
Cellular and Molecular Mechanism of Pulmonary Fibrosis Post-COVID-19: Focus on Galectin-1, -3, -8, -9

 , , and
, , and 
Abstract
:1. Introduction
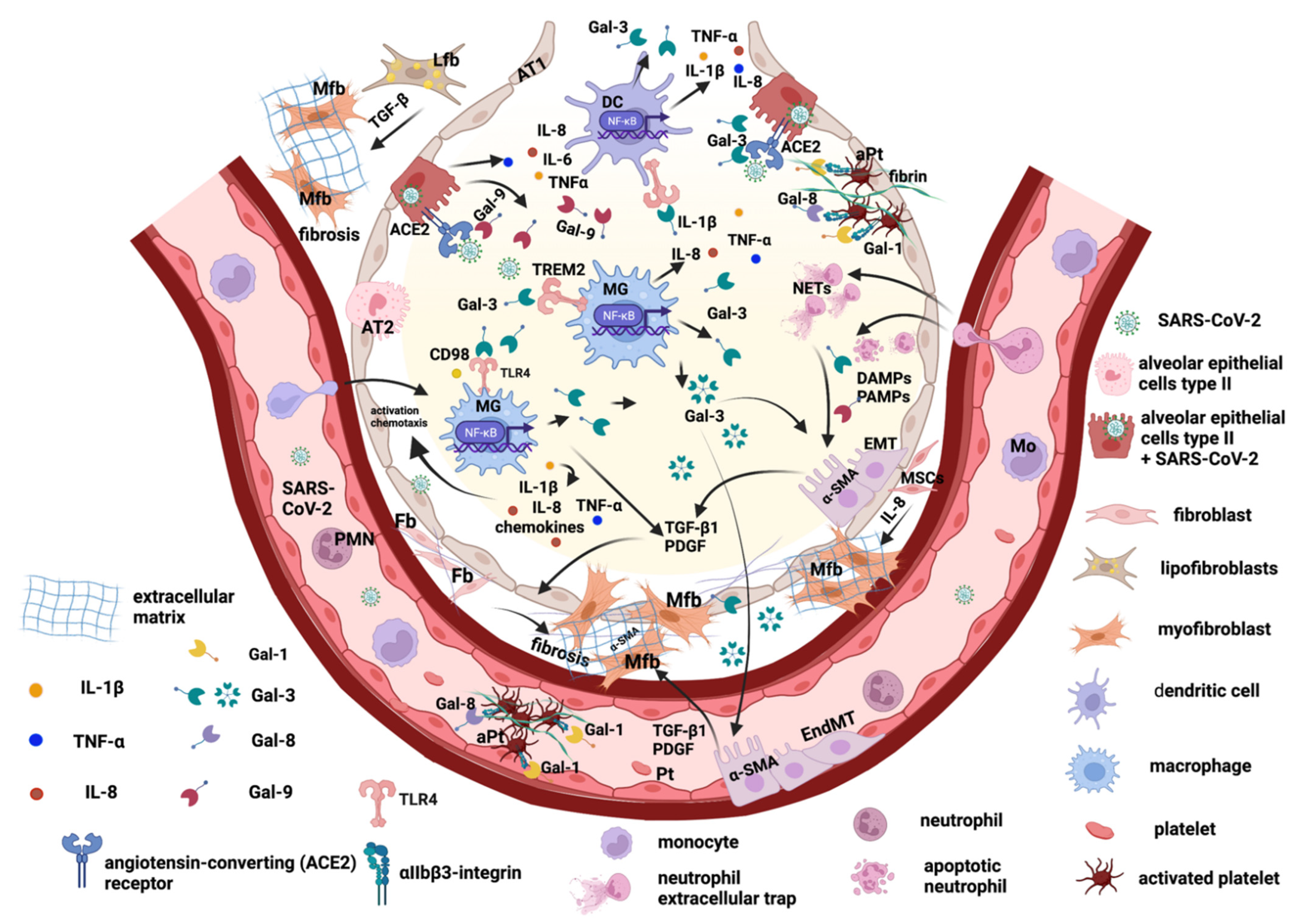
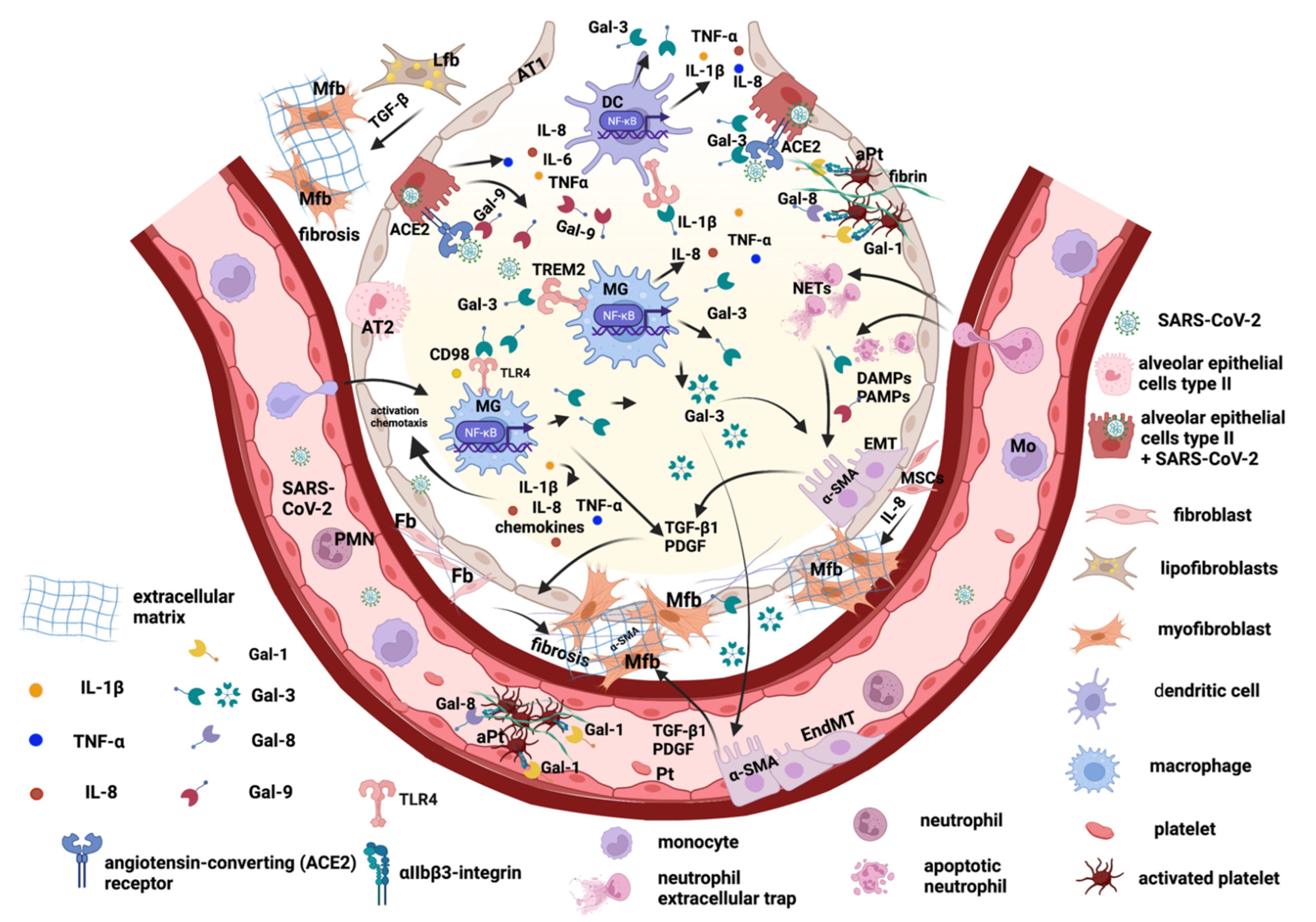
2. The Main Key Players in Promoting Pulmonary Fibrosis after SARS-CoV-2 Viral Infection
2.1. Lung Epithelial Cells and Epithelial–Mesenchymal Transition
2.2. Endothelial Cells and Trans-Differentiation to Myofibroblasts
2.3. Lung Fibroblasts and Trans-Differentiation to Myofibroblasts
2.4. Lung Lipofibroblasts
2.5. Lung Macrophages
3. Galectins Promote Lung Tissue Remodeling and Fibrosis Post-COVID-19
3.1. Galectin-1 and -8
3.2. Galectin-3
3.3. Galectin-9
4. Therapeutic Strategies Based on Galectin Inhibitors
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Todd, N.W.; Luzina, I.G.; Atamas, S.P. Molecular and Cellular Mechanisms of Pulmonary Fibrosis. Fibrogenesis Tissue Repair 2012, 5, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, L.T. Healing after COVID-19: Are Survivors at Risk for Pulmonary Fibrosis? Am. J. Physiol. Lung Cell. Mol. Physiol. 2021, 320, L257–L265. [Google Scholar] [CrossRef] [PubMed]
- Tale, S.; Ghosh, S.; Meitei, S.P.; Kolli, M.; Garbhapu, A.K.; Pudi, S. Post-COVID-19 Pneumonia Pulmonary Fibrosis. QJM Int. J. Med. 2020, 113, 837–838. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, A.; Chauhan, P.; Saha, B.; Jafarzadeh, S.; Nemati, M. Contribution of Monocytes and Macrophages to the Local Tissue Inflammation and Cytokine Storm in COVID-19: Lessons from SARS and MERS, and Potential Therapeutic Interventions. Life Sci. 2020, 257, 118102. [Google Scholar] [CrossRef]
- Tanni, S.E.; Fabro, A.T.; de Albuquerque, A.; Ferreira, E.V.M.; Verrastro, C.G.Y.; Sawamura, M.V.Y.; Ribeiro, S.M.; Baldi, B.G. Pulmonary Fibrosis Secondary to COVID-19: A Narrative Review. Expert Rev. Respir. Med. 2021, 15, 791–803. [Google Scholar] [CrossRef]
- Rai, D.K.; Sharma, P.; Kumar, R. Post COVID 19 Pulmonary Fibrosis. Is It Real Threat? Indian J. Tuberc. 2021, 68, 330–333. [Google Scholar] [CrossRef]
- McGroder, C.F.; Zhang, D.; Choudhury, M.A.; Salvatore, M.M.; D’Souza, B.M.; Hoffman, E.A.; Wei, Y.; Baldwin, M.R.; Garcia, C.K. Pulmonary Fibrosis 4 Months after COVID-19 Is Associated with Severity of Illness and Blood Leucocyte Telomere Length. Thorax 2021, 76, 1242–1245. [Google Scholar] [CrossRef]
- Patil, S.V.; Gondhali, G.; Patil, R.; Kasture, L. Post-COVID-19 Lung Fibrosis: Study of 600 Cases in Tertiary Care Setting in India. In Proceedings of the TP48. TP048 COVID: ARDS Clinical Studies; American Thoracic Society: New York, NY, USA, 2021; p. A2502. [Google Scholar]
- Baldi, B.G.; Tanni, S.E. Pulmonary Fibrosis and Follow-up of COVID-19 Survivors: An Urgent Need for Clarification. J. Bras. Pneumol. 2021, 47, e20210213. [Google Scholar] [CrossRef]
- Gentile, F.; Aimo, A.; Forfori, F.; Catapano, G.; Clemente, A.; Cademartiri, F.; Emdin, M.; Giannoni, A. COVID-19 and Risk of Pulmonary Fibrosis: The Importance of Planning Ahead. Eur. J. Prev. Cardiol. 2020, 27, 1442–1446. [Google Scholar] [CrossRef]
- Ngai, J.C.; Ko, F.W.; Ng, S.S.; To, K.; Tong, M.; Hui, D.S. The Long-term Impact of Severe Acute Respiratory Syndrome on Pulmonary Function, Exercise Capacity and Health Status. Respirology 2010, 15, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Y.Y.; Veldhuizen, R.A.W.; Neumann, A.W.; Petersen, N.O.; Possmayer, F. Current Perspectives in Pulmonary Surfactant—Inhibition, Enhancement and Evaluation. Biochim. Biophys. Acta Biomembr. 2008, 1778, 1947–1977. [Google Scholar] [CrossRef] [Green Version]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L.M. Type 2 Alveolar Cells Are Stem Cells in Adult Lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Camelo, A.; Dunmore, R.; Sleeman, M.A.; Clarke, D.L. The Epithelium in Idiopathic Pulmonary Fibrosis: Breaking the Barrier. Front. Pharmacol. 2014, 4, 173. [Google Scholar] [CrossRef] [Green Version]
