
A Second Life for MAP, a Model Amphipathic Peptide



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. MAPs as Delivery Carrier
3. MAP’s Intrinsic Activity
4. Functionalization of MAPs into Nanoparticles
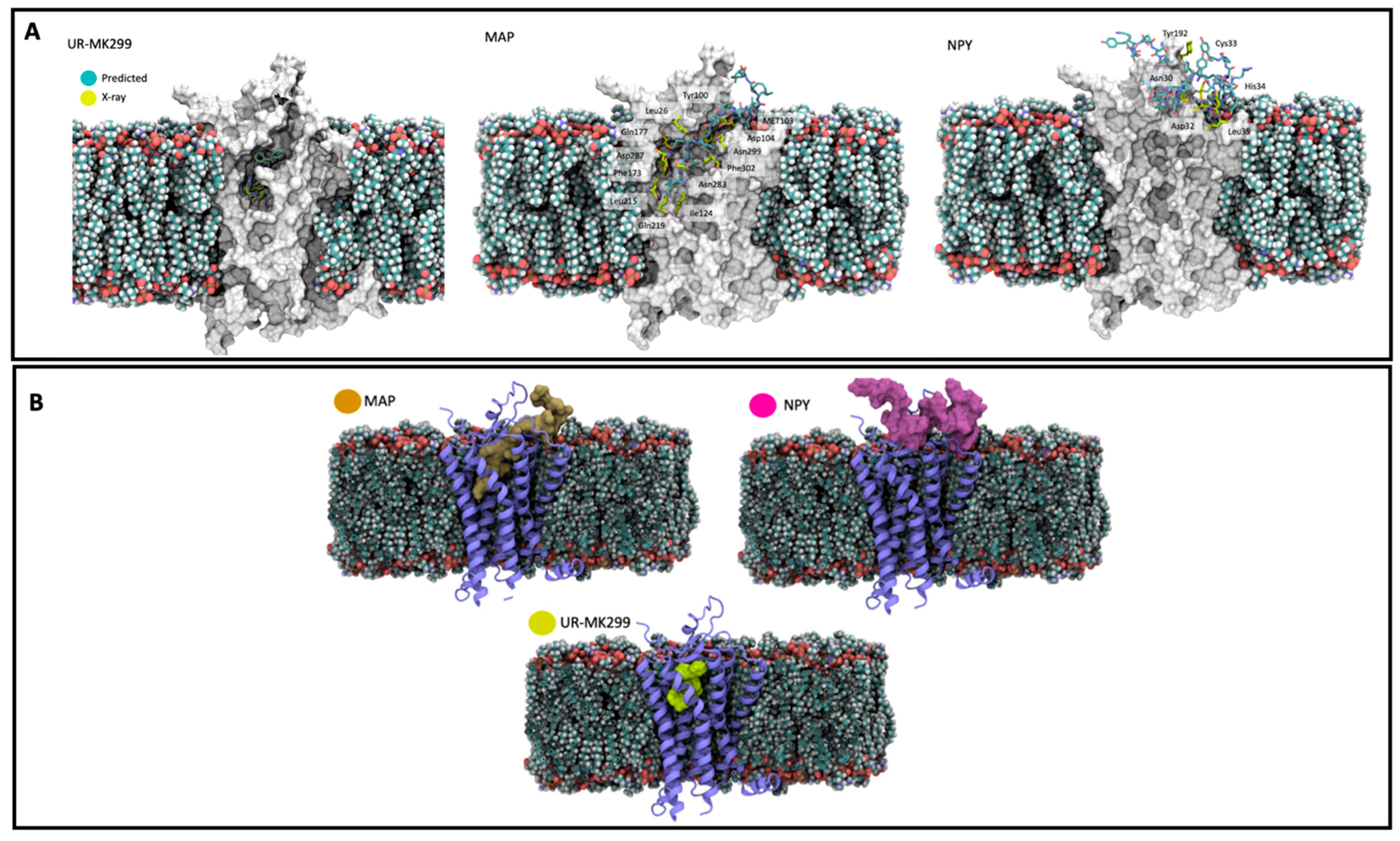
5. In Silico Studies Involving MAPs for New Directions in Biomedicine
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lindgren, M.; Langel, Ü. Classes and Prediction of Cell-Penetrating Peptides. In Methods in Molecular Biology (Clifton, N.J.); Humana Press: Totowa, NJ, USA, 2011; Volume 683, pp. 3–19. ISBN 9781607619192. [Google Scholar]
- Frankel, A.D.; Pabo, C.O.; Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Vivès, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Bi, Y.; Zhang, H.; Dong, S.; Teng, L.; Lee, R.J.; Yang, Z. Cell-Penetrating Peptides in Diagnosis and Treatment of Human Diseases: From Preclinical Research to Clinical Application. Front. Pharmacol. 2020, 11, 697. [Google Scholar] [CrossRef]
- Borrelli, A.; Tornesello, A.L.; Tornesello, M.L.; Buonaguro, F.M. Cell penetrating peptides as molecular carriers for anti-cancer agents. Molecules 2018, 23, 295. [Google Scholar] [CrossRef] [Green Version]
- Mäger, I.; Eiríksdóttir, E.; Langel, K.; EL Andaloussi, S.; Langel, Ü. Assessing the uptake kinetics and internalization mechanisms of cell-penetrating peptides using a quenched fluorescence assay. Biochim. Biophys. Acta-Biomembr. 2010, 1798, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Kerth, A.; Erbe, A.; Dathe, M.; Blume, A. Infrared reflection absorption spectroscopy of amphipathic model peptides at the air/water interface. Biophys. J. 2004, 86, 3750–3758. [Google Scholar] [CrossRef] [Green Version]
- Fotin-Mleczek, M.; Welte, S.; Mader, O.; Duchardt, F.; Fischer, R.; Hufnagel, H.; Scheurich, P.; Brock, R. Cationic cell-penetrating peptides interfere with TNF signalling by induction of TNF receptor internalization. J. Cell Sci. 2005, 118, 3339–3351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reissmann, S. Cell penetration: Scope and limitations by the application of cell-penetrating peptides. J. Pept. Sci. 2014, 20, 760–784. [Google Scholar] [CrossRef]
- Melikov, K.; Chernomordik, L.V. Arginine-rich cell penetrating peptides: From endosomal uptake to nuclear delivery. Cell. Mol. Life Sci. 2005, 62, 2739–2749. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Vives, E.; Ramos, C.; Verbeure, B.; Gait, M.J.; Chernomordik, L.V.; Lebleu, B. Cell-penetrating peptides: A reevaluation of the mechanism of cellular uptake. J. Biol. Chem. 2003, 278, 585–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, R.; Fotin-Mleczek, M.; Hufnagel, H.; Brock, R. Break on through to the Other Side—Biophysics and Cell Biology Shed Light on Cell-Penetrating Peptides. ChemBioChem 2005, 6, 2126–2142. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, W.B.; Fuselier, T.; He, J.; Wimley, W.C. Mechanism Matters: A Taxonomy of Cell Penetrating Peptides. Trends Biochem. Sci. 2015, 40, 749–764. [Google Scholar] [CrossRef] [Green Version]
