
Microglia as Therapeutic Target for Radiation-Induced Brain Injury



Abstract
:1. Introduction
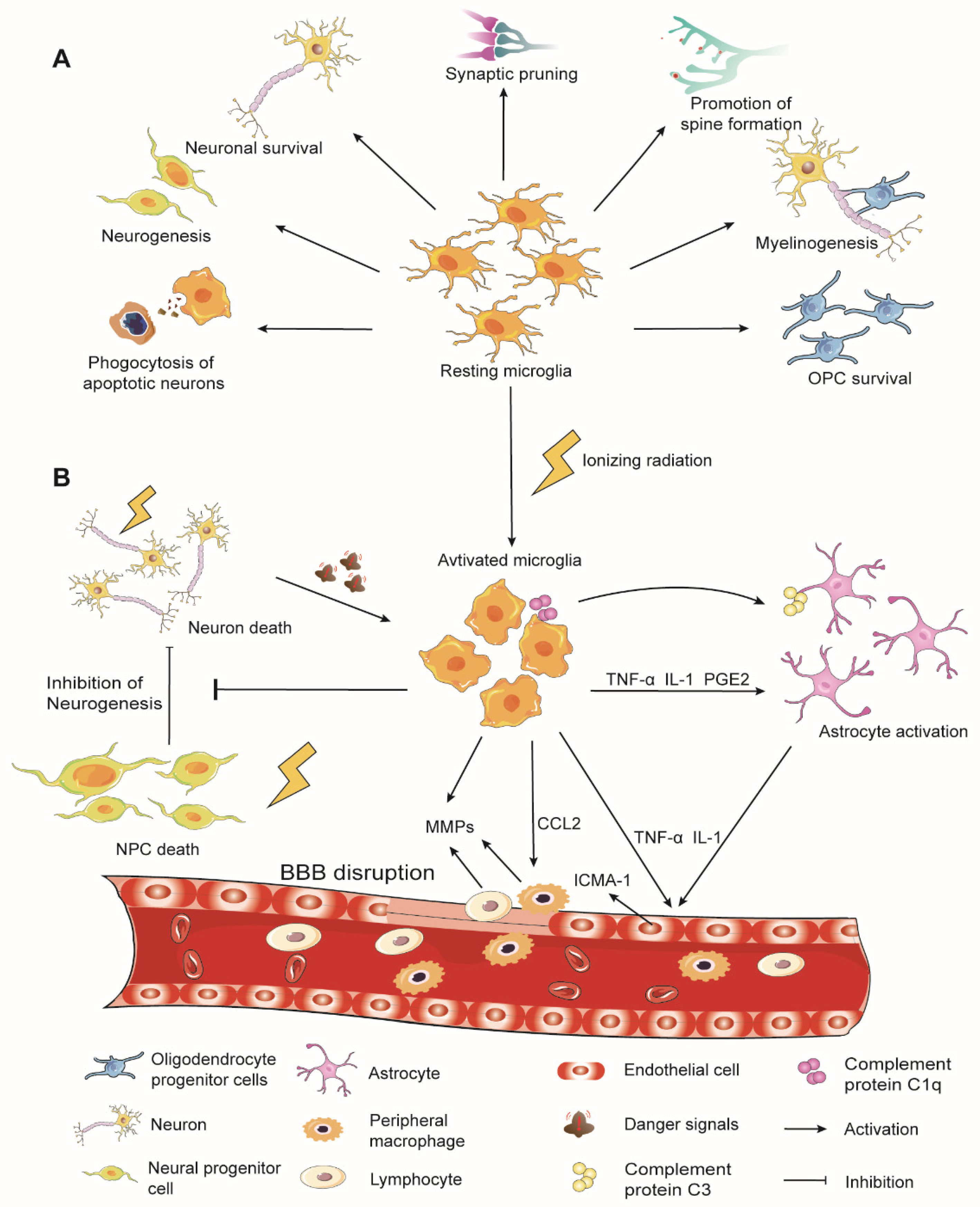
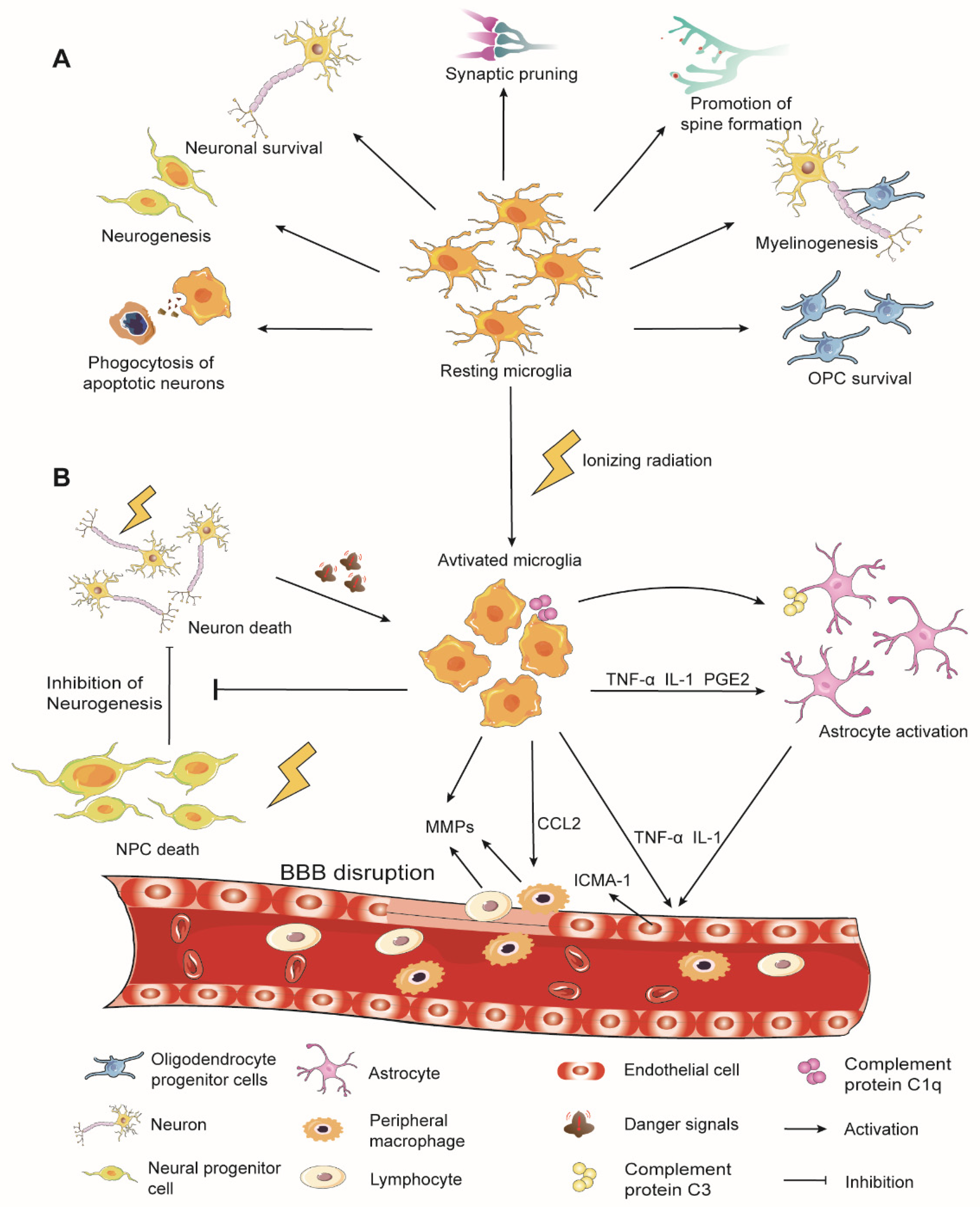
2. Overview of Microglia Physiological Functions
3. Microglia in Radiation-Induced Brain Injury
3.1. Microglial Activation
3.2. ROS/RNS Production and Oxidative Stress
3.3. Regulation of BBB Integrity
3.4. Immune Cell Infiltration in the Brain
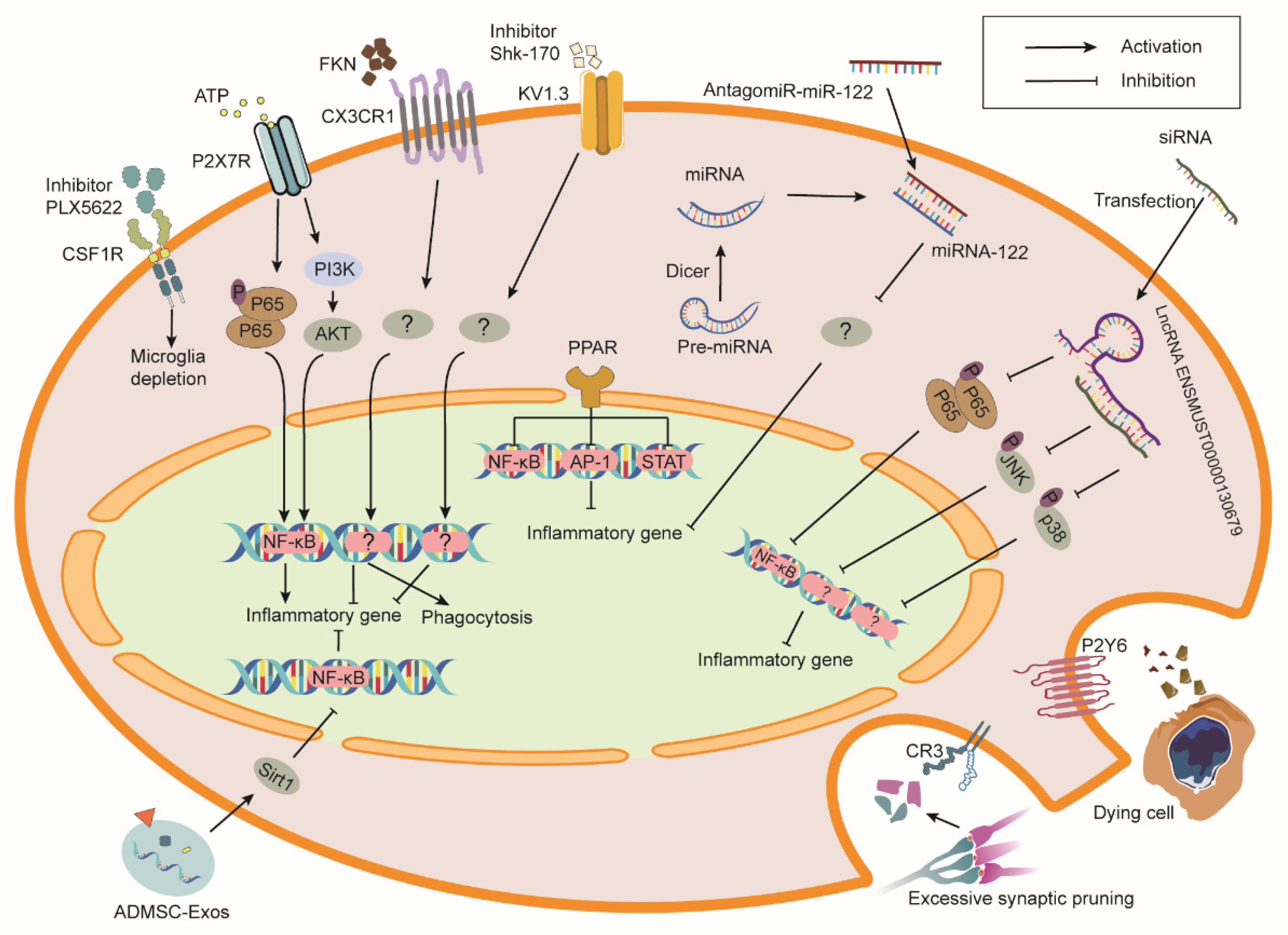
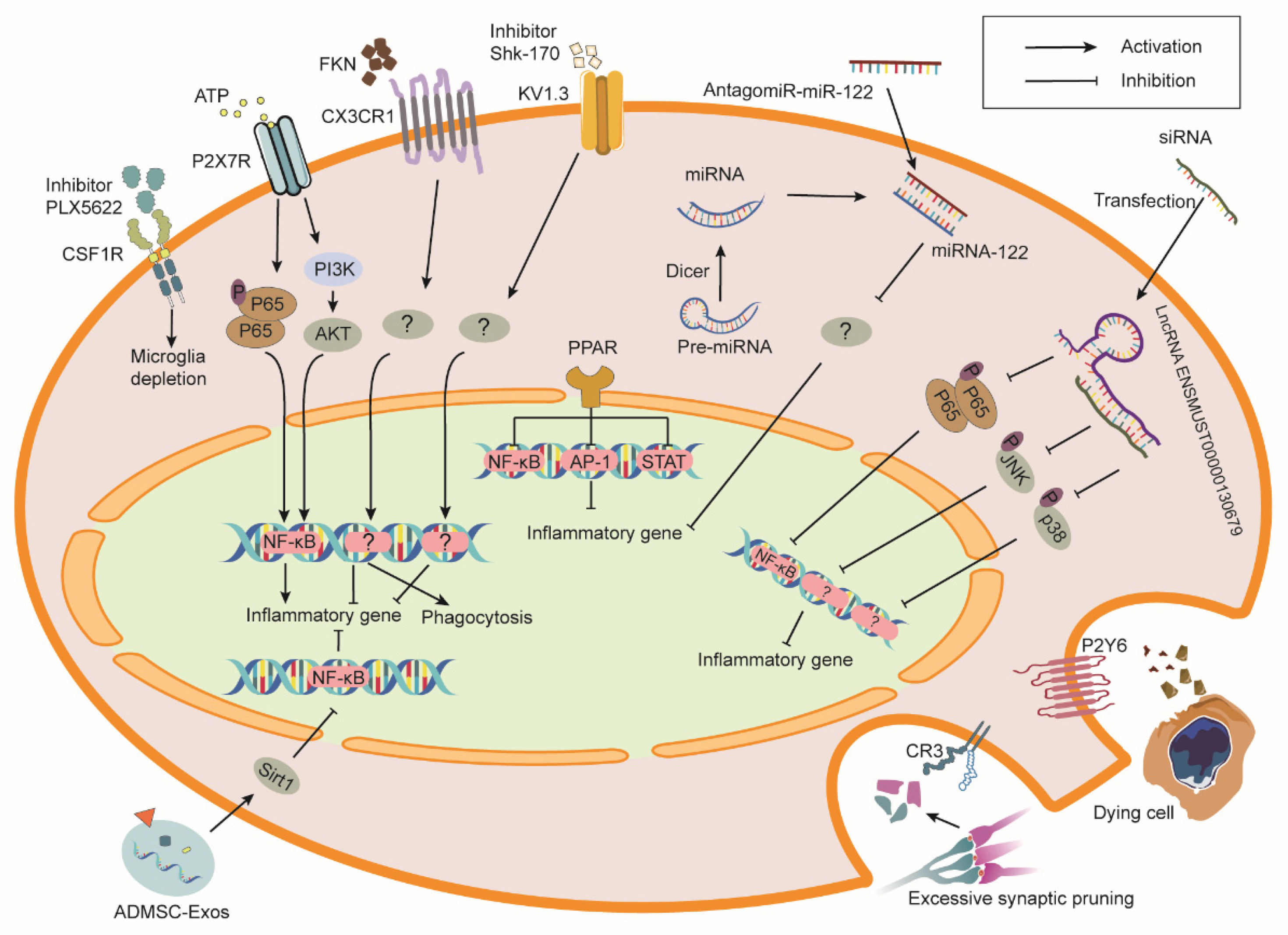
4. Modulation of Microglia for RIBI Therapy
4.1. Colony Stimulating Factor 1 Receptor (CSF1R)
4.2. Complement Receptors and Complement Components
4.3. Purinergic Receptors
4.4. CX3CR1
4.5. Peroxisome Proliferator-Activated Receptors (PPARs)
4.6. Kv1.3 Channel
4.7. MicroRNAs (miRNAs)
4.8. Long Non-Coding RNAs (lncRNAs)
4.9. Extracellular Vesicles (EVs)

{kind=link}
{kind=link}
| Targets | Animal/Cell Model | Source Dose and Dose Rate | Irradiated Site | Time Point after Radiation | Intervention Effect in Irradiation Models | Reference |
|---|---|---|---|---|---|---|
| CSFR1 | C57BL/6J mouse | X-ray with 9 cGy (1.10 Gy/min) | whole brain | 3 days, 2 weeks, 6 weeks | CSFR1 inhibition reduces the increase in mRNA of inflammation markers (TLR9, SYK, CCL6, CD14, CLECL5a, TSLP, CCL5) and the number of activated microglia in hippocampus and ameliorates cognitive dysfunction. | [141] |
| 4He particles with 30 cGy (15–25 cGy/min) | 4–6 weeks | CSFR1 inhibition ameliorates cognitive dysfunction, reduces activated microglia population, and attenuates the increase in PSD-95 puncta but does not affect morphologic and electrophysiologic features of neurons. | [143] | |||
| 4He particles with 15 cGy (16.37 cGy/min) 50 cGy (16.95 cGy/min) 100 cGy (18.07 cGy/min) | 18–21 days and 90–100 days | CSFR1 inhibition improves long-term cognitive impairment and inflammatory response, decreases C5aR and LAMP-1, and increases synapsin-1. | [144] | |||
| γ ray with three fractions of 3.3 Gy | 1, 3 months | CSFR1 blockade reduces the numbers of activated microglia, suppresses monocyte accumulation in brain, and ameliorates cognitive dysfunction. | [142] | |||
| C1q | C57BL/6 mouse | γ-ray with 9 Gy (1.2 Gy/min) | whole brain | 2, 24, 48 h; 1, 2, 3, 4 weeks | Deletion of C1q in microglia protects synaptic loss and reduces activation of microglia and astrocytes, as well as protein levels of TNF-a, IL-1ß, IL-6, IL-1α, CCL2, IL-18, and TLR4. | [71] |
| C3 | C57BL/6 mouse | X-ray with 8 Gy (2.3 Gy/min) | whole brain | 6 h; 7 days; 2, 3, 4 weeks | C3 knockout improves task performance and increases activated microglia and proliferating cells in the granule cell layer. | [165] |
| C3R | C57BL/6J mouse | γ-ray with 10 Gy (1.17 Gy/min) | whole brain | 30 days | CR3 blockade ameliorates behavior deficits in novel object recognition and the Lashley III maze, prevents dendritic spine loss, and increases CD11-positive microglia in hippocampus. | [166] |
| 30, 45 days | CR3 knockout prevents dendritic spine loss and increases activated microglia in hippocampus. | [145] | ||||
| P2Y6 | Balb/c mouse | β-ray with 30 Gy (3 Gy/min) | whole brain | 1, 14, 30 days | P2Y6 receptor antagonism suppresses phagocytosis of irradiated microglia and increases the number of apoptotic neurons. | [182] |
| Primary microglia | β-ray with 8 Gy | 4, 12, 48 h | P2Y6 receptor antagonism suppresses phagocytosis of irradiated microglia and has no effect on the production of inflammatory mediators (TNF-α, IL-1β, IL-6, iNOS). | [182] | ||
| P2X7 | Balb/c mouse | β-ray with 30 Gy (3 Gy/min) | whole brain | 3, 7, 14 days; 8 weeks | P2X7R blockade reduces the activated microglia population and neuron loss in the cortex. | [69] |
| Primary microglia | β-ray with 10 Gy (6 MeV/min) | 24, 48 h | P2X7R blockade reduces the activated microglia population and mRNA expression levels of IL-6, TNF-α, and COX-2. | [69] | ||
| CX3CR1 | C57BL/6J mouse | γ-ray with 10 Gy (2 Gy/min) | whole brain | 3, 6, 12, 24, 48, 72 h; 1, 2, 4 weeks | FKN overexpression promotes M2 phenotypic polarization, reverses the reduced neural stem cell in hippocampus, decreases the TNF-α level, and increases the IL-10 level in the blood. | [20] |
| BV-2 | γ-ray with 10 Gy (2.0 Gy/min) | 1.5, 6 h | FKN promotes microglial phagocytosis and M2 polarization, decreases TNF-α and IL-1β mRNA levels, and increases IL-10 mRNA levels. CX3CR1 knockdown reverses these effects. | [20] | ||
| PPARα | BV-2 | γ-ray with 10 Gy (4.0 Gy/min) | 1, 3, 7, 12, 24 h | PPARα activation prevents the increase in IL-1, and TNF-α mRNA levels, and COX-2 protein via inhibition of p65 translocation and jun phosphorylation. | [83] | |
| 129S1/SvImJ mouse | γ-ray with 10 Gy (3.33 Gy/min) | whole brain | 1 week, 2 months | PPARα activation promotes newborn neuron survival and prevents microglial activation. PPARα knockout abolishes the neuroprotection of fenofibrate. | [201] | |
| Fischer 344 × Brown Norway rats | γ-ray with four fractions of 10 Gy (4 Gy/min) | whole brain | 26, 29 weeks | PPARα activation prevents perirhinal cortex-dependent cognitive impairment without a decrease in microglial activation and an increase in immature neurons. | [78] | |
| PPARδ | BV-2 | γ-ray with 10 Gy (3.56 Gy/min) | 30 min; 7, 24 h | PPARδ activation downregulates ROS production, IL-1 and TNF-α expression, and COX-2 and MCP-1 proteins by inhibiting NF-κB and PKCα/MEK1/2/ERK1/2/AP pathways. | [18] | |
| C57BL/6J | γ-ray with 10 Gy (5 Gy/min) | whole brain | 3 h; 1, 2 weeks | PPARδ activation prevents the increase in IL-1 gene expression and pERK protein but does not rescue neurogenesis and hippocampal-dependent cognitive impairment. | [203] | |
| PPARγ | Fischer 344 rat | γ-ray with nine fractions of 5 Gy (4.41 Gy/min) | whole brain | 50, 54 weeks | PPARγ activation prevents cognitive impairment. | [206] |
| Kv 1.3 | Balb/c mouse | ß-ray with 30 Gy (3 Gy/min) | whole brain | 3, 14 days; 8 weeks | Kv 1.3 blockade prevents neuronal loss and increases activated microglial in hippocampus and cerebral cortex and improves spatial learning and cerebral cortex atrophy in mice. | [74] |
| BV-2 | ß-ray with 10 Gy (3 Gy/min) | 4, 12 h; 1, 2 days | Kv 1.3 blockade or knockdown decreases protein and mRNA level of TNF-α, IL-6, and COX-2 in microglia and inhibits apoptosis of co-cultured primary hippocampal neurons. | [74] | ||
| miR-124 | C57BL/6J mouse | γ-ray with 10 Gy (2.07 Gy/min) | whole brain | 5 weeks | miR-124 overexpression prevents microglia activation and ameliorates cognitive impairment. | [231] |
| miR-741-3p | C57BL/6J mouse | ß-ray with 30 Gy (2.5 Gy/min) | whole brain | 1, 6 weeks | miR-741-3p inhibition resists cognitive dysfunction, hippocampal neuronal injury, and microglia activation and decreases the expression level of IL-6 and TNF-a. | [229] |
| miR-122-5p | C57BL/6J mouse | ß-ray with 30 Gy (3 Gy/min) | whole brain | 6 weeks, 48–50 days | miR-122-5p inhibition prevents cognitive impairment, neuronal damage, microglia activation, and production of TNF-a, IL-6, and IL-1ß in hippocampus. | [230] |
| BV-2 | ß-ray with 10 Gy | 8, 24 h | miR-122-5p inhibition alleviates the decrease in cell viability and increase in the release of TNF-a, IL-6, and IL-1ß in BV2; restores BV2 branching morphogenesis and phagocytosis; and reduces co-cultured SH-SY5Y cell apoptosis. | [230] | ||
| lncRNA ENSMUST00000130679 | BV-2 | X-ray with 10 Gy (2 Gy/min) | 1, 24 h | lncRNA ENSMUST00000130679 knockdown suppresses DDR; phosphorylation of p65, JNK, and p38; and release of TNF-a, IL-6, and IL-1ß in BV2. | [66] | |
| lncRNA ENSMUST00000190863 | BV-2 | X-ray with 10 Gy (2 Gy/min) | 1, 24 h | lncRNA ENSMUST00000190863 knockdown suppresses DDR, phosphorylation of p65, and release of TNF-a in BV2. | [66] | |
| hNSC-derived MV | athymic nude rats | X-ray with 10 Gy (1 Gy/min) | whole brain | 4–7 weeks | MV transplantation into the bilateral hippocampus reduces the number of activated microglia in the hippocampus, neocortex (layer II/III), and amygdala; recovers the complexity of neuronal architecture; and ameliorates cognitive impairment. | [244] |
| 1 month | MV transplantation into the unilateral hippocampus reduces the number of activated microglia in the ipsilateral hippocampus; bilateral or unilateral transplantation increases GDNF and restores PSD-95 protein level in bilateral hippocampus; neither bilateral nor unilateral transplantation protects dendritic spine density. | [243] | ||||
| hNSC-derived EV | C57BL/6J mouse | γ-ray with 10 Gy (2.07 Gy/minute) | whole brain | 5 weeks, 6 months | EV transplantation into the bilateral hippocampus prevents microglia activation in the hippocampus and ameliorates cognitive impairment. | [231] |