- Thompson, B.T.; Chambers, R.C.; Liu, K.D. Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2017, 377, 562–572. [Google Scholar] [CrossRef]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, N.R.; King, L.S.; D’Alessio, F.R. Diverse Macrophage Populations Mediate Acute Lung Inflammation and Resolution. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L709–L725. [Google Scholar] [CrossRef]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. The Leading Role of Epithelial Cells in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Cell Signal 2020, 66, 109482. [Google Scholar] [CrossRef]
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2020, 180, 107–121.e17. [Google Scholar] [CrossRef]
- Li, M.; Krishnaveni, M.S.; Li, C.; Zhou, B.; Xing, Y.; Banfalvi, A.; Li, A.; Lombardi, V.; Akbari, O.; Borok, Z.; et al. Epithelium-Specific Deletion of TGF-β Receptor Type II Protects Mice from Bleomycin-Induced Pulmonary Fibrosis. J. Clin. Investig. 2011, 121, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Mu, D.; Cambier, S.; Fjellbirkeland, L.; Baron, J.L.; Munger, J.S.; Kawakatsu, H.; Sheppard, D.; Broaddus, V.C.; Nishimura, S.L. The Integrin Alpha(v)Beta8 Mediates Epithelial Homeostasis through MT1-MMP-Dependent Activation of TGF-Beta1. J. Cell. Biol. 2002, 157, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Bonniaud, P.; Martin, G.; Margetts, P.J.; Ask, K.; Robertson, J.; Gauldie, J.; Kolb, M. Connective Tissue Growth Factor Is Crucial to Inducing a Profibrotic Environment in “Fibrosis-Resistant” BALB/c Mouse Lungs. Am. J. Respir. Cell. Mol. Biol. 2004, 31, 510–516. [Google Scholar] [CrossRef] [Green Version]
- Watts, K.L.; Spiteri, M.A. Connective Tissue Growth Factor Expression and Induction by Transforming Growth Factor-Beta Is Abrogated by Simvastatin via a Rho Signaling Mechanism. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L1323–L1332. [Google Scholar] [CrossRef] [Green Version]
- Lasky, J.A.; Ortiz, L.A.; Tonthat, B.; Hoyle, G.W.; Corti, M.; Athas, G.; Lungarella, G.; Brody, A.; Friedman, M. Connective Tissue Growth Factor MRNA Expression Is Upregulated in Bleomycin-Induced Lung Fibrosis. Am. J. Physiol. 1998, 275, L365–L371. [Google Scholar] [CrossRef]
- Pan, L.H.; Yamauchi, K.; Uzuki, M.; Nakanishi, T.; Takigawa, M.; Inoue, H.; Sawai, T. Type II Alveolar Epithelial Cells and Interstitial Fibroblasts Express Connective Tissue Growth Factor in IPF. Eur. Respir. J. 2001, 17, 1220–1227. [Google Scholar] [CrossRef] [Green Version]
- Giacomelli, C.; Piccarducci, R.; Marchetti, L.; Romei, C.; Martini, C. Pulmonary Fibrosis from Molecular Mechanisms to Therapeutic Interventions: Lessons from Post-COVID-19 Patients. Biochem. Pharmacol. 2021, 193, 114812. [Google Scholar] [CrossRef]
- Bartis, D.; Mise, N.; Mahida, R.Y.; Eickelberg, O.; Thickett, D.R. Epithelial–Mesenchymal Transition in Lung Development and Disease: Does It Exist and Is It Important? Thorax 2014, 69, 760–765. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar Epithelial Cell Mesenchymal Transition Develops in Vivo during Pulmonary Fibrosis and Is Regulated by the Extracellular Matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [Green Version]
- Pandolfi, L.; Bozzini, S.; Frangipane, V.; Percivalle, E.; De Luigi, A.; Violatto, M.B.; Lopez, G.; Gabanti, E.; Carsana, L.; D’Amato, M.; et al. Neutrophil Extracellular Traps Induce the Epithelial-Mesenchymal Transition: Implications in Post-COVID-19 Fibrosis. Front. Immunol. 2021, 12, 663303. [Google Scholar] [CrossRef]