- Holm, T.; Langel, Ü.; El-Andaloussi, S.; Holm, T.; Langel, U. Cell-Penetrating Peptides: Mechanisms and Applications. Curr. Pharm. Des. 2005, 11, 3597–3611. [Google Scholar] [CrossRef]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.W.; Christison, R.; Bundell, K.; Voyce, C.J.; Brockbank, S.M.V.; Newham, P.; Lindsay, M.A. Characterisation of cell-penetrating peptide-mediated peptide delivery. Br. J. Pharmacol. 2005, 145, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Gazit, E.; Shai, Y.; Lee, W.J.; Brey, P.T. Mode of Action of the Antibacterial Cecropin B2: A Spectrofluorometric Study. Biochemistry 1994, 33, 10681–10692. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef]
- Kaplan, I.M.; Wadia, J.S.; Dowdy, S.F. Cationic TAT peptide transduction domain enters cells by macropinocytosis. J. Control. Release 2005, 102, 247–253. [Google Scholar] [CrossRef]
- Lindgren, M.E.; Hallbrink, M.M.; Elmquist, A.M.; Langel, U.; Hällbrink, M.M.; Elmquist, A.M.; Langel, U. Passage of cell-penetrating peptides across a human epithelial cell layer in vitro. Biochem. J. 2004, 377, 69–76. [Google Scholar] [CrossRef]
- Yamashita, H.; Kato, T.; Oba, M.; Misawa, T.; Hattori, T.; Ohoka, N.; Tanaka, M.; Naito, M.; Kurihara, M.; Demizu, Y. Development of a Cell-penetrating Peptide that Exhibits Responsive Changes in its Secondary Structure in the Cellular Environment. Sci. Rep. 2016, 6, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitz, T.; Iten, R.; Gardiner, J.; Namoto, K.; Walde, P.; Seebach, D. Interaction of α-and β-oligoarginine-acids and amides with anionic lipid vesicles: A mechanistic and thermodynamic study. Biochemistry 2006, 45, 5817–5829. [Google Scholar] [CrossRef]
- Snyder, E.L.; Dowdy, S.F. Cell Penetrating Peptides in Drug Delivery; Springer: Berlin/Heidelberg, Germany, 2004; Volume 21, pp. 389–393. [Google Scholar]
- Sakurai, Y.; Hatakeyama, H.; Sato, Y.; Akita, H.; Takayama, K.; Kobayashi, S.; Futaki, S.; Harashima, H. Endosomal escape and the knockdown efficiency of liposomal-siRNA by the fusogenic peptide shGALA. Biomaterials 2011, 32, 5733–5742. [Google Scholar] [CrossRef] [Green Version]
- Cameron, A.; Appel, J.; Houghten, R.A.; Lindberg, I. Polyarginines Are Potent Furin Inhibitors*. J. Biol. Chem. 2000, 275, 36741–36749. [Google Scholar] [CrossRef] [Green Version]
- Kuo, J.S.; Jan, M.; Lin, Y.-L.; Lin, C. Interactions between octaarginine and U-937 human macrophages: Global gene expression profiling, superoxide anion content, and cytokine production. J. Control. Release 2009, 139, 197–204. [Google Scholar] [CrossRef]
