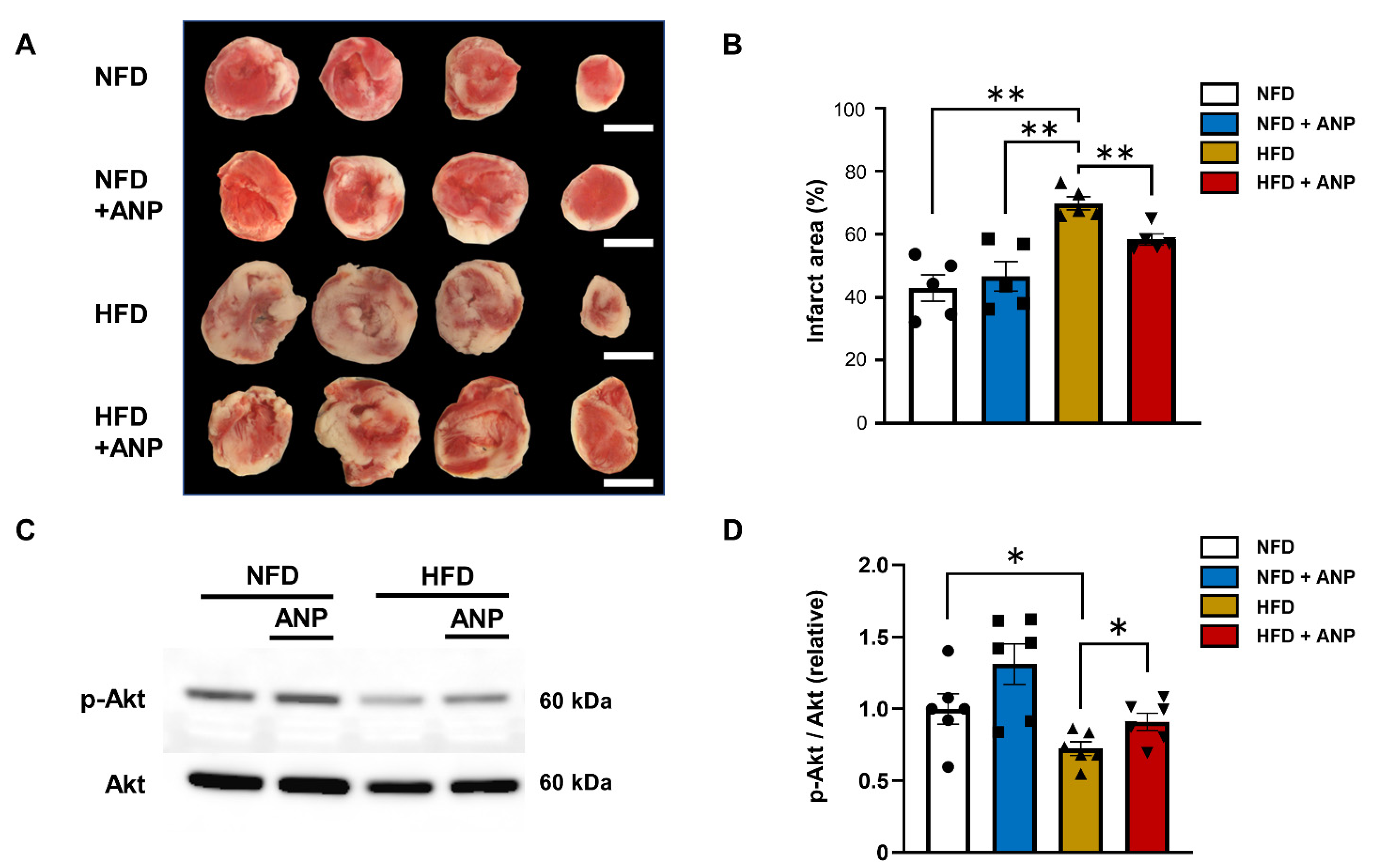
2.4. Effects of ANP Treatment on Cardiac Injury after Ischemia–Reperfusion
2.5. Effects of ANP Treatment on Insulin Signaling during IRI
2.6. Effects of ANP Treatment on Myocardial Ultrastructure
2.7. Effects of ANP Treatment on Mitochondrial Damage after IRI
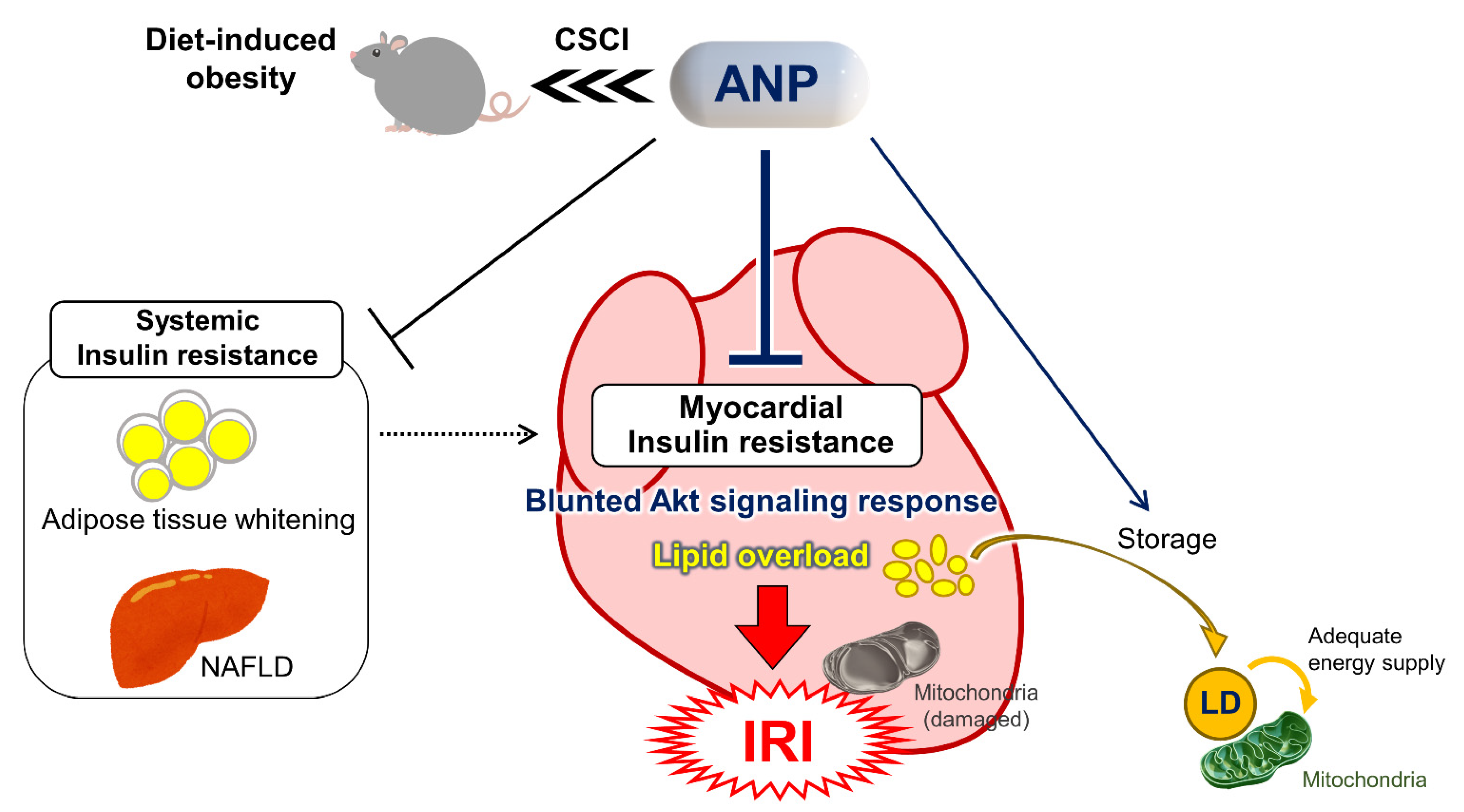
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. Echocardiography
4.3. Insulin Sensitivity Test
4.4. Experiments in Langendorff Hearts
4.5. Ischemia–Reperfusion Model
4.6. Immunoblotting
4.7. RNA Isolation, Reverse Transcription (RT) and Real-Time Polymerase Chain Reaction (PCR)
4.8. Electronic Microscope
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ANP | A-type natriuretic peptide |
| ATGL | adipose triglyceride lipase |
| BNP | B-type natriuretic peptide |
| DIO | diet-induced obesity |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| HFD | high-fat diet |
| HSL | hormone-sensitive lipase |
| IRI | ischemia–reperfusion injury |
| LD | lipid droplet |
| LVDP | left ventricular-developed pressure |
| NFD | normal-fat diet |
| NPR-A | natriuretic peptide receptor A |
| NPs | natriuretic peptides |
| Plin5 | perilipin 5 |
| PPARα | peroxisome-proliferator-activated receptor alpha |
| TTC | triphenyltetrazolium chloride |
References
- Forte, M.; Madonna, M.; Schiavon, S.; Valenti, V.; Versaci, F.; Zoccai, G.B.; Frati, G.; Sciarretta, S. Cardiovascular Pleiotropic Effects of Natriuretic Peptides. Int. J. Mol. Sci. 2019, 20, 3874. [Google Scholar] [CrossRef] [PubMed]
- Yasue, H.; Yoshimura, M.; Sumida, H.; Kikuta, K.; Kugiyama, K.; Jougasaki, M.; Ogawa, H.; Okumura, K.; Mukoyama, M.; Nakao, K. Localization and mechanism of secretion of B-type natriuretic peptide in comparison with those of A-type natriuretic peptide in normal subjects and patients with heart failure. Circulation 1994, 90, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Tamura, N.; Ogawa, Y.; Chusho, H.; Nakamura, K.; Nakao, K.; Suda, M.; Kasahara, M.; Hashimoto, R.; Katsuura, G.; Mukoyama, M.; et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc. Natl. Acad. Sci. USA 2000, 97, 4239–4244. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, K.; Nakagawa, Y.; Nishikimi, T. Cutting Edge of Brain Natriuretic Peptide (BNP) Research- The Diversity of BNP Immunoreactivity and Its Clinical Relevance. Circ. J. 2018, 82, 2455–2461. [Google Scholar] [CrossRef]