- Ye, Z.; Hu, Y. TGF-β1: Gentlemanly Orchestrator in Idiopathic Pulmonary Fibrosis (Review). Int. J. Mol. Med. 2021, 48, 132. [Google Scholar] [CrossRef]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of Canonical Wnt Signalling Is Required for TGF-β-Mediated Fibrosis. Nat. Commun. 2012, 3, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Liu, Y.; Kahn, M.; Ann, D.K.; Han, A.; Wang, H.; Nguyen, C.; Flodby, P.; Zhong, Q.; Krishnaveni, M.S.; et al. Interactions Between β-Catenin and Transforming Growth Factor-β Signaling Pathways Mediate Epithelial-Mesenchymal Transition and Are Dependent on the Transcriptional Co-Activator CAMP-Response Element-Binding Protein (CREB)-Binding Protein (CBP). J. Biol. Chem. 2012, 287, 7026–7038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, C.; Li, J.; Liu, D.; Conforti, F.; Brereton, C.J.; Yao, L.; Zhou, Y.; Alzetani, A.; Chee, S.J.; Marshall, B.G.; et al. Autophagy Inhibition-Mediated Epithelial–Mesenchymal Transition Augments Local Myofibroblast Differentiation in Pulmonary Fibrosis. Cell. Death Dis. 2019, 10, 591. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Frank, D.B.; Kadzik, R.S.; Morley, M.P.; Rathi, K.S.; Wang, T.; Zhou, S.; Cheng, L.; Lu, M.M.; Morrisey, E.E. Hedgehog Actively Maintains Adult Lung Quiescence and Regulates Repair and Regeneration. Nature 2015, 526, 578–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Cassandras, M.; Peng, T. The Role of Hedgehog Signaling in Adult Lung Regeneration and Maintenance. J. Dev. Biol. 2019, 7, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolaños, A.L.; Milla, C.M.; Lira, J.C.; Ramírez, R.; Checa, M.; Barrera, L.; García-Alvarez, J.; Carbajal, V.; Becerril, C.; Gaxiola, M.; et al. Role of Sonic Hedgehog in Idiopathic Pulmonary Fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L978–L990. [Google Scholar] [CrossRef] [Green Version]
- Fitch, P.M.; Howie, S.E.M.; Wallace, W.A.H. Oxidative Damage and TGF-β Differentially Induce Lung Epithelial Cell Sonic Hedgehog and Tenascin-C Expression: Implications for the Regulation of Lung Remodelling in Idiopathic Interstitial Lung Disease: SHH and Tenascin-C in Type-II Alveolar Cells. Int. J. Exp. Pathol. 2011, 92, 8–17. [Google Scholar] [CrossRef]
- Stewart, G.A.; Hoyne, G.F.; Ahmad, S.A.; Jarman, E.; Wallace, W.A.; Harrison, D.J.; Haslett, C.; Lamb, J.R.; Howie, S.E. Expression of the Developmental Sonic Hedgehog (Shh) Signalling Pathway Is up-Regulated in Chronic Lung Fibrosis and the Shh Receptor Patched 1 Is Present in Circulating T Lymphocytes. J. Pathol. 2003, 199, 488–495. [Google Scholar] [CrossRef]
- Lawson, W.E.; Cheng, D.-S.; Degryse, A.L.; Tanjore, H.; Polosukhin, V.V.; Xu, X.C.; Newcomb, D.C.; Jones, B.R.; Roldan, J.; Lane, K.B.; et al. Endoplasmic Reticulum Stress Enhances Fibrotic Remodeling in the Lungs. Proc. Natl. Acad. Sci. USA 2011, 108, 10562–10567. [Google Scholar] [CrossRef] [Green Version]
- Heindryckx, F.; Binet, F.; Ponticos, M.; Rombouts, K.; Lau, J.; Kreuger, J.; Gerwins, P. Endoplasmic Reticulum Stress Enhances Fibrosis through IRE 1α-mediated Degradation of MiR-150 and XBP -1 Splicing. EMBO Mol. Med. 2016, 8, 729–744. [Google Scholar] [CrossRef]
- Clere, C.; Renault, S.; Corre, I. Endothelial-to-Mesenchymal Transition in Cancer. Front. Cell Dev. Biol. 2020, 8, 747. [Google Scholar] [CrossRef]
- Gaikwad, A.V.; Eapen, M.S.; McAlinden, K.D.; Chia, C.; Larby, J.; Myers, S.; Dey, S.; Haug, G.; Markos, J.; Glanville, A.R.; et al. Endothelial to Mesenchymal Transition (EndMT) and Vascular Remodeling in Pulmonary Hypertension and Idiopathic Pulmonary Fibrosis. Expert Rev. Respir. Med. 2020, 14, 1027–1043. [Google Scholar] [CrossRef]
- Abdollahi, A.; Li, M.; Ping, G.; Plathow, C.; Domhan, S.; Kiessling, F.; Lee, L.B.; McMahon, G.; Gröne, H.-J.; Lipson, K.E.; et al. Inhibition of Platelet-Derived Growth Factor Signaling Attenuates Pulmonary Fibrosis. J. Exp. Med. 2005, 201, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; King, T.E.; Barker, E.; Daniloff, E.; Newman, L.S. Basic Fibroblast Growth Factor and Its Receptors in Idiopathic Pulmonary Fibrosis and Lymphangioleiomyomatosis. Am. J. Respir. Crit. Care Med. 2002, 166, 765–773. [Google Scholar] [CrossRef]