- Ward, B.; Seal, B.L.; Brophy, C.M.; Panitch, A. Design of a bioactive cell-penetrating peptide: When a transduction domain does more than transduce. J. Pept. Sci. 2009, 15, 668–674. [Google Scholar] [CrossRef] [Green Version]
- Kilk, K.; Mahlapuu, R.; Soomets, U.; Langel, Ü. Analysis of in vitro toxicity of five cell-penetrating peptides by metabolic profiling. Toxicology 2009, 265, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Moutal, A.; François-Moutal, L.; Brittain, J.M.; Khanna, M.; Khanna, R. Differential neuroprotective potential of CRMP2 peptide aptamers conjugated to cationic, hydrophobic, and amphipathic cell penetrating peptides. Front. Cell. Neurosci. 2015, 8, 471. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Carneado, J.; Kogan, M.J.; Pujals, S.; Giralt, E. Amphipathic peptides and drug delivery. Biopolymers 2004, 76, 196–203. [Google Scholar] [CrossRef]
- Steiner, V.; Schär, M.; Börnsen, K.O.; Mutter, M. Retention behaviour of a template-assembled synthetic protein and its amphiphilic building blocks on reversed-phase columns. J. Chromatogr. A 1991, 586, 43–50. [Google Scholar] [CrossRef]
- Deli, M.A. Potential use of tight junction modulators to reversibly open membranous barriers and improve drug delivery. Biochim. Biophys. Acta-Biomembr. 2009, 1788, 892–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-C.; Eiting, K.; Cui, K.; Leonard, A.K.; Morris, D.; Li, C.-Y.; Farber, K.; Sileno, A.P.; Houston, M.E.; Johnson, P.H.; et al. Therapeutic utility of a novel tight junction modulating peptide for enhancing intranasal drug delivery. J. Pharm. Sci. 2006, 95, 1364–1371. [Google Scholar] [CrossRef]
- Johnson, P.H.; Quay, S.C. Advances in nasal drug delivery through tight junction technology. Expert Opin. Drug Deliv. 2005, 2, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Deshayes, S.; Morris, M.C.; Divita, G.; Heitz, F. Cell-penetrating peptides: Tools for intracellular delivery of therapeutics. Cell. Mol. Life Sci. 2005, 62, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Bocsik, A.; Gróf, I.; Kiss, L.; Ötvös, F.; Zsíros, O.; Daruka, L.; Fülöp, L.; Vastag, M.; Kittel, Á.; Imre, N.; et al. Dual action of the PN159/KLAL/MAP peptide: Increase of drug penetration across caco-2 intestinal barrier model by modulation of tight junctions and plasma membrane permeability. Pharmaceutics 2019, 11, 73. [Google Scholar] [CrossRef] [Green Version]
- Janek, K.; Rothemund, S.; Gast, K.; Beyermann, M.; Zipper, J.; Fabian, H.; Bienert, M.; Krause, E. Study of the Conformational Transition of Aβ(1−42) Using D-Amino Acid Replacement Analogues. Biochemistry 2001, 40, 5457–5463. [Google Scholar] [CrossRef]
- Oehlke, J.; Scheller, A.; Janek, K.; Wiesner, B.; Krause, E.; Beyermann, M.; Bienert, M. Rapid translocation of amphipathic βhelical and β-sheet-forming peptides through plasma membranes of endothelial cells. In Peptide Science—Present and Future; Shimonishi, Y., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2006; pp. 782–783. ISBN 978-0-306-46864-3. [Google Scholar]