| ADMSC-Exos | Sprague–Dawley rats | γ-ray with 30 Gy (1.59 Gy/min) | whole brain | 24 h; 3, 7 days | Tail vein injection pf ADMSC-Exos decreases the levels of caspase-3, MDA, 8-OHdG, TNF-α, IL-4, and SIRT1 and promotes recovery of SOD, CAT, IL-4, and IL-10 levels and suppresses microglial infiltration. | [73] |
| primary microglia | γ-ray with 30 Gy (3 MeV/min) | 24 h | Tail vein injection of ADMSC-Exos decreases the levels of caspase-3, MDA, 8-OHdG, TNF-α, IL-4, and SIRT1 and promotes the recovery of SOD, CAT, IL-4, and IL-10 levels and suppresses microglial activation. The above effects of ADMSC-Exos are inhibited by the SIRT-1 inhibitor EX527. | [73] |
5. Conclusions and Open Questions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ali, F.S.; Arevalo, O.; Zorofchian, S.; Patrizz, A.; Riascos, R.; Tandon, N.; Blanco, A.; Ballester, L.Y.; Esquenazi, Y. Cerebral Radiation Necrosis: Incidence, Pathogenesis, Diagnostic Challenges, and Future Opportunities. Curr. Oncol. Rep. 2019, 21, 66. [Google Scholar] [CrossRef] [PubMed]
- Greene-Schloesser, D.; Robbins, M.E. Radiation-induced cognitive impairment--from bench to bedside. Neuro Oncol. 2012, 14 (Suppl. S4), iv37–iv44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hladik, D.; Tapio, S. Effects of ionizing radiation on the mammalian brain. Mutat Res. Rev. Mutat Res. 2016, 770, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.D.; Jaeckle, K.; Ballman, K.V.; Farace, E.; Cerhan, J.H.; Anderson, S.K.; Carrero, X.W.; Barker, F.G., 2nd; Deming, R.; Burri, S.H.; et al. Effect of Radiosurgery Alone vs Radiosurgery With Whole Brain Radiation Therapy on Cognitive Function in Patients With 1 to 3 Brain Metastases: A Randomized Clinical Trial. JAMA 2016, 316, 401–409. [Google Scholar] [CrossRef]
- Chang, E.L.; Wefel, J.S.; Hess, K.R.; Allen, P.K.; Lang, F.F.; Kornguth, D.G.; Arbuckle, R.B.; Swint, J.M.; Shiu, A.S.; Maor, M.H.; et al. Neurocognition in patients with brain metastases treated with radiosurgery or radiosurgery plus whole-brain irradiation: A randomised controlled trial. Lancet Oncol. 2009, 10, 1037–1044. [Google Scholar] [CrossRef]
- Turnquist, C.; Harris, B.T.; Harris, C.C. Radiation-induced brain injury: Current concepts and therapeutic strategies targeting neuroinflammation. Neurooncol. Adv. 2020, 2, vdaa057. [Google Scholar] [CrossRef]
- Gutierrez-Quintana, R.; Walker, D.J.; Williams, K.J.; Forster, D.M.; Chalmers, A.J. Radiation-induced neuroinflammation: A potential protective role for poly(ADP-ribose) polymerase inhibitors? Neurooncol. Adv. 2022, 4, vdab190. [Google Scholar] [CrossRef]
- Gondi, V.; Hermann, B.P.; Mehta, M.P.; Tome, W.A. Hippocampal dosimetry predicts neurocognitive function impairment after fractionated stereotactic radiotherapy for benign or low-grade adult brain tumors. Int J. Radiat. Oncol. Biol. Phys. 2012, 83, e487–e493. [Google Scholar] [CrossRef] [Green Version]
- Florijn, M.A.; Sharfo, A.W.M.; Wiggenraad, R.G.J.; van Santvoort, J.P.C.; Petoukhova, A.L.; Hoogeman, M.S.; Mast, M.E.; Dirkx, M.L.P. Lower doses to hippocampi and other brain structures for skull-base meningiomas with intensity modulated proton therapy compared to photon therapy. Radiother. Oncol. 2020, 142, 147–153. [Google Scholar] [CrossRef]
- Montay-Gruel, P.; Acharya, M.M.; Petersson, K.; Alikhani, L.; Yakkala, C.; Allen, B.D.; Ollivier, J.; Petit, B.; Jorge, P.G.; Syage, A.R.; et al. Long-term neurocognitive benefits of FLASH radiotherapy driven by reduced reactive oxygen species. Proc. Natl. Acad. Sci. USA 2019, 116, 10943–10951. [Google Scholar] [CrossRef] [Green Version]
- Montay-Gruel, P.; Acharya, M.M.; Goncalves Jorge, P.; Petit, B.; Petridis, I.G.; Fuchs, P.; Leavitt, R.; Petersson, K.; Gondre, M.; Ollivier, J.; et al. Hypofractionated FLASH-RT as an Effective Treatment against Glioblastoma that Reduces Neurocognitive Side Effects in Mice. Clin. Cancer Res. 2021, 27, 775–784. [Google Scholar] [CrossRef]
- Prinz, M.; Masuda, T.; Wheeler, M.A.; Quintana, F.J. Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation. Annu. Rev. Immunol. 2021, 39, 251–277. [Google Scholar] [CrossRef]
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007, 30, 596–602. [Google Scholar] [CrossRef]
- Hu, X.; Liou, A.K.; Leak, R.K.; Xu, M.; An, C.; Suenaga, J.; Shi, Y.; Gao, Y.; Zheng, P.; Chen, J. Neurobiology of microglial action in CNS injuries: Receptor-mediated signaling mechanisms and functional roles. Prog. Neurobiol. 2014, 119–120, 60–84. [Google Scholar] [CrossRef] [Green Version]
- Mott, R.T.; Ait-Ghezala, G.; Town, T.; Mori, T.; Vendrame, M.; Zeng, J.; Ehrhart, J.; Mullan, M.; Tan, J. Neuronal expression of CD22: Novel mechanism for inhibiting microglial proinflammatory cytokine production. Glia 2004, 46, 369–379. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Korimerla, N.; Wahl, D.R. A Complementary Strategy to Mitigate Radiation-Induced Cognitive Decline. Cancer Res. 2021, 81, 1635–1636. [Google Scholar] [CrossRef]
- Schnegg, C.I.; Kooshki, M.; Hsu, F.C.; Sui, G.; Robbins, M.E. PPARdelta prevents radiation-induced proinflammatory responses in microglia via transrepression of NF-kappaB and inhibition of the PKCalpha/MEK1/2/ERK1/2/AP-1 pathway. Free Radic. Biol. Med. 2012, 52, 1734–1743. [Google Scholar] [CrossRef] [Green Version]
- Bachiller, S.; Jimenez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Pan, H.; Lin, Z.; Xiong, C.; Wei, C.; Li, H.; Tong, F.; Dong, X. Neuroprotective Effect of Fractalkine on Radia-tion-induced Brain Injury Through Promoting the M2 Polarization of Microglia. Mol. Neurobiol. 2021, 58, 1074–1087. [Google Scholar] [CrossRef]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swinnen, N.; Smolders, S.; Avila, A.; Notelaers, K.; Paesen, R.; Ameloot, M.; Brone, B.; Legendre, P.; Rigo, J.M. Complex invasion pattern of the cerebral cortex bymicroglial cells during development of the mouse embryo. Glia 2013, 61, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Menassa, D.A.; Gomez-Nicola, D. Microglial Dynamics during Human Brain Development. Front. Immunol. 2018, 9, 1014. [Google Scholar] [CrossRef] [Green Version]
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep. 2017, 18, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Low, D.; Ginhoux, F. Recent advances in the understanding of microglial development and homeostasis. Cell Immunol. 2018, 330, 68–78. [Google Scholar] [CrossRef]
- Bruttger, J.; Karram, K.; Wortge, S.; Regen, T.; Marini, F.; Hoppmann, N.; Klein, M.; Blank, T.; Yona, S.; Wolf, Y.; et al. Genetic Cell Ablation Reveals Clusters of Local Self-Renewing Microglia in the Mammalian Central Nervous System. Immunity 2015, 43, 92–106. [Google Scholar] [CrossRef] [Green Version]
- Chitu, V.; Gokhan, S.; Nandi, S.; Mehler, M.F.; Stanley, E.R. Emerging Roles for CSF-1 Receptor and its Ligands in the Nervous System. Trends Neurosci. 2016, 39, 378–393. [Google Scholar] [CrossRef] [Green Version]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Holscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Tan, Y.L.; Yuan, Y.; Tian, L. Microglial regional heterogeneity and its role in the brain. Mol. Psychiatry 2020, 25, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Bottcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Vargas Aguilar, S.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; Zelada Gonzalez, F.; Perrin, P.; et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, aad8670. [Google Scholar] [CrossRef]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223.e10. [Google Scholar] [CrossRef] [Green Version]
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Moller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Kreisel, T.; Wolf, B.; Keshet, E.; Licht, T. Unique role for dentate gyrus microglia in neuroblast survival and in VEGF-induced activation. Glia 2019, 67, 594–618. [Google Scholar] [CrossRef]
- Ziv, Y.; Ron, N.; Butovsky, O.; Landa, G.; Sudai, E.; Greenberg, N.; Cohen, H.; Kipnis, J.; Schwartz, M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat. Neurosci. 2006, 9, 268–275. [Google Scholar] [CrossRef]
- De Lucia, C.; Rinchon, A.; Olmos-Alonso, A.; Riecken, K.; Fehse, B.; Boche, D.; Perry, V.H.; Gomez-Nicola, D. Microglia regulate hippocampal neurogenesis during chronic neurodegeneration. Brain Behav. Immun. 2016, 55, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Kohman, R.A.; DeYoung, E.K.; Bhattacharya, T.K.; Peterson, L.N.; Rhodes, J.S. Wheel running attenuates microglia proliferation and increases expression of a proneurogenic phenotype in the hippocampus of aged mice. Brain Behav. Immun. 2012, 26, 803–810. [Google Scholar] [CrossRef] [Green Version]