- Romero, Y.; Bueno, M.; Ramirez, R.; Álvarez, D.; Sembrat, J.C.; Goncharova, E.A.; Rojas, M.; Selman, M.; Mora, A.L.; Pardo, A. MTORC1 Activation Decreases Autophagy in Aging and Idiopathic Pulmonary Fibrosis and Contributes to Apoptosis Resistance in IPF Fibroblasts. Aging Cell 2016, 15, 1103–1112. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, G.; Wang, Z.; Qin, H.; Mo, B.; Wang, C. Elongation Factor-2 Kinase Acts Downstream of P38 MAPK to Regulate Proliferation, Apoptosis and Autophagy in Human Lung Fibroblasts. Exp. Cell. Res. 2018, 363, 291–298. [Google Scholar] [CrossRef]
- Youk, J.; Kim, T.; Evans, K.V.; Jeong, Y.-I.; Hur, Y.; Hong, S.P.; Kim, J.H.; Yi, K.; Kim, S.Y.; Na, K.J.; et al. Three-Dimensional Human Alveolar Stem Cell Culture Models Reveal Infection Response to SARS-CoV-2. Cell Stem Cell 2020, 27, 905–919.e10. [Google Scholar] [CrossRef]
- Kruglikov, I.L.; Scherer, P.E. The Role of Adipocytes and Adipocyte-Like Cells in the Severity of COVID-19 Infections. Obesity 2020, 28, 1187–1190. [Google Scholar] [CrossRef]
- Nouri-Keshtkar, M.; Taghizadeh, S.; Farhadi, A.; Ezaddoustdar, A.; Vesali, S.; Hosseini, R.; Totonchi, M.; Kouhkan, A.; Chen, C.; Zhang, J.-S.; et al. Potential Impact of Diabetes and Obesity on Alveolar Type 2 (AT2)-Lipofibroblast (LIF) Interactions After COVID-19 Infection. Front. Cell Dev. Biol. 2021, 9, 676150. [Google Scholar] [CrossRef]
- Rehan, V.K.; Torday, J.S. The Lung Alveolar Lipofibroblast: An Evolutionary Strategy Against Neonatal Hyperoxic Lung Injury. Antioxid. Redox Signal. 2014, 21, 1893–1904. [Google Scholar] [CrossRef] [Green Version]
- El Agha, E.; Moiseenko, A.; Kheirollahi, V.; De Langhe, S.; Crnkovic, S.; Kwapiszewska, G.; Szibor, M.; Kosanovic, D.; Schwind, F.; Schermuly, R.T.; et al. Two-Way Conversion between Lipogenic and Myogenic Fibroblastic Phenotypes Marks the Progression and Resolution of Lung Fibrosis. Cell Stem Cell 2017, 20, 261–273.e3. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Yang, L.; Huang, K. COVID-19 and Obesity: Epidemiology, Pathogenesis and Treatment. DMSO 2020, 13, 4953–4959. [Google Scholar] [CrossRef]
- Amici, S.A.; Dong, J.; Guerau-de-Arellano, M. Molecular Mechanisms Modulating the Phenotype of Macrophages and Microglia. Front. Immunol. 2017, 8, 1520. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, T.; Shichino, S.; Ueha, S.; Matsushima, K. Macrophages in Lung Fibrosis. Int. Immunol. 2021, 33, 665–671. [Google Scholar] [CrossRef]
- Phan, T.H.G.; Paliogiannis, P.; Nasrallah, G.K.; Giordo, R.; Eid, A.H.; Fois, A.G.; Zinellu, A.; Mangoni, A.A.; Pintus, G. Emerging Cellular and Molecular Determinants of Idiopathic Pulmonary Fibrosis. Cell. Mol. Life Sci. 2021, 78, 2031–2057. [Google Scholar] [CrossRef]
- Heukels, P.; Moor, C.C.; von der Thüsen, J.H.; Wijsenbeek, M.S.; Kool, M. Inflammation and Immunity in IPF Pathogenesis and Treatment. Respir. Med. 2019, 147, 79–91. [Google Scholar] [CrossRef]
- Craig, V.J.; Zhang, L.; Hagood, J.S.; Owen, C.A. Matrix Metalloproteinases as Therapeutic Targets for Idiopathic Pulmonary Fibrosis. Am. J. Respir. Cell. Mol. Biol. 2015, 53, 585–600. [Google Scholar] [CrossRef] [Green Version]
- Gharib, S.A.; Johnston, L.K.; Huizar, I.; Birkland, T.P.; Hanson, J.; Wang, Y.; Parks, W.C.; Manicone, A.M. MMP28 Promotes Macrophage Polarization toward M2 Cells and Augments Pulmonary Fibrosis. J. Leukoc. Biol. 2014, 95, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Byrne, A.J.; Mathie, S.A.; Gregory, L.G.; Lloyd, C.M. Pulmonary Macrophages: Key Players in the Innate Defence of the Airways. Thorax 2015, 70, 1189–1196. [Google Scholar] [CrossRef] [Green Version]