- Sarko, D.; Beijer, B.; Boy, R.G.; Nothelfer, E.M.; Leotta, K.; Eisenhut, M.; Altmann, A.; Haberkorn, U.; Mier, W. The pharmacokinetics of cell-penetrating peptides. Mol. Pharm. 2010, 7, 2224–2231. [Google Scholar] [CrossRef]
- Scheller, A.; Oehlke, J.; Wiesner, B.; Dathe, M.; Krause, E.; Beyermann, M.; Melzig, M.; Bienert, M. Structural requirements for cellular uptake of α-helical amphipathic peptides. J. Pept. Sci. 1999, 5, 185–194. [Google Scholar] [CrossRef]
- Oehlke, J.; Krause, E.; Wiesner, B.; Beyermann, M.; Bienert, M. Extensive cellular uptake into endothelial cells of an amphipathic β-sheet forming peptide. FEBS Lett. 1997, 415, 196–199. [Google Scholar] [CrossRef] [Green Version]
- Oehlke, J.; Lorenz, D.; Wiesner, B.; Bienert, M. Studies on the cellular uptake of substance P and lysine-rich, KLA-derived model peptides. J. Mol. Recognit. 2005, 18, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Scheller, A.; Wiesner, B.; Melzig, M.; Bienert, M.; Oehlke, J. Evidence for an amphipathicity independent cellular uptake of amphipathic cell-penetrating peptides. Eur. J. Biochem. 2000, 267, 6043–6050. [Google Scholar] [CrossRef] [Green Version]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Oehlke, J.; Scheller, A.; Wiesner, B.; Krause, E.; Beyermann, M.; Klauschenz, E.; Melzig, M.; Bienert, M. Cellular uptake of an α-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim. Biophys. Acta-Biomembr. 1998, 1414, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Kenien, R.; Shen, W.C.; Zaro, J.L. Vesicle-to-cytosol transport of disulfide-linked cargo mediated by an amphipathic cell-penetrating peptide. J. Drug Target. 2012, 20, 793–800. [Google Scholar] [CrossRef]
- Bocsik, A.; Walter, F.R.; Gyebrovszki, A.; Fülöp, L.; Blasig, I.; Dabrowski, S.; Ötvös, F.; Tóth, A.; Rákhely, G.; Veszelka, S.; et al. Reversible Opening of Intercellular Junctions of Intestinal Epithelial and Brain Endothelial Cells with Tight Junction Modulator Peptides. J. Pharm. Sci. 2016, 105, 754–765. [Google Scholar] [CrossRef] [Green Version]
- Hällbrink, M.; Oehlke, J.; Papsdorf, G.; Bienert, M. Uptake of cell-penetrating peptides is dependent on peptide-to-cell ratio rather than on peptide concentration. Biochim. Biophys. Acta-Biomembr. 2004, 1667, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadhwani, P.; Bürck, J.; Strandberg, E.; Mink, C.; Afonin, S.; Ulrich, A.S. Using a Sterically Restrictive Amino Acid as a 19 F NMR label To Monitor and To Control Peptide Aggregation in Membranes. J. Am. Chem. Soc. 2008, 130, 16515–16517. [Google Scholar] [CrossRef]