- Goetze, J.P.; Bruneau, B.G.; Ramos, H.R.; Ogawa, T.; de Bold, M.K.; de Bold, A.J. Cardiac natriuretic peptides. Nat. Rev. Cardiol. 2020, 17, 698–717. [Google Scholar] [CrossRef]
- Kimura, H.; Nagoshi, T.; Oi, Y.; Yoshii, A.; Tanaka, Y.; Takahashi, H.; Kashiwagi, Y.; Tanaka, T.D.; Yoshimura, M. Treatment with atrial natriuretic peptide induces adipose tissue browning and exerts thermogenic actions in vivo. Sci. Rep. 2021, 11, 17466. [Google Scholar] [CrossRef]
- Cannone, V.; Cabassi, A.; Volpi, R.; Burnett, J.C., Jr. Atrial Natriuretic Peptide: A Molecular Target of Novel Therapeutic Approaches to Cardio-Metabolic Disease. Int. J. Mol. Sci. 2019, 20, 3265. [Google Scholar] [CrossRef]
- Coue, M.; Badin, P.M.; Vila, I.K.; Laurens, C.; Louche, K.; Marques, M.A.; Bourlier, V.; Mouisel, E.; Tavernier, G.; Rustan, A.C.; et al. Defective Natriuretic Peptide Receptor Signaling in Skeletal Muscle Links Obesity to Type 2 Diabetes. Diabetes 2015, 64, 4033–4045. [Google Scholar] [CrossRef]
- Song, E.; Da Eira, D.; Jani, S.; Sepa-Kishi, D.; Vu, V.; Hunter, H.; Lai, M.; Wheeler, M.B.; Ceddia, R.B.; Sweeney, G. Cardiac Autophagy Deficiency Attenuates ANP Production and Disrupts Myocardial-Adipose Cross Talk, Leading to Increased Fat Accumulation and Metabolic Dysfunction. Diabetes 2021, 70, 51–61. [Google Scholar] [CrossRef]
- Collins, S. A heart-adipose tissue connection in the regulation of energy metabolism. Nat. Rev. Endocrinol. 2014, 10, 157–163. [Google Scholar] [CrossRef]
- Coue, M.; Moro, C. Natriuretic peptide control of energy balance and glucose homeostasis. Biochimie 2016, 124, 84–91. [Google Scholar] [CrossRef]
- Jordan, J.; Birkenfeld, A.L.; Melander, O.; Moro, C. Natriuretic Peptides in Cardiovascular and Metabolic Crosstalk: Implications for Hypertension Management. Hypertension 2018, 72, 270–276. [Google Scholar] [CrossRef]
- Bartels, E.D.; Guo, S.; Kousholt, B.S.; Larsen, J.R.; Hasenkam, J.M.; Burnett, J., Jr.; Nielsen, L.B.; Ashina, M.; Goetze, J.P. High doses of ANP and BNP exacerbate lipolysis in humans and the lipolytic effect of BNP is associated with cardiac triglyceride content in pigs. Peptides 2019, 112, 43–47. [Google Scholar] [CrossRef]
- Miyashita, K.; Itoh, H.; Tsujimoto, H.; Tamura, N.; Fukunaga, Y.; Sone, M.; Yamahara, K.; Taura, D.; Inuzuka, M.; Sonoyama, T.; et al. Natriuretic peptides/cGMP/cGMP-dependent protein kinase cascades promote muscle mitochondrial biogenesis and prevent obesity. Diabetes 2009, 58, 2880–2892. [Google Scholar] [CrossRef]