- Wendisch, D.; Dietrich, O.; Mari, T.; von Stillfried, S.; Ibarra, I.L.; Mittermaier, M.; Mache, C.; Chua, R.L.; Knoll, R.; Timm, S.; et al. SARS-CoV-2 Infection Triggers Profibrotic Macrophage Responses and Lung Fibrosis. Cell 2021, 184, 6243–6261.e27. [Google Scholar] [CrossRef]
- Chen, S.T.; Park, M.D.; Del Valle, D.M.; Buckup, M.; Tabachnikova, A.; Simons, N.W.; Mouskas, K.; Lee, B.; Geanon, D.; D’Souza, D.; et al. Shift of Lung Macrophage Composition Is Associated with COVID-19 Disease Severity and Recovery. Immunology 2022. [Google Scholar] [CrossRef]
- Moin, A.S.M.; Sathyapalan, T.; Atkin, S.L.; Butler, A.E. Pro-Fibrotic M2 Macrophage Markers May Increase the Risk for COVID19 in Type 2 Diabetes with Obesity. Metabolism 2020, 112, 154374. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.-Y.; Rabinovich, G.A.; Liu, F.-T. Galectins: Structure, Function and Therapeutic Potential. Expert Rev. Mol. Med. 2008, 10, e17. [Google Scholar] [CrossRef] [PubMed]
- Cousin, J.; Cloninger, M. The Role of Galectin-1 in Cancer Progression, and Synthetic Multivalent Systems for the Study of Galectin-1. Int. J. Mol. Sci. 2016, 17, 1566. [Google Scholar] [CrossRef] [Green Version]
- Fajka-Boja, R.; Urbán, V.S.; Szebeni, G.J.; Czibula, Á.; Blaskó, A.; Kriston-Pál, É.; Makra, I.; Hornung, Á.; Szabó, E.; Uher, F.; et al. Galectin-1 Is a Local but Not Systemic Immunomodulatory Factor in Mesenchymal Stromal Cells. Cytotherapy 2016, 18, 360–370. [Google Scholar] [CrossRef]
- Sundblad, V.; Morosi, L.G.; Geffner, J.R.; Rabinovich, G.A. Galectin-1: A Jack-of-All-Trades in the Resolution of Acute and Chronic Inflammation. J. Immunol. 2017, 199, 3721–3730. [Google Scholar] [CrossRef] [Green Version]
- Frame, M.C.; Patel, H.; Serrels, B.; Lietha, D.; Eck, M.J. The FERM Domain: Organizing the Structure and Function of FAK. Nat. Rev. Mol. Cell. Biol. 2010, 11, 802–814. [Google Scholar] [CrossRef]
- Kathiriya, J.J.; Nakra, N.; Nixon, J.; Patel, P.S.; Vaghasiya, V.; Alhassani, A.; Tian, Z.; Allen-Gipson, D.; Davé, V. Galectin-1 Inhibition Attenuates Profibrotic Signaling in Hypoxia-Induced Pulmonary Fibrosis. Cell. Death Discov. 2017, 3, 17010. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Markovic, S.S.; Gajovic, N.; Jurisevic, M.; Jovanovic, M.; Jovicic, B.P.; Arsenijevic, N.; Mijailovic, Z.; Jovanovic, M.; Dolicanin, Z.; Jovanovic, I. Galectin-1 as the New Player in Staging and Prognosis of COVID-19. Sci. Rep. 2022, 12, 1272. [Google Scholar] [CrossRef]
- Arciniegas, E.; Carrillo, L.M.; Salgado, A. Potential Role of Galectin-Glycan Lattices in SARS-CoV-2 Infection and Pathogenesis: A Hypothesis. Explor. Res. Hypothesis Med. 2021, 6, 142–145. [Google Scholar] [CrossRef]
- Jurk, K.; Kehrel, B.E. Platelets: Physiology and Biochemistry. Semin. Thromb. Hemost. 2005, 31, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Schattner, M. Platelets and Galectins. Ann. Transl. Med. 2014, 2, 85. [Google Scholar] [CrossRef]
- Romaniuk, M.A.; Croci, D.O.; Lapponi, M.J.; Tribulatti, M.V.; Negrotto, S.; Poirier, F.; Campetella, O.; Rabinovich, G.A.; Schattner, M. Binding of Galectin-1 to αIIbβ3 Integrin Triggers “Outside-in” Signals, Stimulates Platelet Activation, and Controls Primary Hemostasis. FASEB J. 2012, 26, 2788–2798. [Google Scholar] [CrossRef]
- Schattner, M.; Rabinovich, G.A. Galectins: New Agonists of Platelet Activation. Biol. Chem. 2013, 394, 857–863. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.S.; Peyvandi, F.; Martin-Loeches, I. Pulmonary Immuno-Thrombosis in COVID-19 ARDS Pathogenesis. Intensive Care Med. 2021, 47, 899–902. [Google Scholar] [CrossRef]
- Caniglia, J.L.; Asuthkar, S.; Tsung, A.J.; Guda, M.R.; Velpula, K.K. Immunopathology of Galectin-3: An Increasingly Promising Target in COVID-19. F1000Research 2020, 9, 1078. [Google Scholar] [CrossRef]
- Caniglia, J.L.; Guda, M.R.; Asuthkar, S.; Tsung, A.J.; Velpula, K.K. A Potential Role for Galectin-3 Inhibitors in the Treatment of COVID-19. PeerJ 2020, 8, e9392. [Google Scholar] [CrossRef]