- Strandberg, E.; Tiltak, D.; Ieronimo, M.; Kanithasen, N.; Wadhwani, P.; Ulrich, A.S. Influence of C-terminal amidation on the antimicrobial and hemolytic activities of cationic α-helical peptides. Pure Appl. Chem. 2007, 79, 717–728. [Google Scholar] [CrossRef]
- Pierigè, F.; Serafini, S.; Rossi, L.; Magnani, M. Cell-based drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 286–295. [Google Scholar] [CrossRef]
- Zorko, M.; Langel, U. Cell-penetrating peptides: Mechanism and kinetics of cargo delivery. Adv. Drug Deliv. Rev. 2005, 57, 529–545. [Google Scholar] [CrossRef]
- Oehlke, J.; Wallukat, G.; Wolf, Y.; Ehrlich, A.; Wiesner, B.; Berger, H.; Bienert, M. Enhancement of intracellular concentration and biological activity of PNA after conjugation with a cell-penetrating synthetic model peptide. Eur. J. Biochem. 2004, 271, 3043–3049. [Google Scholar] [CrossRef] [PubMed]
- Säälik, P.; Niinep, A.; Pae, J.; Hansen, M.; Lubenets, D.; Langel, Ü.; Pooga, M. Penetration without cells: Membrane translocation of cell-penetrating peptides in the model giant plasma membrane vesicles. J. Control. Release 2011, 153, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Pae, J.; Säälik, P.; Liivamägi, L.; Lubenets, D.; Arukuusk, P.; Langel, Ü.; Pooga, M. Translocation of cell-penetrating peptides across the plasma membrane is controlled by cholesterol and microenvironment created by membranous proteins. J. Control. Release 2014, 192, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Swiecicki, J.-M.; Bartsch, A.; Tailhades, J.; Di Pisa, M.; Heller, B.; Chassaing, G.; Mansuy, C.; Burlina, F.; Lavielle, S. The Efficacies of Cell-Penetrating Peptides in Accumulating in Large Unilamellar Vesicles Depend on their Ability To Form Inverted Micelles. ChemBioChem 2014, 15, 884–891. [Google Scholar] [CrossRef]
- Sakamoto, K.; Morishita, T.; Aburai, K.; Sakai, K.; Abe, M.; Nakase, I.; Futaki, S.; Sakai, H. Key Process and Factors Controlling the Direct Translocation of Cell-Penetrating Peptide through Bio-Membrane. Int. J. Mol. Sci. 2020, 21, 5466. [Google Scholar]
- Bhatia, T.; Husen, P.; Brewer, J.; Bagatolli, L.A.; Hansen, P.L.; Ipsen, J.H.; Mouritsen, O.G. Preparing giant unilamellar vesicles (GUVs) of complex lipid mixtures on demand: Mixing small unilamellar vesicles of compositionally heterogeneous mixtures. Biochim. Biophys. Acta-Biomembr. 2015, 1848, 3175–3180. [Google Scholar] [CrossRef] [Green Version]
- Kenien, R.; Zaro, J.L.; Shen, W.-C. MAP-mediated nuclear delivery of a cargo protein. J. Drug Target. 2012, 20, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Alves, C.; Sharma, V.; Lázaro, D.F.; Silva, S.; Gomes, P.; Outeiro, T.F. A new MAP-Rasagiline conjugate reduces α-synuclein inclusion formation in a cell model. Pharmacol. Rep. 2020, 72, 456–464. [Google Scholar] [CrossRef]
- Zemel, A.; Fattal, D.R.; Ben-Shaul, A. Energetics and Self-Assembly of Amphipathic Peptide Pores in Lipid Membranes. Biophys. J. 2003, 84, 2242–2255. [Google Scholar] [CrossRef] [Green Version]