- Bordicchia, M.; Liu, D.; Amri, E.Z.; Ailhaud, G.; Dessi-Fulgheri, P.; Zhang, C.; Takahashi, N.; Sarzani, R.; Collins, S. Cardiac natriuretic peptides act via p38 MAPK to induce the brown fat thermogenic program in mouse and human adipocytes. J. Clin. Investig. 2012, 122, 1022–1036. [Google Scholar] [CrossRef]
- Plante, E.; Menaouar, A.; Danalache, B.A.; Broderick, T.L.; Jankowski, M.; Gutkowska, J. Treatment with brain natriuretic peptide prevents the development of cardiac dysfunction in obese diabetic db/db mice. Diabetologia 2014, 57, 1257–1267. [Google Scholar] [CrossRef]
- Wu, W.; Shi, F.; Liu, D.; Ceddia, R.P.; Gaffin, R.; Wei, W.; Fang, H.; Lewandowski, E.D.; Collins, S. Enhancing natriuretic peptide signaling in adipose tissue, but not in muscle, protects against diet-induced obesity and insulin resistance. Sci. Signal. 2017, 10, eaam6870. [Google Scholar] [CrossRef]
- Kimura, H.; Nagoshi, T.; Yoshii, A.; Kashiwagi, Y.; Tanaka, Y.; Ito, K.; Yoshino, T.; Tanaka, T.D.; Yoshimura, M. The thermogenic actions of natriuretic peptide in brown adipocytes: The direct measurement of the intracellular temperature using a fluorescent thermoprobe. Sci. Rep. 2017, 7, 12978. [Google Scholar] [CrossRef]
- Kang, R.; Nagoshi, T.; Kimura, H.; Tanaka, T.D.; Yoshii, A.; Inoue, Y.; Morimoto, S.; Ogawa, K.; Minai, K.; Ogawa, T.; et al. Possible association between body temperature and B-type natriuretic peptide in patients with cardiovascular diseases. J. Card. Fail. 2021, 27, 75–82. [Google Scholar] [CrossRef]
- Uno, G.; Nagoshi, T.; Yoshii, A.; Inoue, Y.; Tanaka, Y.; Kimura, H.; Ito, S.; Ogawa, K.; Tanaka, T.D.; Minai, K.; et al. Collaborative Activities of Noradrenaline and Natriuretic Peptide for Glucose Utilization in Patients with Acute Coronary Syndrome. Sci. Rep. 2019, 9, 7822. [Google Scholar] [CrossRef]
- Nagoshi, T.; Yoshimura, M.; Rosano, G.M.; Lopaschuk, G.D.; Mochizuki, S. Optimization of Cardiac Metabolism in Heart Failure. Curr. Pharm. Des. 2011, 17, 3846–3853. [Google Scholar] [CrossRef] [PubMed]
- Nagoshi, T.; Matsui, T.; Aoyama, T.; Leri, A.; Anversa, P.; Li, L.; Ogawa, W.; del Monte, F.; Gwathmey, J.K.; Grazette, L.; et al. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J. Clin. Investig. 2005, 115, 2128–2138. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Kajstura, J.; Discher, D.J.; Wasserlauf, B.J.; Bishopric, N.H.; Anversa, P.; Webster, K.A. Reperfusion-activated Akt kinase prevents apoptosis in transgenic mouse hearts overexpressing insulin-like growth factor-1. Circ. Res. 2001, 88, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, A.K.; Sack, M.N.; Mjos, O.D.; Yellon, D.M. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ. Res. 2001, 89, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Ouwens, D.M.; Boer, C.; Fodor, M.; de Galan, P.; Heine, R.J.; Maassen, J.A.; Diamant, M. Cardiac dysfunction induced by high-fat diet is associated with altered myocardial insulin signalling in rats. Diabetologia 2005, 48, 1229–1237. [Google Scholar] [CrossRef]
- Noyan-Ashraf, M.H.; Shikatani, E.A.; Schuiki, I.; Mukovozov, I.; Wu, J.; Li, R.K.; Volchuk, A.; Robinson, L.A.; Billia, F.; Drucker, D.J.; et al. A glucagon-like peptide-1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation 2013, 127, 74–85. [Google Scholar] [CrossRef]
- Zheng, P.; Xie, Z.; Yuan, Y.; Sui, W.; Wang, C.; Gao, X.; Zhao, Y.; Zhang, F.; Gu, Y.; Hu, P.; et al. Plin5 alleviates myocardial ischaemia/reperfusion injury by reducing oxidative stress through inhibiting the lipolysis of lipid droplets. Sci. Rep. 2017, 7, 42574. [Google Scholar] [CrossRef]
- Coue, M.; Barquissau, V.; Morigny, P.; Louche, K.; Lefort, C.; Mairal, A.; Carpene, C.; Viguerie, N.; Arner, P.; Langin, D.; et al. Natriuretic peptides promote glucose uptake in a cGMP-dependent manner in human adipocytes. Sci. Rep. 2018, 8, 1097. [Google Scholar] [CrossRef]
- Kato, T.; Muraski, J.; Chen, Y.; Tsujita, Y.; Wall, J.; Glembotski, C.C.; Schaefer, E.; Beckerle, M.; Sussman, M.A. Atrial natriuretic peptide promotes cardiomyocyte survival by cGMP-dependent nuclear accumulation of zyxin and Akt. J. Clin. Investig. 2005, 115, 2716–2730. [Google Scholar] [CrossRef]
- Breivik, L.; Jensen, A.; Guvag, S.; Aarnes, E.K.; Aspevik, A.; Helgeland, E.; Hovland, S.; Brattelid, T.; Jonassen, A.K. B-type natriuretic peptide expression and cardioprotection is regulated by Akt dependent signaling at early reperfusion. Peptides 2015, 66, 43–50. [Google Scholar] [CrossRef][Green Version]
- Aoyagi, T.; Higa, J.K.; Aoyagi, H.; Yorichika, N.; Shimada, B.K.; Matsui, T. Cardiac mTOR rescues the detrimental effects of diet-induced obesity in the heart after ischemia–reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1530–H1539. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, A.; Nagoshi, T.; Kashiwagi, Y.; Kimura, H.; Tanaka, Y.; Oi, Y.; Ito, K.; Yoshino, T.; Tanaka, T.D.; Yoshimura, M. Cardiac ischemia–reperfusion injury under insulin-resistant conditions: SGLT1 but not SGLT2 plays a compensatory protective role in diet-induced obesity. Cardiovasc. Diabetol. 2019, 18, 85. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, Y.; Nagoshi, T.; Yoshino, T.; Tanaka, T.D.; Ito, K.; Harada, T.; Takahashi, H.; Ikegami, M.; Anzawa, R.; Yoshimura, M. Expression of SGLT1 in Human Hearts and Impairment of Cardiac Glucose Uptake by Phlorizin during Ischemia–reperfusion Injury in Mice. PLoS ONE 2015, 10, e0130605. [Google Scholar] [CrossRef] [PubMed]
- Osumi, T.; Kuramoto, K. Heart lipid droplets and lipid droplet-binding proteins: Biochemistry, physiology, and pathology. Exp. Cell Res. 2016, 340, 198–204. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Cardiomyopathy in obesity, insulin resistance and diabetes. J. Physiol. 2020, 598, 2977–2993. [Google Scholar] [CrossRef]
- Mizuno, M.; Kuno, A.; Yano, T.; Miki, T.; Oshima, H.; Sato, T.; Nakata, K.; Kimura, Y.; Tanno, M.; Miura, T. Empagliflozin normalizes the size and number of mitochondria and prevents reduction in mitochondrial size after myocardial infarction in diabetic hearts. Physiol. Rep. 2018, 6, e13741. [Google Scholar] [CrossRef]
- Barger, P.M.; Kelly, D.P. PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc. Med. 2000, 10, 238–245. [Google Scholar] [CrossRef]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy revisited. Circulation 2007, 115, 3213–3223. [Google Scholar] [CrossRef]
- Kintscher, U.; Foryst-Ludwig, A.; Haemmerle, G.; Zechner, R. The Role of Adipose Triglyceride Lipase and Cytosolic Lipolysis in Cardiac Function and Heart Failure. Cell Rep. Med. 2020, 1, 100001. [Google Scholar] [CrossRef]
- Kolleritsch, S.; Kien, B.; Schoiswohl, G.; Diwoky, C.; Schreiber, R.; Heier, C.; Maresch, L.K.; Schweiger, M.; Eichmann, T.O.; Stryeck, S.; et al. Low cardiac lipolysis reduces mitochondrial fission and prevents lipotoxic heart dysfunction in Perilipin 5 mutant mice. Cardiovasc. Res. 2020, 116, 339–352. [Google Scholar] [CrossRef]
- Li, X.; Wu, Y.; Zhao, J.; Wang, H.; Tan, J.; Yang, M.; Li, Y.; Deng, S.; Gao, S.; Li, H.; et al. Distinct cardiac energy metabolism and oxidative stress adaptations between obese and non-obese type 2 diabetes mellitus. Theranostics 2020, 10, 2675–2695. [Google Scholar] [CrossRef]