- Elola, M.T.; Ferragut, F.; Méndez-Huergo, S.P.; Croci, D.O.; Bracalente, C.; Rabinovich, G.A. Galectins: Multitask Signaling Molecules Linking Fibroblast, Endothelial and Immune Cell Programs in the Tumor Microenvironment. Cell. Immunol. 2018, 333, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Nangia-Makker, P.; Hogan, V.; Raz, A. Galectin-3 and Cancer Stemness. Glycobiology 2018, 28, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Alvarez, L.; Ortega, E. The Many Roles of Galectin-3, a Multifaceted Molecule, in Innate Immune Responses against Pathogens. Mediat. Inflamm. 2017, 2017, 9247574. [Google Scholar] [CrossRef] [Green Version]
- Mackinnon, A.C.; Gibbons, M.A.; Farnworth, S.L.; Leffler, H.; Nilsson, U.J.; Delaine, T.; Simpson, A.J.; Forbes, S.J.; Hirani, N.; Gauldie, J.; et al. Regulation of Transforming Growth Factor-Β1-Driven Lung Fibrosis by Galectin-3. Am. J. Respir. Crit. Care Med. 2012, 185, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Kasper, M.; Hughes, R.C. Immunocytochemical evidence for a modulation of galectin 3 (mac-2), a carbohydrate binding protein, in pulmonary fibrosis. J. Pathol. 1996, 179, 309–316. [Google Scholar] [CrossRef]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.-I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Behloul, N.; Baha, S.; Shi, R.; Meng, J. Role of the GTNGTKR Motif in the N-Terminal Receptor-Binding Domain of the SARS-CoV-2 Spike Protein. Virus Res. 2020, 286, 198058. [Google Scholar] [CrossRef]
- Garcia-Revilla, J.; Deierborg, T.; Venero, J.L.; Boza-Serrano, A. Hyperinflammation and Fibrosis in Severe COVID-19 Patients: Galectin-3, a Target Molecule to Consider. Front. Immunol. 2020, 11, 2069. [Google Scholar] [CrossRef]
- Sato, S.; St-Pierre, C.; Bhaumik, P.; Nieminen, J. Galectins in Innate Immunity: Dual Functions of Host Soluble Beta-Galactoside-Binding Lectins as Damage-Associated Molecular Patterns (DAMPs) and as Receptors for Pathogen-Associated Molecular Patterns (PAMPs). Immunol. Rev. 2009, 230, 172–187. [Google Scholar] [CrossRef]
- De Biasi, S.; Meschiari, M.; Gibellini, L.; Bellinazzi, C.; Borella, R.; Fidanza, L.; Gozzi, L.; Iannone, A.; Lo Tartaro, D.; Mattioli, M.; et al. Marked T Cell Activation, Senescence, Exhaustion and Skewing towards TH17 in Patients with COVID-19 Pneumonia. Nat. Commun. 2020, 11, 3434. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, M.; Chen, X.; Montaner, L.J. Cytokine Storm and Leukocyte Changes in Mild versus Severe SARS-CoV-2 Infection: Review of 3939 COVID-19 Patients in China and Emerging Pathogenesis and Therapy Concepts. J. Leukoc. Biol. 2020, 108, 17–41. [Google Scholar] [CrossRef]
- Portacci, A.; Diaferia, F.; Santomasi, C.; Dragonieri, S.; Boniello, E.; Di Serio, F.; Carpagnano, G.E. Galectin-3 as Prognostic Biomarker in Patients with COVID-19 Acute Respiratory Failure. Respir. Med. 2021, 187, 106556. [Google Scholar] [CrossRef]
- Xu, Z.; Li, X.; Huang, Y.; Mao, P.; Wu, S.; Yang, B.; Yang, Y.; Chen, K.; Liu, X.; Li, Y. The Predictive Value of Plasma Galectin-3 for Ards Severity and Clinical Outcome. Shock 2017, 47, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Sureshbabu, A.; Tonner, E.; Allan, G.J.; Flint, D.J. Relative Roles of TGF-β and IGFBP-5 in Idiopathic Pulmonary Fibrosis. Pulm. Med. 2011, 2011, 517687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurotsu, S.; Tanaka, K.; Niino, T.; Asano, T.; Sugizaki, T.; Azuma, A.; Suzuki, H.; Mizushima, T. Ameliorative Effect of Mepenzolate Bromide against Pulmonary Fibrosis. J. Pharmacol. Exp. Ther. 2014, 350, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollin, L.; Maillet, I.; Quesniaux, V.; Holweg, A.; Ryffel, B. Antifibrotic and Anti-Inflammatory Activity of the Tyrosine Kinase Inhibitor Nintedanib in Experimental Models of Lung Fibrosis. J. Pharmacol. Exp. Ther. 2014, 349, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Nishi, Y.; Sano, H.; Kawashima, T.; Okada, T.; Kuroda, T.; Kikkawa, K.; Kawashima, S.; Tanabe, M.; Goto, T.; Matsuzawa, Y.; et al. Role of Galectin-3 in Human Pulmonary Fibrosis. Allergol. Int. 2007, 56, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Chen, L.