- Dathe, M.; Meyer, J.; Beyermann, M.; Maul, B.; Hoischen, C.; Bienert, M. General aspects of peptide selectivity towards lipid bilayers and cell membranes studied by variation of the structural parameters of amphipathic helical model peptides. Biochim. Biophys. Acta-Biomembr. 2002, 1558, 171–186. [Google Scholar] [CrossRef] [Green Version]
- Leuschner, C.; Hansel, W. Membrane Disrupting Lytic Peptides for Cancer Treatments. Curr. Pharm. Des. 2005, 10, 2299–2310. [Google Scholar] [CrossRef] [PubMed]
- Saar, K.; Lindgren, M.; Hansen, M.; Eiríksdóttir, E.; Jiang, Y. Cell-penetrating peptides: A comparative membrane toxicity study. Anal. Biochem. 2005, 345, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Alves, C.; Duarte, D.; Costa, A.; Sarmento, B.; Almeida, A.J.; Gomes, P.; Vale, N. Model Amphipathic Peptide Coupled with Tacrine to Improve Its Antiproliferative Activity. Int. J. Mol. Sci. 2020, 22, 242. [Google Scholar] [CrossRef]
- Silva, S.; Marto, J.; Gonçalves, L.M.; Duarte, D.; Soares, O.S.G.P.; Vasques-Nóvoa, F.; Almeida, A.J.; Vale, N. New Peptide Functionalized Nanostructured Lipid Carriers with CNS Drugs and Evaluation Anti-proliferative Activity. Int. J. Mol. Sci. 2022, 23, 7109. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Almeida, A.; Vale, N. Combination of Cell-Penetrating Peptides with Nanoparticles for Therapeutic Application: A Review. Biomolecules 2019, 9, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaro, J.L.; Fei, L.; Shen, W.-C. Recombinant peptide constructs for targeted cell penetrating peptide-mediated delivery. J. Control. Release 2012, 158, 357–361. [Google Scholar] [CrossRef]
- Ward, C.; Meehan, J.; Gray, M.E.; Murray, A.F.; Argyle, D.J.; Kunkler, I.H.; Langdon, S.P. The impact of tumour pH on cancer progression: Strategies for clinical intervention. Explor. Target. Anti-Tumor Ther. 2020, 1, 71–100. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Herrero, E.; Fernández-Medarde, A. The reversed intra- and extracellular pH in tumors as a unified strategy to chemotherapeutic delivery using targeted nanocarriers. Acta Pharm. Sin. B 2021, 11, 2243–2264. [Google Scholar] [CrossRef] [PubMed]
- Silva, S.; Marto, J.; Gonçalves, L.M.; Fernandes, H.S.; Sousa, S.F.; Almeida, A.J.; Vale, N. Development of Neuropeptide Y and Cell-Penetrating Peptide MAP Adsorbed onto Lipid Nanoparticle Surface. Molecules 2022, 27, 2734. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M.F. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform. Chem. 2016, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Terstappen, G.C.; Reggiani, A. In silico research in drug discovery. Trends Pharmacol. Sci. 2001, 22, 23–26. [Google Scholar] [CrossRef]