- Thored, P.; Heldmann, U.; Gomes-Leal, W.; Gisler, R.; Darsalia, V.; Taneera, J.; Nygren, J.M.; Jacobsen, S.E.; Ekdahl, C.T.; Kokaia, Z.; et al. Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia 2009, 57, 835–849. [Google Scholar] [CrossRef]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef]
- Meucci, O.; Fatatis, A.; Simen, A.A.; Miller, R.J. Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc. Natl. Acad. Sci. USA 2000, 97, 8075–8080. [Google Scholar] [CrossRef] [Green Version]
- Bachstetter, A.D.; Morganti, J.M.; Jernberg, J.; Schlunk, A.; Mitchell, S.H.; Brewster, K.W.; Hudson, C.E.; Cole, M.J.; Harrison, J.K.; Bickford, P.C.; et al. Fractalkine and CX 3 CR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol. Aging 2011, 32, 2030–2044. [Google Scholar] [CrossRef] [Green Version]
- Vukovic, J.; Colditz, M.J.; Blackmore, D.G.; Ruitenberg, M.J.; Bartlett, P.F. Microglia modulate hippocampal neural precursor activity in response to exercise and aging. J. Neurosci. 2012, 32, 6435–6443. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro Xavier, A.L.; Kress, B.T.; Goldman, S.A.; Lacerda de Menezes, J.R.; Nedergaard, M. A Distinct Population of Microglia Supports Adult Neurogenesis in the Subventricular Zone. J. Neurosci. 2015, 35, 11848–11861. [Google Scholar] [CrossRef]
- Riccomagno, M.M.; Kolodkin, A.L. Sculpting neural circuits by axon and dendrite pruning. Annu. Rev. Cell Dev. Biol 2015, 31, 779–805. [Google Scholar] [CrossRef] [Green Version]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991.e978. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Weinhard, L.; di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1228. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Yue, H.; Hu, Z.; Shen, Y.; Ma, J.; Li, J.; Wang, X.D.; Wang, L.; Sun, B.; Shi, P.; et al. Microglia mediate forgetting via complement-dependent synaptic elimination. Science 2020, 367, 688–694. [Google Scholar] [CrossRef]
- Basilico, B.; Pagani, F.; Grimaldi, A.; Cortese, B.; Di Angelantonio, S.; Weinhard, L.; Gross, C.; Limatola, C.; Maggi, L.; Ragozzino, D. Microglia shape presynaptic properties at developing glutamatergic synapses. Glia 2019, 67, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, Y.; Paolicelli, R.C.; Sforazzini, F.; Weinhard, L.; Bolasco, G.; Pagani, F.; Vyssotski, A.L.; Bifone, A.; Gozzi, A.; Ragozzino, D.; et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci. 2014, 17, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, A.; Wake, H.; Ishikawa, A.W.; Eto, K.; Shibata, K.; Murakoshi, H.; Koizumi, S.; Moorhouse, A.J.; Yoshimura, Y.; Nabekura, J. Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 2016, 7, 12540. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, P.T.; Dorman, L.C.; Pan, S.; Vainchtein, I.D.; Han, R.T.; Nakao-Inoue, H.; Taloma, S.E.; Barron, J.J.; Molofsky, A.B.; Kheirbek, M.A.; et al. Microglial Remodeling of the Extracellular Matrix Promotes Synapse Plasticity. Cell 2020, 182, 388–403.e315. [Google Scholar] [CrossRef]
- Lim, S.H.; Park, E.; You, B.; Jung, Y.; Park, A.R.; Park, S.G.; Lee, J.R. Neuronal synapse formation induced by microglia and interleukin 10. PLoS ONE 2013, 8, e81218. [Google Scholar]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., 3rd; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, A.N.; Appel, B. Microglia phagocytose myelin sheaths to modify developmental myelination. Nat. Neurosci. 2020, 23, 1055–1066. [Google Scholar] [CrossRef]
- Lloyd, A.F.; Miron, V.E. The pro-remyelination properties of microglia in the central nervous system. Nat. Rev. Neurol. 2019, 15, 447–458. [Google Scholar] [CrossRef]
- Hagemeyer, N.; Hanft, K.M.; Akriditou, M.A.; Unger, N.; Park, E.S.; Stanley, E.R.; Staszewski, O.; Dimou, L.; Prinz, M. Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol. 2017, 134, 441–458. [Google Scholar] [CrossRef] [Green Version]
- Voss, E.V.; Skuljec, J.; Gudi, V.; Skripuletz, T.; Pul, R.; Trebst, C.; Stangel, M. Characterisation of microglia during de- and remyelination: Can they create a repair promoting environment? Neurobiol. Dis. 2012, 45, 519–528. [Google Scholar] [CrossRef]
- Nemes-Baran, A.D.; White, D.R.; DeSilva, T.M. Fractalkine-Dependent Microglial Pruning of Viable Oligodendrocyte Progenitor Cells Regulates Myelination. Cell Rep. 2020, 32, 108047. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Jung, J.S.; Kim, T.H.; Lim, S.J.; Oh, E.S.; Kim, J.Y.; Ji, K.A.; Joe, E.H.; Cho, K.H.; Han, I.O. Ionizing radiation induces astrocyte gliosis through microglia activation. Neurobiol. Dis. 2006, 21, 457–467. [Google Scholar] [CrossRef]
- Lumniczky, K.; Szatmari, T.; Safrany, G. Ionizing Radiation-Induced Immune and Inflammatory Reactions in the Brain. Front. Immunol. 2017, 8, 517. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, Y.; Sejimo, Y.; Kurachi, M.; Ishizaki, Y.; Nakano, T.; Takahashi, A. X-ray irradiation induces disruption of the blood-brain barrier with localized changes in claudin-5 and activation of microglia in the mouse brain. Neurochem. Int. 2018, 119, 199–206. [Google Scholar] [CrossRef]
- Monje, M.L.; Mizumatsu, S.; Fike, J.R.; Palmer, T.D. Irradiation induces neural precursor-cell dysfunction. Nat. Med. 2002, 8, 955–962. [Google Scholar] [CrossRef]
- Xu, A.; Li, R.; Ren, A.; Jian, H.; Huang, Z.; Zeng, Q.; Wang, B.; Zheng, J.; Chen, X.; Zheng, N.; et al. Regulatory coupling between long noncoding RNAs and senescence in irradiated microglia. J. Neuroinflamm. 2020, 17, 321. [Google Scholar] [CrossRef]
- Jezkova, L.; Zadneprianetc, M.; Kulikova, E.; Smirnova, E.; Bulanova, T.; Depes, D.; Falkova, I.; Boreyko, A.; Krasavin, E.; Davidkova, M.; et al. Particles with similar LET values generate DNA breaks of different complexity and reparability: A high-resolution microscopy analysis of gammaH2AX/53BP1 foci. Nanoscale 2018, 10, 1162–1179. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Dong, J.H.; Huang, G.D.; Qu, X.F.; Wu, G.; Dong, X.R. NF-kappaB signaling modulates radiationinduced microglial activation. Oncol. Rep. 2014, 31, 2555–2560. [Google Scholar] [CrossRef]
- Xu, P.; Xu, Y.; Hu, B.; Wang, J.; Pan, R.; Murugan, M.; Wu, L.J.; Tang, Y. Extracellular ATP enhances radiation-induced brain injury through microglial activation and paracrine signaling via P2X7 receptor. Brain Behav. Immun. 2015, 50, 87–100. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Huang, H.; Liu, T.; Yang, T.; Yi, X. Exposure to X-rays Causes Depression-like Behaviors in Mice via HMGB1-mediated Pyroptosis. Neuroscience 2022, 481, 99–110. [Google Scholar] [CrossRef]
- Markarian, M.; Krattli, R.P., Jr.; Baddour, J.D.; Alikhani, L.; Giedzinski, E.; Usmani, M.T.; Agrawal, A.; Baulch, J.E.; Tenner, A.J.; Acharya, M.M. Glia-Selective Deletion of Complement C1q Prevents Radiation-Induced Cognitive Deficits and Neuroinflammation. Cancer Res. 2021, 81, 1732–1744. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.; Byrne, S.; Middleton, R.J.; Banati, R.B.; Liu, G.J. Control of Neuroinflammation through Radiation-Induced Microglial Changes. Cells 2021, 10, 2381. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yang, Y.; Zhao, B.; Yang, Y.; Wang, J.; Shen, K.; Yang, X.; Hu, D.; Zheng, G.; Han, J. Exosomes Derived From Adipose-Derived Mesenchymal Stem Cells Ameliorate Radiation-Induced Brain Injury by Activating the SIRT1 Pathway. Front. Cell Dev. Biol 2021, 9, 693782. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Lu, K.; Li, Z.; Zhao, Y.; Wang, Y.; Hu, B.; Xu, P.; Shi, X.; Zhou, B.; Pennington, M.; et al. Blockade of Kv1.3 channels ameliorates radiation-induced brain injury. Neuro Oncol. 2014, 16, 528–539. [Google Scholar] [CrossRef] [Green Version]
- Kalm, M.; Fukuda, A.; Fukuda, H.; Ohrfelt, A.; Lannering, B.; Bjork-Eriksson, T.; Blennow, K.; Marky, I.; Blomgren, K. Transient inflammation in neurogenic regions after irradiation of the developing brain. Radiat. Res. 2009, 171, 66–76. [Google Scholar] [CrossRef]
- Chen, H.; Chong, Z.Z.; De Toledo, S.M.; Azzam, E.I.; Elkabes, S.; Souayah, N. Delayed activation of human microglial cells by high dose ionizing radiation. Brain Res. 2016, 1646, 193–198. [Google Scholar] [CrossRef]
- Hong, J.H.; Chiang, C.S.; Campbell, I.L.; Sun, J.R.; Withers, H.R.; McBride, W.H. Induction of acute phase gene expression by brain irradiation. Int. J. Radiat. Oncol. Biol. Phys. 1995, 33, 619–626. [Google Scholar] [CrossRef]
