
Neuroimmune Crosstalk in Rheumatoid Arthritis


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Affective Disturbance in RA
2.1. Psychiatric Comorbidities
2.2. Depression
2.3. Anxiety
2.4. Schizophrenia
2.5. Bipolar Disorder
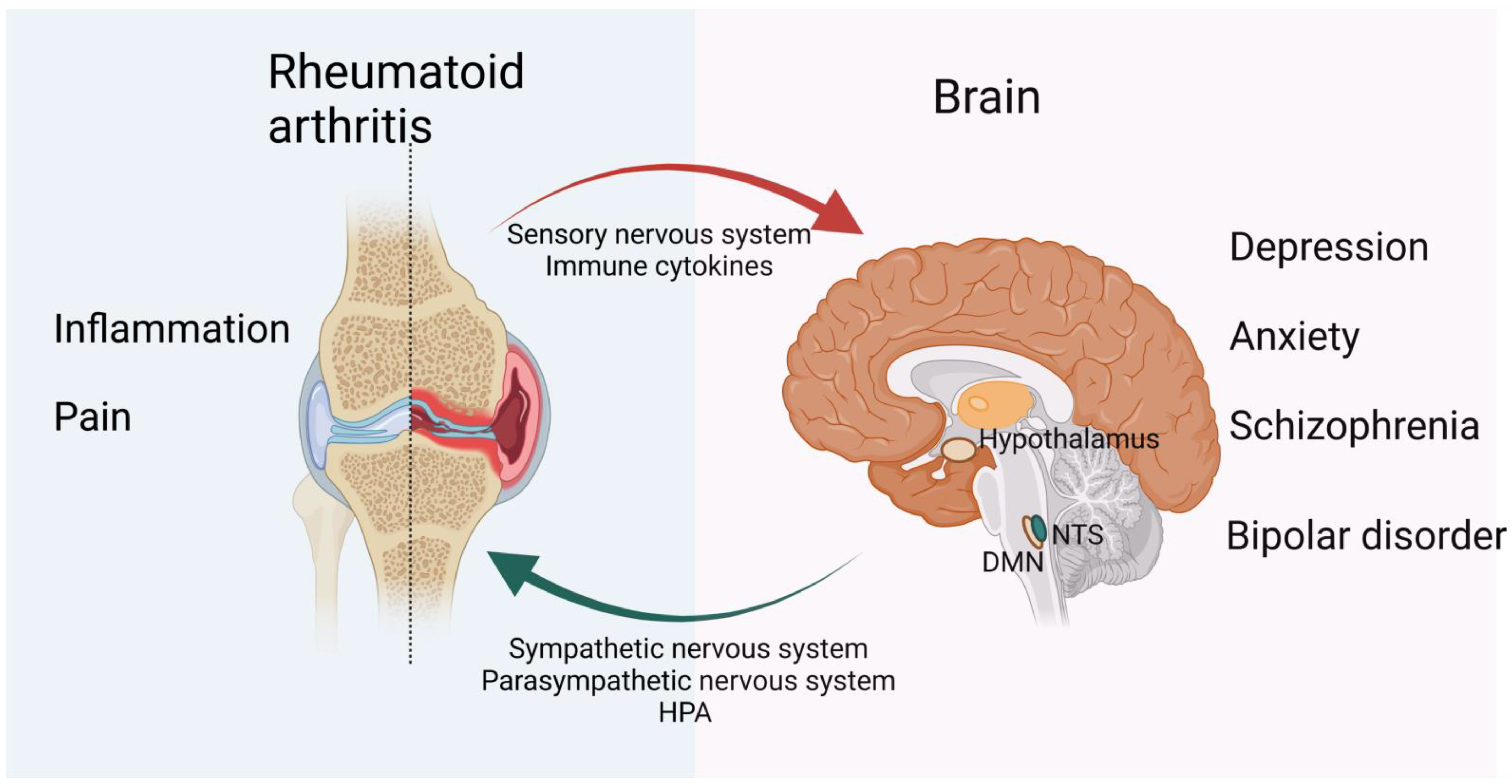
3. CNS Modulation of RA
4. The Regulatory Role of the Peripheral Nervous System in the Development of RA
4.1. Sensory Nervous System Interactions with Immune Cells in RA
4.2. The Sympathetic Nervous System in RA
4.3. Parasympathetic Nervous System
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sturgeon, J.A.; Finan, P.H.; Zautra, A.J. Affective disturbance in rheumatoid arthritis: Psychological and disease-related pathways. Nat. Rev. Rheumatol. 2016, 12, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Figus, F.A.; Piga, M.; Azzolin, I.; McConnell, R.; Iagnocco, A. Rheumatoid arthritis: Extra-articular manifestations and comorbidities. Autoimmun. Rev. 2021, 20, 102776. [Google Scholar] [CrossRef] [PubMed]
- Joaquim, A.F.; Appenzeller, S. Neuropsychiatric manifestations in rheumatoid arthritis. Autoimmun. Rev. 2015, 14, 1116–1122. [Google Scholar] [CrossRef]
- Kabata, H.; Artis, D. Neuro-immune crosstalk and allergic inflammation. J. Clin. Investig. 2019, 129, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.; Sinniger, V.; Pellissier, S. Anti-inflammatory properties of the vagus nerve: Potential therapeutic implications of vagus nerve stimulation. J. Physiol. 2016, 594, 5781–5790. [Google Scholar] [CrossRef]
- Vasconcelos, D.P.; Jabangwe, C.; Lamghari, M.; Alves, C.J. The Neuroimmune Interplay in Joint Pain: The Role of Macrophages. Front. Immunol. 2022, 13, 812962. [Google Scholar] [CrossRef]
- Mohanta, S.K.; Peng, L.; Li, Y.; Lu, S.; Sun, T.; Carnevale, L.; Perrotta, M.; Ma, Z.; Förstera, B.; Stanic, K.; et al. Neuroimmune cardiovascular interfaces control atherosclerosis. Nature 2022, 605, 152–159. [Google Scholar] [CrossRef]
- Raoof, R.; Willemen, H.L.D.M.; Eijkelkamp, N. Divergent roles of immune cells and their mediators in pain. Rheumatology 2018, 57, 429–440. [Google Scholar] [CrossRef]
- Dou, B.; Li, Y.; Ma, J.; Xu, Z.; Fan, W.; Tian, L.; Chen, Z.; Li, N.; Gong, Y.; Lyu, Z.; et al. Role of Neuroimmune Crosstalk in Mediating the Anti-inflammatory and Analgesic Effects of Acupuncture on Inflammatory Pain. Front. Neurosci. 2021, 15, 695670. [Google Scholar] [CrossRef]
- Wang, T.; Ma, C. Peripheral Nociceptors as Immune Sensors in the Development of Pain and Itch; Springer: Dordrecht, The Netherlands, 2016; pp. 77–85. [Google Scholar]
- Van, L.; Verdurmen, J.; Have, T. The association between arthritis and psychiatric disorders; results from a longitudinal population-based study. J. Psychosom. Res. 2010, 68, 187–193. [Google Scholar] [CrossRef]