- Ussher, J.R.; Campbell, J.E.; Mulvihill, E.E.; Baggio, L.L.; Bates, H.E.; McLean, B.A.; Gopal, K.; Capozzi, M.; Yusta, B.; Cao, X.; et al. Inactivation of the Glucose-Dependent Insulinotropic Polypeptide Receptor Improves Outcomes following Experimental Myocardial Infarction. Cell Metab. 2018, 27, 450–460. [Google Scholar] [CrossRef]
- Wang, T.J.; Larson, M.G.; Levy, D.; Benjamin, E.J.; Leip, E.P.; Wilson, P.W.; Vasan, R.S. Impact of obesity on plasma natriuretic peptide levels. Circulation 2004, 109, 594–600. [Google Scholar] [CrossRef]
- Bachmann, K.N.; Gupta, D.K.; Xu, M.; Brittain, E.; Farber-Eger, E.; Arora, P.; Collins, S.; Wells, Q.S.; Wang, T.J. Unexpectedly Low Natriuretic Peptide Levels in Patients with Heart Failure. JACC Heart Fail. 2021, 9, 192–200. [Google Scholar] [CrossRef]
- Standeven, K.F.; Hess, K.; Carter, A.M.; Rice, G.I.; Cordell, P.A.; Balmforth, A.J.; Lu, B.; Scott, D.J.; Turner, A.J.; Hooper, N.M.; et al. Neprilysin, obesity and the metabolic syndrome. Int. J. Obes. 2011, 35, 1031–1040. [Google Scholar] [CrossRef]
- Parcha, V.; Patel, N.; Musunuru, K.; Margulies, K.B.; Cappola, T.P.; Halade, G.; Wang, T.J.; Arora, G.; Arora, P. Natriuretic Peptide Deficiency in Obese Individuals: Mechanistic Insights from Healthy Organ Donor Cohort. J. Am. Coll. Cardiol. 2021, 77, 3138–3140. [Google Scholar] [CrossRef]
- Arora, P.; Wu, C.; Hamid, T.; Arora, G.; Agha, O.; Allen, K.; Tainsh, R.E.; Hu, D.; Ryan, R.A.; Domian, I.J.; et al. Acute Metabolic Influences on the Natriuretic Peptide System in Humans. J. Am. Coll. Cardiol. 2016, 67, 804–812. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Nishikimi, T.; Kuwahara, K.; Fujishima, A.; Oka, S.; Tsutamoto, T.; Kinoshita, H.; Nakao, K.; Cho, K.; Inazumi, H.; et al. MiR30-GALNT1/2 Axis-Mediated Glycosylation Contributes to the Increased Secretion of Inactive Human Prohormone for Brain Natriuretic Peptide (proBNP) From Failing Hearts. J. Am. Heart Assoc. 2017, 6, e003601. [Google Scholar] [CrossRef]
- Burley, D.S.; Baxter, G.F. B-type natriuretic peptide at early reperfusion limits infarct size in the rat isolated heart. Basic Res. Cardiol. 2007, 102, 529–541. [Google Scholar] [CrossRef]
- Nagoshi, T.; Date, T.; Fujisaki, M.; Yoshino, T.; Sekiyama, H.; Ogawa, K.; Kayama, Y.; Minai, K.; Komukai, K.; Ogawa, T.; et al. Biphasic Action of Aldosterone on Akt Signaling in Cardiomyocytes. Horm. Metab. Res. 2012, 44, 931–937. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. New directions for protecting the heart against ischaemia-reperfusion injury: Targeting the Reperfusion Injury Salvage Kinase (RISK)-pathway. Cardiovasc. Res. 2004, 61, 448–460. [Google Scholar] [CrossRef]