- Greene-Schloesser, D.; Payne, V.; Peiffer, A.M.; Hsu, F.C.; Riddle, D.R.; Zhao, W.; Chan, M.D.; Metheny-Barlow, L.; Robbins, M.E. The peroxisomal proliferator-activated receptor (PPAR) alpha agonist, fenofibrate, prevents fractionated whole-brain irradiation-induced cognitive impairment. Radiat. Res. 2014, 181, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Jenrow, K.A.; Brown, S.L.; Lapanowski, K.; Naei, H.; Kolozsvary, A.; Kim, J.H. Selective inhibition of microglia-mediated neuroinflammation mitigates radiation-induced cognitive impairment. Radiat. Res. 2013, 179, 549–556. [Google Scholar] [CrossRef] [Green Version]
- Casciati, A.; Dobos, K.; Antonelli, F.; Benedek, A.; Kempf, S.J.; Belles, M.; Balogh, A.; Tanori, M.; Heredia, L.; Atkinson, M.J.; et al. Age-related effects of X-ray irradiation on mouse hippocampus. Oncotarget 2016, 7, 28040–28058. [Google Scholar] [CrossRef] [Green Version]
- Betlazar, C.; Middleton, R.J.; Banati, R.B.; Liu, G.J. The impact of high and low dose ionising radiation on the central nervous system. Redox Biol. 2016, 9, 144–156. [Google Scholar] [CrossRef] [Green Version]
- Claude, J.; Linnartz-Gerlach, B.; Kudin, A.P.; Kunz, W.S.; Neumann, H. Microglial CD33-related Siglec-E inhibits neurotoxicity by preventing the phagocytosis-associated oxidative burst. J. Neurosci. 2013, 33, 18270–18276. [Google Scholar] [CrossRef]
- Ramanan, S.; Kooshki, M.; Zhao, W.; Hsu, F.C.; Robbins, M.E. PPARalpha ligands inhibit radiation-induced microglial inflammatory responses by negatively regulating NF-kappaB and AP-1 pathways. Free Radic. Biol. Med. 2008, 45, 1695–1704. [Google Scholar] [CrossRef] [Green Version]
- Leach, J.K.; Van Tuyle, G.; Lin, P.S.; Schmidt-Ullrich, R.; Mikkelsen, R.B. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001, 61, 3894–3901. [Google Scholar]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Cho, H.J.; Lee, W.H.; Hwang, O.M.H.; Sonntag, W.E.; Lee, Y.W. Role of NADPH oxidase in radiation-induced pro-oxidative and pro-inflammatory pathways in mouse brain. Int J. Radiat. Biol. 2017, 93, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Sui, G.; Rosa, P.M.; Zhao, W. Radiation-induced c-Jun activation depends on MEK1-ERK1/2 signaling pathway in microglial cells. PLoS ONE 2012, 7, e36739. [Google Scholar] [CrossRef] [Green Version]
- Han, J.E.; Choi, J.W. Control of JNK for an activation of NADPH oxidase in LPS-stimulated BV2 microglia. Arch. Pharm Res. 2012, 35, 709–715. [Google Scholar] [CrossRef]
- Betlazar, C.; Middleton, R.J.; Howell, N.; Storer, B.; Davis, E.; Davies, J.; Banati, R.; Liu, G.J. Mitochondrial Translocator Protein (TSPO) Expression in the Brain After Whole Body Gamma Irradiation. Front. Cell Dev. Biol. 2021, 9, 715444. [Google Scholar] [CrossRef]
- Anholt, R.R.; Pedersen, P.L.; De Souza, E.B.; Snyder, S.H. The peripheral-type benzodiazepine receptor. Localization to the mitochondrial outer membrane. J. Biol. Chem. 1986, 261, 576–583. [Google Scholar] [CrossRef]
- Choi, J.; Ifuku, M.; Noda, M.; Guilarte, T.R. Translocator protein (18 kDa)/peripheral benzodiazepine receptor specific ligands induce microglia functions consistent with an activated state. Glia 2011, 59, 219–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, A.; Herb, M.; Schramm, M.; Langmann, T. The TSPO-NOX1 axis controls phagocyte-triggered pathological angiogenesis in the eye. Nat. Commun. 2020, 11, 2709. [Google Scholar] [CrossRef]
- Loth, M.K.; Guariglia, S.R.; Re, D.B.; Perez, J.; de Paiva, V.N.; Dziedzic, J.L.; Chambers, J.W.; Azzam, D.J.; Guilarte, T.R. A Novel Interaction of Translocator Protein 18 kDa (TSPO) with NADPH Oxidase in Microglia. Mol. Neurobiol. 2020, 57, 4467–4487. [Google Scholar] [CrossRef]
- Lee, J.W.; Kim, L.E.; Shim, H.J.; Kim, E.K.; Hwang, W.C.; Min, D.S.; Yu, S.W. A translocator protein 18 kDa ligand, Ro5-4864, inhibits ATP-induced NLRP3 inflammasome activation. Biochem. Biophys. Res. Commun. 2016, 474, 587–593. [Google Scholar] [CrossRef]
- Batarseh, A.; Li, J.; Papadopoulos, V. Protein kinase C epsilon regulation of translocator protein (18 kDa) Tspo gene expression is mediated through a MAPK pathway targeting STAT3 and c-Jun transcription factors. Biochemistry 2010, 49, 4766–4778. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.Y.; Yu, J.Z.; Li, Q.Y.; Ma, C.G.; Lu, C.Z.; Xiao, B.G. TSPO-specific ligand vinpocetine exerts a neuroprotective effect by suppressing microglial inflammation. Neuron Glia Biol. 2011, 7, 187–197. [Google Scholar] [CrossRef]
- Choi, H.B.; Khoo, C.; Ryu, J.K.; van Breemen, E.; Kim, S.U.; McLarnon, J.G. Inhibition of lipopolysaccharide-induced cyclooxygenase-2, tumor necrosis factor-alpha and [Ca2+]i responses in human microglia by the peripheral benzodiazepine receptor ligand PK11195. J. Neurochem. 2002, 83, 546–555. [Google Scholar] [CrossRef]
- Lee, J.W.; Nam, H.; Yu, S.W. Systematic Analysis of Translocator Protein 18 kDa (TSPO) Ligands on Toll-like Receptors-mediated Pro-inflammatory Responses in Microglia and Astrocytes. Exp. Neurobiol. 2016, 25, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Betlazar, C.; Middleton, R.J.; Banati, R.; Liu, G.J. The Translocator Protein (TSPO) in Mitochondrial Bioenergetics and Immune Processes. Cells. 2020, 9, 512. [Google Scholar] [CrossRef] [Green Version]
- Azrad, M.; Zeineh, N.; Weizman, A.; Veenman, L.; Gavish, M. The TSPO Ligands 2-Cl-MGV-1, MGV-1, and PK11195 Differentially Suppress the Inflammatory Response of BV-2 Microglial Cell to LPS. Int. J. Mol. Sci. 2019, 20, 594. [Google Scholar] [CrossRef] [Green Version]
- Monga, S.; Nagler, R.; Amara, R.; Weizman, A.; Gavish, M. Inhibitory Effects of the Two Novel TSPO Ligands 2-Cl-MGV-1 and MGV-1 on LPS-induced Microglial Activation. Cells. 2019, 8, 486. [Google Scholar] [CrossRef] [Green Version]
- Dringen, R. Oxidative and antioxidative potential of brain microglial cells. Antioxid. Redox Signal. 2005, 7, 1223–1233. [Google Scholar] [CrossRef]
- Ismail, A.F.; El-Sonbaty, S.M. Fermentation enhances Ginkgo biloba protective role on gamma-irradiation induced neuroinflammatory gene expression and stress hormones in rat brain. J. Photochem. Photobiol. B 2016, 158, 154–163. [Google Scholar] [CrossRef]
- Fishman, K.; Baure, J.; Zou, Y.; Huang, T.T.; Andres-Mach, M.; Rola, R.; Suarez, T.; Acharya, M.; Limoli, C.L.; Lamborn, K.R.; et al. Radiation-induced reductions in neurogenesis are ameliorated in mice deficient in CuZnSOD or MnSOD. Free Radic. Biol. Med. 2009, 47, 1459–1467. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Aid, S.; Kim, H.W.; Jackson, S.H.; Bosetti, F. Inhibition of NADPH oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. J. Neurochem. 2012, 120, 292–301. [Google Scholar] [CrossRef]
- Bhat, S.A.; Sood, A.; Shukla, R.; Hanif, K. AT2R Activation Prevents Microglia Pro-inflammatory Activation in a NOX-Dependent Manner: Inhibition of PKC Activation and p47(phox) Phosphorylation by PP2A. Mol. Neurobiol. 2019, 56, 3005–3023. [Google Scholar] [CrossRef]
- Vilhardt, F.; Haslund-Vinding, J.; Jaquet, V.; McBean, G. Microglia antioxidant systems and redox signalling. Br. J. Pharmacol. 2017, 174, 1719–1732. [Google Scholar] [CrossRef] [Green Version]
- Hamada, N.; Matsumoto, H.; Hara, T.; Kobayashi, Y. Intercellular and intracellular signaling pathways mediating ionizing radiation-induced bystander effects. J. Radiat. Res. 2007, 48, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Haslund-Vinding, J.; McBean, G.; Jaquet, V.; Vilhardt, F. NADPH oxidases in oxidant production by microglia: Activating receptors, pharmacology and association with disease. Br. J. Pharmacol. 2017, 174, 1733–1749. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Qian, L.; Flood, P.M.; Shi, J.S.; Hong, J.S.; Gao, H.M. Inhibition of IkappaB kinase-beta protects dopamine neurons against lipopolysaccharide-induced neurotoxicity. J. Pharmacol. Exp. Ther. 2010, 333, 822–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo, A.I.; McBean, G.; Cindric, M.; Egea, J.; Lopez, M.G.; Rada, P.; Zarkovic, N.; Cuadrado, A. Redox control of microglial function: Molecular mechanisms and functional significance. Antioxid Redox Signal. 2014, 21, 1766–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Min, J.S.; Kim, B.; Chae, U.B.; Yun, J.W.; Choi, M.S.; Kong, I.K.; Chang, K.T.; Lee, D.S. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-kappaB pathways. Neurosci. Lett. 2015, 584, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Mehrabadi, A.R.; Korolainen, M.A.; Odero, G.; Miller, D.W.; Kauppinen, T.M. Poly(ADP-ribose) polymerase-1 regulates microglia mediated decrease of endothelial tight junction integrity. Neurochem. Int. 2017, 108, 266–271. [Google Scholar] [CrossRef]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Gaber, M.W.; McColgan, T.; Naimark, M.D.; Kiani, M.F.; Merchant, T.E. Radiation-induced permeability and leukocyte adhesion in the rat blood-brain barrier: Modulation with anti-ICAM-1 antibodies. Brain Res. 2003, 969, 59–69. [Google Scholar] [CrossRef]