-P.; Yuan, G.; Zhang, A.-M.; Jiang, Y.; Li, S. Differences of TNF-α, IL-6 and Gal-3 in Lobar Pneumonia and Bronchial Pneumonia Caused by Mycoplasma Pneumoniae. Technol. Health Care 2020, 28, 711–719. [Google Scholar] [CrossRef]
- Henderson, N.C.; Collis, E.A.; Mackinnon, A.C.; Simpson, K.J.; Haslett, C.; Zent, R.; Ginsberg, M.; Sethi, T. CD98hc (SLC3A2) Interaction with Beta 1 Integrins Is Required for Transformation. J. Biol. Chem. 2004, 279, 54731–54741. [Google Scholar] [CrossRef] [Green Version]
- Rintoul, R.C.; Buttery, R.C.; Mackinnon, A.C.; Wong, W.S.; Mosher, D.; Haslett, C.; Sethi, T. Cross-Linking CD98 Promotes Integrin-like Signaling and Anchorage-Independent Growth. Mol. Biol. Cell 2002, 13, 2841–2852. [Google Scholar] [CrossRef] [Green Version]
- Burguillos, M.A.; Svensson, M.; Schulte, T.; Boza-Serrano, A.; Garcia-Quintanilla, A.; Kavanagh, E.; Santiago, M.; Viceconte, N.; Oliva-Martin, M.J.; Osman, A.M.; et al. Microglia-Secreted Galectin-3 Acts as a Toll-like Receptor 4 Ligand and Contributes to Microglial Activation. Cell. Rep. 2015, 10, 1626–1638. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Wang, W.; Qin, W.; Cheng, K.; Coulup, S.; Chavez, S.; Jiang, S.; Raparia, K.; De Almeida, L.M.V.; Stehlik, C.; et al. TLR4-Dependent Fibroblast Activation Drives Persistent Organ Fibrosis in Skin and Lung. JCI Insight 2018, 3, 98850. [Google Scholar] [CrossRef] [Green Version]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and Mouse Single-Nucleus Transcriptomics Reveal TREM2-Dependent and TREM2-Independent Cellular Responses in Alzheimer’s Disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef]
- Boza-Serrano, A.; Ruiz, R.; Sanchez-Varo, R.; García-Revilla, J.; Yang, Y.; Jimenez-Ferrer, I.; Paulus, A.; Wennström, M.; Vilalta, A.; Allendorf, D.; et al. Galectin-3, a Novel Endogenous TREM2 Ligand, Detrimentally Regulates Inflammatory Response in Alzheimer’s Disease. Acta Neuropathol. 2019, 138, 251–273. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, I.R.; Gilfillan, S.; Cella, M.; Aoshi, T.; Miller, M.; Piccio, L.; Hernandez, M.; Colonna, M. Cutting Edge: TREM-2 Attenuates Macrophage Activation. J. Immunol. 2006, 177, 3520–3524. [Google Scholar] [CrossRef]
- Wu, K.; Byers, D.E.; Jin, X.; Agapov, E.; Alexander-Brett, J.; Patel, A.C.; Cella, M.; Gilfilan, S.; Colonna, M.; Kober, D.L.; et al. TREM-2 Promotes Macrophage Survival and Lung Disease after Respiratory Viral Infection. J. Exp. Med. 2015, 212, 681–697. [Google Scholar] [CrossRef] [Green Version]
- Jia, W.; Wang, Z.; Gao, C.; Wu, J.; Wu, Q. Trajectory Modeling of Endothelial-to-Mesenchymal Transition Reveals Galectin-3 as a Mediator in Pulmonary Fibrosis. Cell. Death Dis. 2021, 12, 327. [Google Scholar] [CrossRef]
- Bozorgmehr, N.; Mashhouri, S.; Perez Rosero, E.; Xu, L.; Shahbaz, S.; Sligl, W.; Osman, M.; Kutsogiannis, D.J.; MacIntyre, E.; O’Neil, C.R.; et al. Galectin-9, a Player in Cytokine Release Syndrome and a Surrogate Diagnostic Biomarker in SARS-CoV-2 Infection. mBio 2021, 12, e00384-21. [Google Scholar] [CrossRef]
- Du, L.; Bouzidi, M.S.; Gala, A.; Deiter, F.; Billaud, J.-N.; Yeung, S.T.; Dabral, P.; Jin, J.; Simmons, G.; Dossani, Z.; et al. Human Galectin-9 Potently Enhances SARS-CoV-2 Replication and Inflammation in Airway Epithelial Cells. Microbiology 2022. [Google Scholar] [CrossRef]