- Neubauer, S. The failing heart—An engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef]
- Moro, C.; Klimcakova, E.; Lolmede, K.; Berlan, M.; Lafontan, M.; Stich, V.; Bouloumie, A.; Galitzky, J.; Arner, P.; Langin, D. Atrial natriuretic peptide inhibits the production of adipokines and cytokines linked to inflammation and insulin resistance in human subcutaneous adipose tissue. Diabetologia 2007, 50, 1038–1047. [Google Scholar] [CrossRef]
- Abel, E.D.; Litwin, S.E.; Sweeney, G. Cardiac remodeling in obesity. Physiol. Rev. 2008, 88, 389–419. [Google Scholar] [CrossRef] [PubMed]
- Thakker, G.D.; Frangogiannis, N.G.; Bujak, M.; Zymek, P.; Gaubatz, J.W.; Reddy, A.K.; Taffet, G.; Michael, L.H.; Entman, M.L.; Ballantyne, C.M. Effects of diet-induced obesity on inflammation and remodeling after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2504–H2514. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Zhang, H.; Gutierrez Cortes, N.; Wu, D.; Wang, P.; Zhang, J.; Mattison, J.A.; Smith, E.; Bettcher, L.F.; Wang, M.; et al. Increased Drp1 Acetylation by Lipid Overload Induces Cardiomyocyte Death and Heart Dysfunction. Circ. Res. 2020, 126, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Bonda, T.A.; Szynaka, B.; Sokolowska, M.; Dziemidowicz, M.; Waszkiewicz, E.; Winnicka, M.M.; Bernaczyk, P.; Wawrusiewicz-Kurylonek, N.; Kaminski, K.A. Interleukin 6 modulates PPARalpha and PGC-1alpha and is involved in high-fat diet induced cardiac lipotoxicity in mouse. Int. J. Cardiol. 2016, 219, 1–8. [Google Scholar] [CrossRef]
- Shono, M.; Yoshimura, M.; Nakayama, M.; Yamamuro, M.; Abe, K.; Suzuki, S.; Mizuno, Y.; Sugiyama, S.; Saito, Y.; Nakao, K.; et al. Predominant effect of A-type natriuretic peptide on reduction of oxidative stress during the treatment of patients with heart failure. Circ. J. 2007, 71, 1040–1046. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Sarzani, R.; Paci, V.M.; Zingaretti, C.M.; Pierleoni, C.; Cinti, S.; Cola, G.; Rappelli, A.; Dessi-Fulgheri, P. Fasting inhibits natriuretic peptides clearance receptor expression in rat adipose tissue. J. Hypertens. 1995, 13, 1241–1246. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nagoshi, T.; Yoshii, A.; Oi, Y.; Takahashi, H.; Kimura, H.; Ito, K.; Kashiwagi, Y.; Tanaka, T.D.; Yoshimura, M. Xanthine oxidase inhibition attenuates doxorubicin-induced cardiotoxicity in mice. Free Radic. Biol. Med. 2021, 162, 298–308. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NFD | NFD+ANP | HFD | HFD+ANP | |