- Mildner, A.; Schmidt, H.; Nitsche, M.; Merkler, D.; Hanisch, U.K.; Mack, M.; Heikenwalder, M.; Bruck, W.; Priller, J.; Prinz, M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 2007, 10, 1544–1553. [Google Scholar] [CrossRef]
- Morganti, J.M.; Jopson, T.D.; Liu, S.; Gupta, N.; Rosi, S. Cranial irradiation alters the brain’s microenvironment and permits CCR2+ macrophage infiltration. PLoS ONE 2014, 9, e93650. [Google Scholar] [CrossRef]
- Yuan, H.; Gaber, M.W.; Boyd, K.; Wilson, C.M.; Kiani, M.F.; Merchant, T.E. Effects of fractionated radiation on the brain vasculature in a murine model: Blood-brain barrier permeability, astrocyte proliferation, and ultrastructural changes. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 860–866. [Google Scholar] [CrossRef]
- Allen, B.D.; Apodaca, L.A.; Syage, A.R.; Markarian, M.; Baddour, A.A.D.; Minasyan, H.; Alikhani, L.; Lu, C.; West, B.L.; Giedzinski, E.; et al. Attenuation of neuroinflammation reverses Adriamycin-induced cognitive impairments. Acta Neuropathol. Commun. 2019, 7, 186. [Google Scholar] [CrossRef]
- Wang, J.J.; Tong, F.; Lin, Z.Y.; Dong, X.R. The Effects of Vascular Endothelial Cells on Regulating Post-Irradiation Microglia Phenotype in Irradiation-Induced Brain Injury. J. Thorac. Oncol. 2021, 16, 71. [Google Scholar] [CrossRef]
- Khan, S.Y.; Awad, E.M.; Oszwald, A.; Mayr, M.; Yin, X.; Waltenberger, B.; Stuppner, H.; Lipovac, M.; Uhrin, P.; Breuss, J.M. Premature senescence of endothelial cells upon chronic exposure to TNFalpha can be prevented by N-acetyl cysteine and plumericin. Sci. Rep. 2017, 7, 39501. [Google Scholar] [CrossRef]
- Kyrkanides, S.; Olschowka, J.A.; Williams, J.P.; Hansen, J.T.; O’Banion, M.K. TNF alpha and IL-1beta mediate intercellular adhesion molecule-1 induction via microglia-astrocyte interaction in CNS radiation injury. J. Neuroimmunol. 1999, 95, 95–106. [Google Scholar] [CrossRef]
- Jang, C.; Kim, J.; Kwon, Y.; Jo, S.A. Telmisartan Inhibits TNFalpha-Induced Leukocyte Adhesion by Blocking ICAM-1 Expression in Astroglial Cells but Not in Endothelial Cells. Biomol. Ther. 2020, 28, 423–430. [Google Scholar] [CrossRef]
- Wilson, C.M.; Gaber, M.W.; Sabek, O.M.; Zawaski, J.A.; Merchant, T.E. Radiation-induced astrogliosis and blood-brain barrier damage can be abrogated using anti-TNF treatment. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 934–941. [Google Scholar] [CrossRef]
- Ronaldson, P.T.; Davis, T.P. Regulation of blood-brain barrier integrity by microglia in health and disease: A therapeutic opportunity. J. Cereb. Blood Flow Metab. 2020, 40, S6–S24. [Google Scholar] [CrossRef]
- Moravan, M.J.; Olschowka, J.A.; Williams, J.P.; O’Banion, M.K. Cranial irradiation leads to acute and persistent neuroinflammation with delayed increases in T-cell infiltration and CD11c expression in C57BL/6 mouse brain. Radiat. Res. 2011, 176, 459–473. [Google Scholar] [CrossRef] [Green Version]
- Burrell, K.; Hill, R.P.; Zadeh, G. High-resolution in-vivo analysis of normal brain response to cranial irradiation. PLoS ONE 2012, 7, e38366. [Google Scholar] [CrossRef]
- Moravan, M.J.; Olschowka, J.A.; Williams, J.P.; O’Banion, M.K. Brain radiation injury leads to a dose- and time-dependent recruitment of peripheral myeloid cells that depends on CCR2 signaling. J. Neuroinflamm. 2016, 13, 30. [Google Scholar] [CrossRef] [Green Version]
- Belarbi, K.; Jopson, T.; Arellano, C.; Fike, J.R.; Rosi, S. CCR2 deficiency prevents neuronal dysfunction and cognitive impairments induced by cranial irradiation. Cancer Res. 2013, 73, 1201–1210. [Google Scholar] [CrossRef] [Green Version]
- Osman, A.M.; Sun, Y.; Burns, T.C.; He, L.; Kee, N.; Oliva-Vilarnau, N.; Alevyzaki, A.; Zhou, K.; Louhivuori, L.; Uhlen, P.; et al. Radiation Triggers a Dynamic Sequence of Transient Microglial Alterations in Juvenile Brain. Cell Rep. 2020, 31, 107699. [Google Scholar] [CrossRef]
- Whitelaw, B.S.; Tanny, S.; Johnston, C.J.; Majewska, A.K.; O’Banion, M.K.; Marples, B. In Vivo Imaging of the Microglial Landscape after Whole Brain Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, 1066–1071. [Google Scholar] [CrossRef]
- Lee, S.W.; Haditsch, U.; Cord, B.J.; Guzman, R.; Kim, S.J.; Boettcher, C.; Priller, J.; Ormerod, B.K.; Palmer, T.D. Absence of CCL2 is sufficient to restore hippocampal neurogenesis following cranial irradiation. Brain Behav. Immun. 2013, 30, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Acharya, M.M.; Patel, N.H.; Craver, B.M.; Tran, K.K.; Giedzinski, E.; Tseng, B.P.; Parihar, V.K.; Limoli, C.L. Consequences of low dose ionizing radiation exposure on the hippocampal microenvironment. PLoS ONE 2015, 10, e0128316. [Google Scholar] [CrossRef]
- Raber, J.; Allen, A.R.; Rosi, S.; Sharma, S.; Dayger, C.; Davis, M.J.; Fike, J.R. Effects of (56)Fe radiation on hippocampal function in mice deficient in chemokine receptor 2 (CCR2). Behav. Brain Res. 2013, 246, 69–75. [Google Scholar] [CrossRef]
- Dietrich, J.; Baryawno, N.; Nayyar, N.; Valtis, Y.K.; Yang, B.; Ly, I.; Besnard, A.; Severe, N.; Gustafsson, K.U.; Andronesi, O.C.; et al. Bone marrow drives central nervous system regeneration after radiation injury. J. Clin. Investig. 2018, 128, 281–293. [Google Scholar] [CrossRef] [Green Version]
- Stanley, E.R.; Chitu, V. CSF-1 receptor signaling in myeloid cells. Cold Spring Harb. Perspect. Biol. 2014, 6, a021857. [Google Scholar] [CrossRef] [Green Version]
- Erblich, B.; Zhu, L.; Etgen, A.M.; Dobrenis, K.; Pollard, J.W. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS ONE 2011, 6, e26317. [Google Scholar] [CrossRef] [Green Version]
- Oosterhof, N.; Chang, I.J.; Karimiani, E.G.; Kuil, L.E.; Jensen, D.M.; Daza, R.; Young, E.; Astle, L.; van der Linde, H.C.; Shivaram, G.M.; et al. Homozygous Mutations in CSF1R Cause a Pediatric-Onset Leukoencephalopathy and Can Result in Congenital Absence of Microglia. Am. J. Hum. Genet. 2019, 104, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [Green Version]
- Acharya, M.M.; Green, K.N.; Allen, B.D.; Najafi, A.R.; Syage, A.; Minasyan, H.; Le, M.T.; Kawashita, T.; Giedzinski, E.; Parihar, V.K.; et al. Elimination of microglia improves cognitive function following cranial irradiation. Sci. Rep. 2016, 6, 31545. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Jopson, T.D.; Paladini, M.S.; Liu, S.; West, B.L.; Gupta, N.; Rosi, S. Colony-stimulating factor 1 receptor blockade prevents fractionated whole-brain irradiation-induced memory deficits. J. Neuroinflamm. 2016, 13, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, B.D.; Syage, A.R.; Maroso, M.; Baddour, A.A.D.; Luong, V.; Minasyan, H.; Giedzinski, E.; West, B.L.; Soltesz, I.; Limoli, C.L.; et al. Mitigation of helium irradiation-induced brain injury by microglia depletion. J. Neuroinflamm. 2020, 17, 159. [Google Scholar] [CrossRef]
- Krukowski, K.; Feng, X.; Paladini, M.S.; Chou, A.; Sacramento, K.; Grue, K.; Riparip, L.K.; Jones, T.; Campbell-Beachler, M.; Nelson, G.; et al. Temporary microglia-depletion after cosmic radiation modifies phagocytic activity and prevents cognitive deficits. Sci. Rep. 2018, 8, 7857. [Google Scholar] [CrossRef] [PubMed]
- Hinkle, J.J.; Olschowka, J.A.; Love, T.M.; Williams, J.P.; O’Banion, M.K. Cranial irradiation mediated spine loss is sex-specific and complement receptor-3 dependent in male mice. Sci. Rep. 2019, 9, 18899. [Google Scholar] [CrossRef] [Green Version]
- Parihar, V.K.; Limoli, C.L. Cranial irradiation compromises neuronal architecture in the hippocampus. Proc. Natl. Acad. Sci. USA 2013, 110, 12822–12827. [Google Scholar] [CrossRef] [Green Version]
- Duman, J.G.; Dinh, J.; Zhou, W.; Cham, H.; Mavratsas, V.C.; Paveskovic, M.; Mulherkar, S.; McGovern, S.L.; Tolias, K.F.; Grosshans, D.R. Memantine prevents acute radiation-induced toxicities at hippocampal excitatory synapses. Neuro-Oncol. 2018, 20, 655–665. [Google Scholar] [CrossRef]
- Parihar, V.K.; Pasha, J.; Tran, K.K.; Craver, B.M.; Acharya, M.M.; Limoli, C.L. Persistent changes in neuronal structure and synaptic plasticity caused by proton irradiation. Brain Struct. Funct. 2015, 220, 1161–1171. [Google Scholar] [CrossRef] [Green Version]
- Parihar, V.K.; Allen, B.D.; Caressi, C.; Kwok, S.; Chu, E.; Tran, K.K.; Chmielewski, N.N.; Giedzinski, E.; Acharya, M.M.; Britten, R.A.; et al. Cosmic radiation exposure and persistent cognitive dysfunction. Sci. Rep. 2016, 6, 34774. [Google Scholar] [CrossRef] [Green Version]
- Meijne, M.G. [Running in high temperatures]. Ned. Tijdschr. Geneeskd. 1988, 132, 440–443. [Google Scholar]