- Martín-Quirós, A.; Maroun-Eid, C.; Avendaño-Ortiz, J.; Lozano-Rodríguez, R.; Valentín Quiroga, J.; Terrón, V.; Montalbán-Hernández, K.; García-Garrido, M.A.; Muñoz del Val, E.; del Balzo-Castillo, Á.; et al. Potential Role of the Galectin-9/TIM-3 Axis in the Disparate Progression of SARS-CoV-2 in a Married Couple: A Case Report. Biomed. Hub. 2021, 6, 48–58. [Google Scholar] [CrossRef]
- Bai, G.; Furushima, D.; Niki, T.; Matsuba, T.; Maeda, Y.; Takahashi, A.; Hattori, T.; Ashino, Y. High Levels of the Cleaved Form of Galectin-9 and Osteopontin in the Plasma Are Associated with Inflammatory Markers That Reflect the Severity of COVID-19 Pneumonia. Int. J. Mol. Sci. 2021, 22, 4978. [Google Scholar] [CrossRef] [PubMed]
- Galectin Therapeutics Inc. Phase 2 Study to Evaluate Non-Invasive Imaging Methods in Efficacy Assessment of GR-MD-02 for the Treatment of Liver Fibrosis in Patients with NASH with Advanced Fibrosis. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT02421094 (accessed on 20 July 2022).
- Galecto Biotech AB. GULLIVER-1—A Randomised, Double-Blind, Placebo Controlled, Phase Ib, 12-Week Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Orally Administered GB1211 in Participants with Suspected or Confirmed Non-Alcoholic Steatohepatitis (NASH) and Liver Fibrosis. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04607655 (accessed on 20 July 2022).
- Galecto Biotech AB; Syneos Health; bioRASI, LLC. A Study to Test the Efficacy and Safety of Inhaled GB0139 in Subjects with Idiopathic Pulmonary Fibrosis (IPF). 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03832946 (accessed on 20 July 2022).
- Galecto Biotech AB. A Placebo-Controlled RCT in HV’s Investigating the Safety, Tolerability and PK (Pharmacokinetic) of TD139, a Galectin-3 Inhibitor, followed by an Expansion Cohort Treating Subjects with Idiopathic Pulmonary Fibrosis (IPF). 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT02257177 (accessed on 20 July 2022).

{kind=link}
| Sponsor | Compound | Proposed Target | Indication | Phase | NCT Number |
|---|---|---|---|---|---|
| Galectin Therapeutics Inc. | GR-MD-02 | Galectin 3 | Non-alcoholic steatohepatitis (NASH) with advance fibrosis | Phase 2 | NCT02421094 |
| Galecto Biotech AB | GB1211 | Galectin 3 | Non-alcoholic steatohepatitis (NASH) and liver fibrosis | Phase 1b/2a | NCT04607655 |
| Galecto Biotech AB | GB0139 | Galectin-3 | Idiopathic Pulmonary Fibrosis (IPF) | Phase 2 | NCT03832946 |
| Galecto Biotech AB | TD139 | Galectin-3 | Idiopathic Pulmonary Fibrosis (IPF) | Phase 1/2 | NCT02257177 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oatis, D.; Simon-Repolski, E.; Balta, C.; Mihu, A.; Pieretti, G.; Alfano, R.; Peluso, L.; Trotta, M.C.; D’Amico, M.; Hermenean, A. Cellular and Molecular Mechanism of Pulmonary Fibrosis Post-COVID-19: Focus on Galectin-1, -3, -8, -9. Int. J. Mol. Sci. 2022, 23, 8210. https://doi.org/10.3390/ijms23158210
Oatis D, Simon-Repolski E, Balta C, Mihu A, Pieretti G, Alfano R, Peluso L, Trotta MC, D’Amico M, Hermenean A. Cellular and Molecular Mechanism of Pulmonary Fibrosis Post-COVID-19: Focus on Galectin-1, -3, -8, -9. International Journal of Molecular Sciences. 2022; 23(15):8210. https://doi.org/10.3390/ijms23158210
Chicago/Turabian StyleOatis, Daniela, Erika Simon-Repolski, Cornel Balta, Alin Mihu, Gorizio Pieretti, Roberto Alfano, Luisa Peluso, Maria Consiglia Trotta, Michele D’Amico, and Anca Hermenean. 2022. "Cellular and Molecular Mechanism of Pulmonary Fibrosis Post-COVID-19: Focus on Galectin-1, -3, -8, -9" International Journal of Molecular Sciences 23, no. 15: 8210. https://doi.org/10.3390/ijms23158210
APA StyleOatis, D., Simon-Repolski, E., Balta, C., Mihu, A., Pieretti, G., Alfano, R., Peluso, L., Trotta, M. C., D’Amico, M., & Hermenean, A. (2022). Cellular and Molecular Mechanism of Pulmonary Fibrosis Post-COVID-19: Focus on Galectin-1, -3, -8, -9. International Journal of Molecular Sciences, 23(15), 8210. https://doi.org/10.3390/ijms23158210