- Mohan, V.; Gibbs, A.; Cummings, M.; Jaeger, E.; DesJarlais, R. Docking: Successes and Challenges. Curr. Pharm. Des. 2005, 11, 323–333. [Google Scholar] [CrossRef] [Green Version]
- McDonald, R.L.; Vaughan, P.F.T.; Beck-Sickinger, A.G.; Peers, C. Inhibition of Ca2+ channel currents in human neuroblastoma (SH-SY5Y) cells by neuropeptide Y and a novel cyclic neuropeptide Y analogue. Neuropharmacology 1995, 34, 1507–1514. [Google Scholar] [CrossRef]
- Croce, N.; Dinallo, V.; Ricci, V.; Federici, G.; Caltagirone, C.; Bernardini, S.; Angelucci, F. Neuroprotective effect of neuropeptide Y against beta-amyloid 25-35 toxicity in SH-SY5Y neuroblastoma cells is associated with increased neurotrophin production. Neurodegener. Dis. 2011, 8, 300–309. [Google Scholar] [CrossRef]
- Croce, N.; Ciotti, M.T.; Gelfo, F.; Cortelli, S.; Federici, G.; Caltagirone, C.; Bernardini, S.; Angelucci, F. Neuropeptide Y Protects Rat Cortical Neurons against β-Amyloid Toxicity and Re-establishes Synthesis and Release of Nerve Growth Factor. ACS Chem. Neurosci. 2012, 3, 312–318. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, S.P.; Williams, J.A. Structural characterization of Y1 and Y2 receptors for neuropeptide Y and peptide YY by affinity cross-linking. J. Biol. Chem. 1990, 265, 8304–8310. [Google Scholar] [CrossRef]
- Regberg, J.; Srimanee, A.; Erlandsson, M.; Sillard, R.; Dobchev, D.A.; Karelson, M.; Langel, Ü. Rational design of a series of novel amphipathic cell-penetrating peptides. Int. J. Pharm. 2014, 464, 111–116. [Google Scholar] [CrossRef]
- Wada, S.; Tsuda, H.; Okada, T.; Urata, H. Cellular uptake of Aib-containing amphipathic helix peptide. Bioorg. Med. Chem. Lett. 2011, 21, 5688–5691. [Google Scholar] [CrossRef]
- Wada, S.; Hashimoto, Y.; Kawai, Y.; Miyata, K.; Tsuda, H.; Nakagawa, O.; Urata, H. Effect of Ala replacement with Aib in amphipathic cell-penetrating peptide on oligonucleotide delivery into cells. Bioorg. Med. Chem. 2013, 21, 7669–7673. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, S.; Kurrikoff, K.; Langel, Ü.; Almeida, A.J.; Vale, N. A Second Life for MAP, a Model Amphipathic Peptide. Int. J. Mol. Sci. 2022, 23, 8322. https://doi.org/10.3390/ijms23158322
Silva S, Kurrikoff K, Langel Ü, Almeida AJ, Vale N. A Second Life for MAP, a Model Amphipathic Peptide. International Journal of Molecular Sciences. 2022; 23(15):8322. https://doi.org/10.3390/ijms23158322
Chicago/Turabian StyleSilva, Sara, Kaido Kurrikoff, Ülo Langel, António J. Almeida, and Nuno Vale. 2022. "A Second Life for MAP, a Model Amphipathic Peptide" International Journal of Molecular Sciences 23, no. 15: 8322. https://doi.org/10.3390/ijms23158322
APA StyleSilva, S., Kurrikoff, K., Langel, Ü., Almeida, A. J., & Vale, N. (2022). A Second Life for MAP, a Model Amphipathic Peptide. International Journal of Molecular Sciences, 23(15), 8322. https://doi.org/10.3390/ijms23158322