- Lowette, K.; Roosen, L.; Tack, J.; Vanden Berghe, P. Effects of high-fructose diets on central appetite signaling and cognitive function. Front. Nutr. 2015, 2, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagher, N.N.; Najafi, A.R.; Kayala, K.M.; Elmore, M.R.; White, T.E.; Medeiros, R.; West, B.L.; Green, K.N. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J. Neuroinflamm. 2015, 12, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Ritzel, R.M.; Khan, N.; Cao, T.; He, J.; Lei, Z.; Matyas, J.J.; Sabirzhanov, B.; Liu, S.; Li, H.; et al. Delayed microglial depletion after spinal cord injury reduces chronic inflammation and neurodegeneration in the brain and improves neurological recovery in male mice. Theranostics 2020, 10, 11376–11403. [Google Scholar] [CrossRef] [PubMed]
- Elmore, M.R.; Lee, R.J.; West, B.L.; Green, K.N. Characterizing newly repopulated microglia in the adult mouse: Impacts on animal behavior, cell morphology, and neuroinflammation. PLoS ONE 2015, 10, e0122912. [Google Scholar] [CrossRef] [PubMed]
- Rosi, S. Colony stimulating factor-1 receptor as a treatment for cognitive deficits postfractionated whole-brain irradiation. Brain Circ. 2017, 3, 180–182. [Google Scholar] [CrossRef]
- Szalay, G.; Martinecz, B.; Lenart, N.; Kornyei, Z.; Orsolits, B.; Judak, L.; Csaszar, E.; Fekete, R.; West, B.L.; Katona, G.; et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun. 2016, 7, 11499. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, Z.; Cao, B.B.; Qiu, Y.H.; Peng, Y.P. TGF-beta1 Neuroprotection via Inhibition of Microglial Activation in a Rat Model of Parkinson’s Disease. J. Neuroimmune Pharmacol. 2017, 12, 433–446. [Google Scholar] [CrossRef]
- Mangale, V.; Syage, A.R.; Ekiz, H.A.; Skinner, D.D.; Cheng, Y.; Stone, C.L.; Brown, R.M.; O’Connell, R.M.; Green, K.N.; Lane, T.E. Microglia influence host defense, disease, and repair following murine coronavirus infection of the central nervous system. Glia 2020, 68, 2345–2360. [Google Scholar] [CrossRef]
- Conigliaro, P.; Triggianese, P.; Ballanti, E.; Perricone, C.; Perricone, R.; Chimenti, M.S. Complement, infection, and autoimmunity. Curr. Opin. Rheumatol. 2019, 31, 532–541. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Liao, J.; Aloor, J.; Nie, H.; Wilson, B.C.; Fessler, M.B.; Gao, H.M.; Hong, J.S. CD11b/CD18 (Mac-1) is a novel surface receptor for extracellular double-stranded RNA to mediate cellular inflammatory responses. J. Immunol. 2013, 190, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, M.I.; Chu, S.H.; Hernandez, M.X.; Fang, M.J.; Modarresi, L.; Selvan, P.; MacGregor, G.R.; Tenner, A.J. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J. Neuroinflamm. 2017, 14, 48. [Google Scholar] [CrossRef] [Green Version]
- Andrews, R.N.; Dugan, G.O.; Peiffer, A.M.; Hawkins, G.A.; Hanbury, D.B.; Bourland, J.D.; Hampson, R.E.; Deadwyler, S.A.; Clinea, J.M. White Matter is the Predilection Site of Late-Delayed Radiation-Induced Brain Injury in Non-Human Primates. Radiat. Res. 2019, 191, 217–231. [Google Scholar] [CrossRef]
- Kalm, M.; Andreasson, U.; Bjork-Eriksson, T.; Zetterberg, H.; Pekny, M.; Blennow, K.; Pekna, M.; Blomgren, K. C3 deficiency ameliorates the negative effects of irradiation of the young brain on hippocampal development and learning. Oncotarget 2016, 7, 19382–19394. [Google Scholar] [CrossRef] [Green Version]
- Hinkle, J.J.; Olschowka, J.A.; Williams, J.P.; O’Banion, M.K. Pharmacologic manipulation of complement receptor 3 prevents dendritic spine loss and cognitive impairment after acute cranial radiation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Haggadone, M.D.; Grailer, J.J.; Fattahi, F.; Zetoune, F.S.; Ward, P.A. Bidirectional Crosstalk between C5a Receptors and the NLRP3 Inflammasome in Macrophages and Monocytes. Mediat. Inflamm. 2016, 2016, 1340156. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, M.X.; Jiang, S.; Cole, T.A.; Chu, S.H.; Fonseca, M.I.; Fang, M.J.; Hohsfield, L.A.; Torres, M.D.; Green, K.N.; Wetsel, R.A.; et al. Prevention of C5aR1 signaling delays microglial inflammatory polarization, favors clearance pathways and suppresses cognitive loss. Mol. Neurodegener. 2017, 12, 66. [Google Scholar] [CrossRef]
- Cardozo, P.L.; de Lima, I.B.Q.; Maciel, E.M.A.; Silva, N.C.; Dobransky, T.; Ribeiro, F.M. Synaptic Elimination in Neurological Disorders. Curr. Neuropharmacol. 2019, 17, 1071–1095. [Google Scholar] [CrossRef]
- Krukowski, K.; Grue, K.; Frias, E.S.; Pietrykowski, J.; Jones, T.; Nelson, G.; Rosi, S. Female mice are protected from space radiation-induced maladaptive responses. Brain Behav. Immun. 2018, 74, 106–120. [Google Scholar] [CrossRef]
- Liu, B.; Hinshaw, R.G.; Le, K.X.; Park, M.A.; Wang, S.; Belanger, A.P.; Dubey, S.; Frost, J.L.; Shi, Q.; Holton, P.; et al. Space-like (56)Fe irradiation manifests mild, early sex-specific behavioral and neuropathological changes in wildtype and Alzheimer’s-like transgenic mice. Sci. Rep. 2019, 9, 12118. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, I.; Willems, J.G.; Kooi, E.J.; van Eden, C.; Gold, S.M.; Geurts, J.J.; Baas, F.; Huitinga, I.; Ramaglia, V. Complement C1q-C3-associated synaptic changes in multiple sclerosis hippocampus. Ann. Neurol. 2015, 77, 1007–1026. [Google Scholar] [CrossRef] [PubMed]
- Bie, B.; Wu, J.; Foss, J.F.; Naguib, M. Activation of mGluR1 Mediates C1q-Dependent Microglial Phagocytosis of Glutamatergic Synapses in Alzheimer’s Rodent Models. Mol. Neurobiol. 2019, 56, 5568–5585. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calovi, S.; Mut-Arbona, P.; Sperlagh, B. Microglia and the Purinergic Signaling System. Neuroscience 2019, 405, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Potucek, Y.D.; Crain, J.M.; Watters, J.J. Purinergic receptors modulate MAP kinases and transcription factors that control microglial inflammatory gene expression. Neurochem. Int. 2006, 49, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Von Kugelgen, I.; Hoffmann, K. Pharmacology and structure of P2Y receptors. Neuropharmacology 2016, 104, 50–61. [Google Scholar] [CrossRef]
- Gu, B.J.; Wiley, J.S. P2X7 as a scavenger receptor for innate phagocytosis in the brain. Br. J. Pharmacol. 2018, 175, 4195–4208. [Google Scholar] [CrossRef]
- Takenouchi, T.; Nakai, M.; Iwamaru, Y.; Sugama, S.; Tsukimoto, M.; Fujita, M.; Wei, J.; Sekigawa, A.; Sato, M.; Kojima, S.; et al. The activation of P2X7 receptor impairs lysosomal functions and stimulates the release of autophagolysosomes in microglial cells. J. Immunol. 2009, 182, 2051–2062. [Google Scholar] [CrossRef] [Green Version]
- Sekar, P.; Huang, D.Y.; Hsieh, S.L.; Chang, S.F.; Lin, W.W. AMPK-dependent and independent actions of P2X7 in regulation of mitochondrial and lysosomal functions in microglia. Cell Commun. Signal. 2018, 16, 83. [Google Scholar] [CrossRef] [Green Version]
- Campagno, K.E.; Mitchell, C.H. The P2X7 Receptor in Microglial Cells Modulates the Endolysosomal Axis, Autophagy, and Phagocytosis. Front. Cell. Neurosci. 2021, 15, 645244. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Hu, W.; Liu, Y.; Xu, P.; Li, Z.; Wu, R.; Shi, X.; Tang, Y. P2Y6 Receptor-Mediated Microglial Phagocytosis in Radiation-Induced Brain Injury. Mol. Neurobiol. 2016, 53, 3552–3564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, R.X.; Shen, H.; Huang, S.X.; Wang, L.P.; Li, Z.W.; Peng, P.; Mamtilahun, M.; Tang, Y.H.; Shen, F.X.; Tian, H.L.; et al. P2Y6 receptor inhibition aggravates ischemic brain injury by reducing microglial phagocytosis. CNS Neurosci. Ther. 2020, 26, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, J.; Zhang, Y.; Huang, Y.; Chen, D.; Shi, Z.; Smith, A.D.; Li, W.; Gao, Y. Central nervous system diseases related to pathological microglial phagocytosis. CNS Neurosci. Ther. 2021, 27, 528–539. [Google Scholar] [CrossRef]
- Puigdellivol, M.; Milde, S.; Vilalta, A.; Cockram, T.O.J.; Allendorf, D.H.; Lee, J.Y.; Dundee, J.M.; Pampuscenko, K.; Borutaite, V.; Nuthall, H.N.; et al. The microglial P2Y6 receptor mediates neuronal loss and memory deficits in neurodegeneration. Cell Rep. 2021, 37, 110148. [Google Scholar] [CrossRef]
- Jones, B.A.; Beamer, M.; Ahmed, S. Fractalkine/CX3CL1: A potential new target for inflammatory diseases. Mol. Interv. 2010, 10, 263–270. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Wang, S.; Pigott, V.M.; Jiang, T.; Foley, L.M.; Mishra, A.; Nayak, R.; Zhu, W.; Begum, G.; Shi, Y.; et al. Selective role of Na(+) /H(+) exchanger in Cx3cr1(+) microglial activation, white matter demyelination, and post-stroke function recovery. Glia 2018, 66, 2279–2298. [Google Scholar] [CrossRef]