|---|---|---|---|---|
| (n = 14) | (n = 14) | (n = 13) | (n = 13) | |
| LVSP, mmHg | 92.5 ± 3.7 | 91.4 ± 3.0 | 121.0 ± 3.7 ‡‡ | 111.4 ± 3.3 ‡‡ |
| LVEDP, mmHg | 8.9 ± 0.7 | 7.8 ± 0.7 | 9.6 ± 0.7 | 9.8 ± 0.7 |
| LVDP, mmHg | 83.6 ± 3.5 | 83.5 ± 2.9 | 105.8 ± 5.2 ‡‡ | 101.6 ± 3.5 ‡‡ |
| +dp/dt, mmHg/s | 2360 ± 169 | 2338 ± 125 | 3214 ± 256 ‡‡ | 2746 ± 136 |
| −dp/dt, mmHg/s | −2227 ± 192 | −2204 ± 153 | −2980 ± 285 ‡ | −2468 ± 126 |
| HR, bpm | 268 ± 17 | 280 ± 19 | 302 ± 16 | 248 ± 18 |
| RPP, mmHg·bpm | 25,314 ± 2422 | 25,981 ± 2454 | 35,733 ± 2564 ‡ | 27,690 ± 2200 |
| Coronary flow, mL/min | 3.06 ± 0.28 | 3.30 ± 0.35 | 4.17 ± 0.31 ‡ | 3.08 ± 0.31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oi, Y.; Nagoshi, T.; Kimura, H.; Tanaka, Y.; Yoshii, A.; Yasutake, R.; Takahashi, H.; Kashiwagi, Y.; Tanaka, T.D.; Tachibana, T.; et al. Exogenous ANP Treatment Ameliorates Myocardial Insulin Resistance and Protects against Ischemia–Reperfusion Injury in Diet-Induced Obesity. Int. J. Mol. Sci. 2022, 23, 8373. https://doi.org/10.3390/ijms23158373
Oi Y, Nagoshi T, Kimura H, Tanaka Y, Yoshii A, Yasutake R, Takahashi H, Kashiwagi Y, Tanaka TD, Tachibana T, et al. Exogenous ANP Treatment Ameliorates Myocardial Insulin Resistance and Protects against Ischemia–Reperfusion Injury in Diet-Induced Obesity. International Journal of Molecular Sciences. 2022; 23(15):8373. https://doi.org/10.3390/ijms23158373
Chicago/Turabian StyleOi, Yuhei, Tomohisa Nagoshi, Haruka Kimura, Yoshiro Tanaka, Akira Yoshii, Rei Yasutake, Hirotake Takahashi, Yusuke Kashiwagi, Toshikazu D. Tanaka, Toshiaki Tachibana, and et al. 2022. "Exogenous ANP Treatment Ameliorates Myocardial Insulin Resistance and Protects against Ischemia–Reperfusion Injury in Diet-Induced Obesity" International Journal of Molecular Sciences 23, no. 15: 8373. https://doi.org/10.3390/ijms23158373
APA StyleOi, Y., Nagoshi, T., Kimura, H., Tanaka, Y., Yoshii, A., Yasutake, R., Takahashi, H., Kashiwagi, Y., Tanaka, T. D., Tachibana, T., & Yoshimura, M. (2022). Exogenous ANP Treatment Ameliorates Myocardial Insulin Resistance and Protects against Ischemia–Reperfusion Injury in Diet-Induced Obesity. International Journal of Molecular Sciences, 23(15), 8373. https://doi.org/10.3390/ijms23158373