- Bolos, M.; Llorens-Martin, M.; Perea, J.R.; Jurado-Arjona, J.; Rabano, A.; Hernandez, F.; Avila, J. Absence of CX3CR1 impairs the internalization of Tau by microglia. Mol. Neurodegener. 2017, 12, 59. [Google Scholar] [CrossRef] [Green Version]
- Tristao, F.S.; Lazzarini, M.; Martin, S.; Amar, M.; Stuhmer, W.; Kirchhoff, F.; Gomes, L.A.; Lanfumey, L.; Prediger, R.D.; Sepulveda, J.E.; et al. CX3CR1 Disruption Differentially Influences Dopaminergic Neuron Degeneration in Parkinsonian Mice Depending on the Neurotoxin and Route of Administration. Neurotox. Res. 2016, 29, 364–380. [Google Scholar] [CrossRef]
- Pawelec, P.; Ziemka-Nalecz, M.; Sypecka, J.; Zalewska, T. The Impact of the CX3CL1/CX3CR1 Axis in Neurological Disorders. Cells 2020, 9, 2277. [Google Scholar] [CrossRef]
- Agarwal, S.; Yadav, A.; Chaturvedi, R.K. Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem. Biophys. Res. Commun. 2017, 483, 1166–1177. [Google Scholar] [CrossRef]
- Grygiel-Gorniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Delerive, P.; De Bosscher, K.; Besnard, S.; Vanden Berghe, W.; Peters, J.M.; Gonzalez, F.J.; Fruchart, J.C.; Tedgui, A.; Haegeman, G.; Staels, B. Peroxisome proliferator-activated receptor alpha negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-kappaB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Joe, E.H.; Jou, I. PPAR-alpha activators suppress STAT1 inflammatory signaling in lipopolysaccharide-activated rat glia. Neuroreport 2005, 16, 829–833. [Google Scholar]
- Korbecki, J.; Bobinski, R.; Dutka, M. Self-regulation of the inflammatory response by peroxisome proliferator-activated receptors. Inflamm. Res. 2019, 68, 443–458. [Google Scholar] [CrossRef] [Green Version]
- Kang, E.S.; Hwang, J.S.; Lee, W.J.; Lee, G.H.; Choi, M.J.; Paek, K.S.; Lim, D.S.; Seo, H.G. Ligand-activated PPARdelta inhibits angiotensin II-stimulated hypertrophy of vascular smooth muscle cells by targeting ROS. PLoS ONE 2019, 14, e0210482. [Google Scholar]
- Strosznajder, A.K.; Wojtowicz, S.; Jezyna, M.J.; Sun, G.Y.; Strosznajder, J.B. Recent Insights on the Role of PPAR-beta/delta in Neuroinflammation and Neurodegeneration, and Its Potential Target for Therapy. Neuromol. Med. 2021, 23, 86–98. [Google Scholar] [CrossRef]
- Chao, X.; Xiong, C.; Dong, W.; Qu, Y.; Ning, W.; Liu, W.; Han, F.; Ma, Y.; Wang, R.; Fei, Z.; et al. Activation of peroxisome proliferator-activated receptor beta/delta attenuates acute ischemic stroke on middle cerebral ischemia occlusion in rats. J. Stroke Cerebrovasc. Dis. 2014, 23, 1396–1402. [Google Scholar] [CrossRef]
- Li, W.; Zhang, Z.; Zhang, K.; Xue, Z.; Li, Y.; Zhang, Z.; Zhang, L.; Gu, C.; Zhang, Q.; Hao, J.; et al. Arctigenin Suppress Th17 Cells and Ameliorates Experimental Autoimmune Encephalomyelitis Through AMPK and PPAR-gamma/ROR-gammat Signaling. Mol. Neurobiol. 2016, 53, 5356–5366. [Google Scholar] [CrossRef]
- Bright, J.J.; Kanakasabai, S.; Chearwae, W.; Chakraborty, S. PPAR Regulation of Inflammatory Signaling in CNS Diseases. PPAR Res. 2008, 2008, 658520. [Google Scholar] [CrossRef] [Green Version]
- Ramanan, S.; Kooshki, M.; Zhao, W.; Hsu, F.C.; Riddle, D.R.; Robbins, M.E. The PPARalpha agonist fenofibrate preserves hippocampal neurogenesis and inhibits microglial activation after whole-brain irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 870–877. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Gao, L.; Cheng, Z.; Cai, J.; Niu, Y.; Meng, W.; Zhao, Q. Kukoamine A Prevents Radiation-Induced Neuroinflammation and Preserves Hippocampal Neurogenesis in Rats by Inhibiting Activation of NF-kappaB and AP-1. Neurotox. Res. 2017, 31, 259–268. [Google Scholar] [CrossRef]
- Schnegg, C.I.; Greene-Schloesser, D.; Kooshki, M.; Payne, V.S.; Hsu, F.C.; Robbins, M.E. The PPARδ agonist GW0742 inhibits neuroinflammation, but does not restore neurogenesis or prevent early delayed hippocampal-dependent cognitive impairment after whole-brain irradiation. Free Radic. Biol. Med. 2013, 61, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.J.; Lee, E.J.; Park, J.S.; Kim, S.N.; Park, E.M.; Kim, H.S. Anti-inflammatory mechanism of galangin in lipopolysaccharide-stimulated microglia: Critical role of PPAR-gamma signaling pathway. Biochem. Pharmacol. 2017, 144, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Wang, H.; Zhang, F.; Li, X.; Xiang, L.; Aiguo, S. PPARγ agonist pioglitazone inhibits microglia inflammation by blocking p38 mitogen-activated protein kinase signaling pathways. Inflamm. Res. 2010, 59, 921–929. [Google Scholar] [CrossRef]
- Zhao, W.; Payne, V.; Tommasi, E.; Diz, D.I.; Hsu, F.C.; Robbins, M.E. Administration of the peroxisomal proliferator-activated receptor gamma agonist pioglitazone during fractionated brain irradiation prevents radiation-induced cognitive impairment. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 6–9. [Google Scholar] [CrossRef]
- Warden, A.; Truitt, J.; Merriman, M.; Ponomareva, O.; Jameson, K.; Ferguson, L.B.; Mayfield, R.D.; Harris, R.A. Localization of PPAR isotypes in the adult mouse and human brain. Sci. Rep. 2016, 6, 27618. [Google Scholar] [CrossRef]
- Chen, Y.J.; Nguyen, H.M.; Maezawa, I.; Jin, L.W.; Wulff, H. Inhibition of the potassium channel Kv1.3 reduces infarction and inflammation in ischemic stroke. Ann. Clin. Transl. Neurol. 2018, 5, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Pannasch, U.; Farber, K.; Nolte, C.; Blonski, M.; Yan Chiu, S.; Messing, A.; Kettenmann, H. The potassium channels Kv1.5 and Kv1.3 modulate distinct functions of microglia. Mol. Cell. Neurosci. 2006, 33, 401–411. [Google Scholar] [CrossRef]
- Wang, X.; Li, G.; Guo, J.; Zhang, Z.; Zhang, S.; Zhu, Y.; Cheng, J.; Yu, L.; Ji, Y.; Tao, J. Kv1.3 Channel as a Key Therapeutic Target for Neuroinflammatory Diseases: State of the Art and Beyond. Front. Neurosci. 2019, 13, 1393. [Google Scholar] [CrossRef] [Green Version]
- Ramesha, S.; Rayaprolu, S.; Bowen, C.A.; Giver, C.R.; Bitarafan, S.; Nguyen, H.M.; Gao, T.; Chen, M.J.; Nwabueze, N.; Dammer, E.B.; et al. Unique molecular characteristics and microglial origin of Kv1.3 channel-positive brain myeloid cells in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2021, 118, e2013545118. [Google Scholar] [CrossRef]
- Maezawa, I.; Nguyen, H.M.; Di Lucente, J.; Jenkins, D.P.; Singh, V.; Hilt, S.; Kim, K.; Rangaraju, S.; Levey, A.I.; Wulff, H.; et al. Kv1.3 inhibition as a potential microglia-targeted therapy for Alzheimer’s disease: Preclinical proof of concept. Brain 2018, 141, 596–612. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Nguyen, H.M.; Malovic, E.; Luo, J.; Langley, M.; Palanisamy, B.N.; Singh, N.; Manne, S.; Neal, M.; Gabrielle, M.; et al. Kv1.3 modulates neuroinflammation and neurodegeneration in Parkinson’s disease. J. Clin. Investig. 2020, 130, 4195–4212. [Google Scholar] [CrossRef]
- Nguyen, H.M.; di Lucente, J.; Chen, Y.J.; Cui, Y.; Ibrahim, R.H.; Pennington, M.W.; Jin, L.W.; Maezawa, I.; Wulff, H. Biophysical basis for Kv1.3 regulation of membrane potential changes induced by P2X4-mediated calcium entry in microglia. Glia 2020, 68, 2377–2394. [Google Scholar] [CrossRef]
- Fomina, A.F.; Nguyen, H.M.; Wulff, H. Kv1.3 inhibition attenuates neuroinflammation through disruption of microglial calcium signaling. Channels 2021, 15, 67–78. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Shukla, G.C.; Singh, J.; Barik, S. MicroRNAs: Processing, Maturation, Target Recognition and Regulatory Functions. Mol. Cell. Pharmacol. 2011, 3, 83–92. [Google Scholar]
- Sabirzhanov, B.; Makarevich, O.; Barrett, J.; Jackson, I.L.; Faden, A.I.; Stoica, B.A. Down-Regulation of miR-23a-3p Mediates Irradiation-Induced Neuronal Apoptosis. Int. J. Mol. Sci. 2020, 21, 3695. [Google Scholar] [CrossRef]
- Segaran, R.C.; Chan, L.Y.; Wang, H.; Sethi, G.; Tang, F.R. Neuronal Development-Related miRNAs as Biomarkers for Alzheimer’s Disease, Depression, Schizophrenia and Ionizing Radiation Exposure. Curr. Med. Chem. 2021, 28, 19–52. [Google Scholar] [CrossRef]
- Gaudet, A.D.; Fonken, L.K.; Watkins, L.R.; Nelson, R.J.; Popovich, P.G. MicroRNAs: Roles in Regulating Neuroinflammation. Neuroscientist 2018, 24, 221–245. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Cui, J.; Gong, Y.; Wei, S.; Wei, Y.; Yi, L. MicroRNA: A novel implication for damage and protection against ionizing radiation. Environ. Sci. Pollut. Res. Int. 2021, 28, 15584–15596. [Google Scholar] [CrossRef]
- Guo, Y.; Hong, W.; Wang, X.; Zhang, P.; Korner, H.; Tu, J.; Wei, W. MicroRNAs in Microglia: How do MicroRNAs Affect Activation, Inflammation, Polarization of Microglia and Mediate the Interaction Between Microglia and Glioma? Front. Mol. Neurosci. 2019, 12, 125. [Google Scholar] [CrossRef]
- Varol, D.; Mildner, A.; Blank, T.; Shemer, A.; Barashi, N.; Yona, S.; David, E.; Boura-Halfon, S.; Segal-Hayoun, Y.; Chappell-Maor, L.; et al. Dicer Deficiency Differentially Impacts Microglia of the Developing and Adult Brain. Immunity 2017, 46, 1030–1044.e8. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; He, Q.; Chen, Y.; Shao, W.; Yuan, C.; Wang, Y. JNK-mediated microglial DICER degradation potentiates inflammatory responses to induce dopaminergic neuron loss. J. Neuroinflamm. 2018, 15, 184. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, Y.; Lv, X.; Xu, B.; Zhang, H.; Yan, J.; Li, H.; Wu, L. Evolution of an X-Linked miRNA Family Predominantly Expressed in Mammalian Male Germ Cells. Mol. Biol. Evol. 2019, 36, 663–678. [Google Scholar] [CrossRef]
- Nie, J.; Li, C.P.; Li, J.H.; Chen, X.; Zhong, X. Analysis of nonalcoholic fatty liver disease microRNA expression spectra in rat liver tissues. Mol. Med. Rep. 2018, 18, 2669–2680. [Google Scholar]
- Tian, T.; Zhang, Y.; Wu, T.; Yang, L.; Chen, C.; Li, N.; Li, Y.; Xu, S.; Fu, Z.; Cui, X.; et al. miRNA profiling in the hippocampus of attention-deficit/hyperactivity disorder rats. J. Cell Biochem. 2019, 120, 3621–3629. [Google Scholar] [CrossRef]
- Wen, X.; Xie, H.; Gui, R.; Nie, X.; Shan, D.; Huang, R.; Deng, H.; Zhang, J. CircRNA-011235 Counteracts The Deleterious Effect of Irradiation Treatment on Bone Mesenchymal Stem Cells by Regulating The miR-741-3p/CDK6 Pathway. Cell J. 2022, 24, 15–21. [Google Scholar]
- Ou, M.; Fan, W.; Sun, F.; Li, M.; Lin, M.; Yu, Y.; Liang, S.; Liao, H.; Jie, W.; Cai, Y.; et al. Nasal Delivery of AntagomiR-741 Protects Against the Radiation-Induced Brain Injury in Mice. Radiat. Res. 2021, 195, 355–365. [Google Scholar] [CrossRef]
- Zhou, H.; Sun, F.; Ou, M.; Zhang, Y.; Lin, M.; Song, L.; Yu, Y.; Liao, H.; Fan, W.; Xing, H.; et al. Prior nasal delivery of antagomiR-122 prevents radiation-induced brain injury. Mol. Ther 2021, 29, 3465–3483. [Google Scholar] [CrossRef]
- Leavitt, R.J.; Acharya, M.M.; Baulch, J.E.; Limoli, C.L. Extracellular Vesicle-Derived miR-124 Resolves Radiation-Induced Brain Injury. Cancer Res. 2020, 80, 4266–4277. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ye, Y.; Kong, C.; Su, X.; Zhang, X.; Bai, W.; He, X. MiR-124 Enriched Exosomes Promoted the M2 Polarization of Microglia and Enhanced Hippocampus Neurogenesis After Traumatic Brain Injury by Inhibiting TLR4 Pathway. Neurochem. Res. 2019, 44, 811–828. [Google Scholar] [CrossRef]
- Huang, S.; Ge, X.; Yu, J.; Han, Z.; Yin, Z.; Li, Y.; Chen, F.; Wang, H.; Zhang, J.; Lei, P. Increased miR-124-3p in microglial exosomes following traumatic brain injury inhibits neuronal inflammation and contributes to neurite outgrowth via their transfer into neurons. FASEB J. 2018, 32, 512–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, A.; Zhang, T.; Duan, H.; Pan, Y.; Zhang, X.; Yang, G.; Wang, J.; Deng, Y.; Yang, Z. MiR-124 contributes to M2 polarization of microglia and confers brain inflammatory protection via the C/EBP-alpha pathway in intracerebral hemorrhage. Immunol. Lett. 2017, 182, 1–11. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, J.X.; Chen, W.K.; Wu, G.C.; Wang, Y.Q.; Zhu, K.Y.; Wang, J. miR-124/VAMP3 is a novel therapeutic target for mitigation of surgical trauma-induced microglial activation. Signal Transduct. Target. Ther. 2019, 4, 27. [Google Scholar] [CrossRef]
- Song, Y.; Li, Z.; He, T.; Qu, M.; Jiang, L.; Li, W.; Shi, X.; Pan, J.; Zhang, L.; Wang, Y.; et al. M2 microglia-derived exosomes protect the mouse brain from ischemia-reperfusion injury via exosomal miR-124. Theranostics 2019, 9, 2910–2923. [Google Scholar] [CrossRef]
- Ge, X.; Guo, M.; Hu, T.; Li, W.; Huang, S.; Yin, Z.; Li, Y.; Chen, F.; Zhu, L.; Kang, C.; et al. Increased Microglial Exosomal miR-124-3p Alleviates Neurodegeneration and Improves Cognitive Outcome after rmTBI. Mol. Ther. 2020, 28, 503–522. [Google Scholar] [CrossRef] [Green Version]
- Cabili, M.N.; Dunagin, M.C.; McClanahan, P.D.; Biaesch, A.; Padovan-Merhar, O.; Regev, A.; Rinn, J.L.; Raj, A. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 2015, 16, 20. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Kertesz, M.; Spitale, R.C.; Segal, E.; Chang, H.Y. Understanding the transcriptome through RNA structure. Nat. Rev. Genet. 2011, 12, 641–655. [Google Scholar] [CrossRef] [Green Version]
- Cuevas-Diaz Duran, R.; Wei, H.; Kim, D.H.; Wu, J.Q. Invited Review: Long non-coding RNAs: Important regulators in the development, function and disorders of the central nervous system. Neuropathol. Appl. Neurobiol. 2019, 45, 538–556. [Google Scholar] [CrossRef] [Green Version]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Reed, S.L.; Escayg, A. Extracellular vesicles in the treatment of neurological disorders. Neurobiol Dis 2021, 157, 105445. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Giedzinski, E.; Angulo, M.C.; Lui, T.; Lu, C.; Park, A.L.; Tang, S.; Martirosian, V.; Ru, N.; Chmielewski, N.N.; et al. Functional equivalence of stem cell and stem cell-derived extracellular vesicle transplantation to repair the irradiated brain. Stem Cells Transl. Med. 2020, 9, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Baulch, J.E.; Acharya, M.M.; Allen, B.D.; Ru, N.; Chmielewski, N.N.; Martirosian, V.; Giedzinski, E.; Syage, A.; Park, A.L.; Benke, S.N.; et al. Cranial grafting of stem cell-derived microvesicles improves cognition and reduces neuropathology in the irradiated brain. Proc. Natl. Acad. Sci. USA 2016, 113, 4836–4841. [Google Scholar] [CrossRef] [Green Version]
- Holm, M.M.; Kaiser, J.; Schwab, M.E. Extracellular Vesicles: Multimodal Envoys in Neural Maintenance and Repair. Trends Neurosci 2018, 41, 360–372. [Google Scholar] [CrossRef]
- Rienecker, K.D.A.; Paladini, M.S.; Grue, K.; Krukowski, K.; Rosi, S. Microglia: Ally and Enemy in Deep Space. Neurosci. Biobehav. Rev. 2021, 126, 509–514. [Google Scholar] [CrossRef]
- Parihar, V.K.; Angulo, M.C.; Allen, B.D.; Syage, A.; Usmani, M.T.; Passerat de la Chapelle, E.; Amin, A.N.; Flores, L.; Lin, X.; Giedzinski, E.; et al. Sex-Specific Cognitive Deficits Following Space Radiation Exposure. Front. Behav. Neurosci. 2020, 14, 535885. [Google Scholar] [CrossRef]
- Han, W.; Umekawa, T.; Zhou, K.; Zhang, X.M.; Ohshima, M.; Dominguez, C.A.; Harris, R.A.; Zhu, C.; Blomgren, K. Cranial irradiation induces transient microglia accumulation, followed by long-lasting inflammation and loss of microglia. Oncotarget 2016, 7, 82305–82323. [Google Scholar] [CrossRef] [Green Version]
- Hua, K.; Schindler, M.K.; McQuail, J.A.; Forbes, M.E.; Riddle, D.R. Regionally distinct responses of microglia and glial progenitor cells to whole brain irradiation in adult and aging rats. PLoS ONE 2012, 7, e52728. [Google Scholar]
- Li, M.D.; Burns, T.C.; Kumar, S.; Morgan, A.A.; Sloan, S.A.; Palmer, T.D. Aging-like changes in the transcriptome of irradiated microglia. Glia 2015, 63, 754–767. [Google Scholar] [CrossRef] [Green Version]
- Stojiljkovic, M.R.; Ain, Q.; Bondeva, T.; Heller, R.; Schmeer, C.; Witte, O.W. Phenotypic and functional differences between senescent and aged murine microglia. Neurobiol. Aging 2019, 74, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Neher, J.J.; Neniskyte, U.; Hornik, T.; Brown, G.C. Inhibition of UDP/P2Y6 purinergic signaling prevents phagocytosis of viable neurons by activated microglia in vitro and in vivo. Glia 2014, 62, 1463–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neher, J.J.; Emmrich, J.V.; Fricker, M.; Mander, P.K.; Thery, C.; Brown, G.C. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, E4098–E4107. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.C. Neuronal Loss after Stroke Due to Microglial Phagocytosis of Stressed Neurons. Int. J. Mol. Sci. 2021, 22, 13442. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Huang, Y.; Duan, M.; Yang, Q.; Ren, B.; Tang, F. Microglia as Therapeutic Target for Radiation-Induced Brain Injury. Int. J. Mol. Sci. 2022, 23, 8286. https://doi.org/10.3390/ijms23158286
Liu Q, Huang Y, Duan M, Yang Q, Ren B, Tang F. Microglia as Therapeutic Target for Radiation-Induced Brain Injury. International Journal of Molecular Sciences. 2022; 23(15):8286. https://doi.org/10.3390/ijms23158286
Chicago/Turabian StyleLiu, Qun, Yan Huang, Mengyun Duan, Qun Yang, Boxu Ren, and Fengru Tang. 2022. "Microglia as Therapeutic Target for Radiation-Induced Brain Injury" International Journal of Molecular Sciences 23, no. 15: 8286. https://doi.org/10.3390/ijms23158286
APA StyleLiu, Q., Huang, Y., Duan, M., Yang, Q., Ren, B., & Tang, F. (2022). Microglia as Therapeutic Target for Radiation-Induced Brain Injury. International Journal of Molecular Sciences, 23(15), 8286. https://doi.org/10.3390/ijms23158286