- Postal, M.; Costallat, L.T.L.; Appenzeller, S. Neuropsychiatric Manifestations in Systemic Lupus Erythematosus. CNS Drugs 2011, 25, 721–736. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Lu, B.; Edwards, R.R.; Wasan, A.D.; Nassikas, N.J.; Clauw, D.J.; Solomon, D.H.; Karlson, E.W. The role of sleep problems in central pain processing in rheumatoid arthritis. Arthritis Rheum. 2014, 65, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Rupp, I.; Boshuizen, H.C.; Roorda, L.D.; Dinant, H.J.; Bos, G.V.D. Poor and good health outcomes in rheumatoid arthritis: The role of comorbidity. J. Rheumatol. 2006, 33, 1488. [Google Scholar]
- Löwe, B.; Willand, L.; Eich, W.; Zipfel, S.; Fiehn, C. Psychiatric comorbidity and work disability in patients with inflammatory rheumatic diseases. Psychosom. Med. 2004, 66, 395. [Google Scholar] [PubMed]
- Marrie, R.A.; Hitchon, C.A.; Walld, R.; Patten, S.B.; Bolton, J.M.; Sareen, J.; Walker, J.R.; Singer, A.; Lix, L.M.; El-Gabalawy, R. Increased Burden of Psychiatric Disorders in Rheumatoid Arthritis. Arthritis Care Res. 2018, 70, 970–978. [Google Scholar] [CrossRef] [PubMed]
- Mattey, D.L.; Dawes, P.T.; Hassell, A.B.; Brownfield, A.; Packham, J.C. Effect of psychological distress on continuation of anti-tumor necrosis factor therapy in patients with rheumatoid arthritis. J. Rheumatol. 2010, 37, 2021–2024. [Google Scholar] [CrossRef]
- Faith, M.; Rebecca, D.; Matthew, H.; Hyrich, K.L.; Sam, N.; Sophia, S.; James, G. The relationship between depression and biologic treatment response in rheumatoid arthritis: An analysis of the British Society for Rheumatology Biologics Register. Rheumatology 2018, 57, 835–843. [Google Scholar]
- Faith, M.; Lauren, R.; Sophia, S.; Matthew, H. The prevalence of depression in rheumatoid arthritis: A systematic review and meta-analysis. Rheumatology 2013, 52, 2136–2148. [Google Scholar]
- Vandyke, M.M.; Parker, J.C.; Smarr, K.L.; Hewett, J.E.; Johnson, G.E.; Slaughter, J.R.; Walker, S.E. Anxiety in rheumatoid arthritis. Arthritis Rheum. 2010, 51, 408–412. [Google Scholar] [CrossRef]
- Covic, T.; Cumming, S.R.; Pallant, J.F.; Manolios, N.; Emery, P.; Conaghan, P.G.; Tennant, A. Depression and Anxiety in Patients with Rheumatoid Arthritis: A comparison of the Depression, Anxiety and Stress Scale (DASS) and the Hospital, Anxiety and Depression Scale (HADS). BMC Psychiatry 2012, 12, 6. [Google Scholar] [CrossRef]
- Geisser, M.E.; Roth, R.S.; Robinson, M.E. Assessing depression among persons with chronic pain using the Center for Epidemiological Studies-Depression Scale and the Beck Depression Inventory: A comparative analysis. Clin. J. Pain 1997, 13, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Ann, M.R.; Randy, W.; Bolton, J.M.; Jitender, S.; Patten, S.B.; Alexander, S.; Lix, L.M.; Hitchon, C.A.; Renée, E.-G.; Alan, K. Psychiatric comorbidity increases mortality in immune-mediated inflammatory diseases. Gen. Hosp. Psychiatry 2018, 53, 65. [Google Scholar]
- Sambamoorthi, U.; Shah, D.; Zhao, X. Healthcare burden of depression in adults with arthritis. Expert Rev. Pharm. Outcomes Res. 2017, 17, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Dimatteo, M.R.; Lepper, H.S.; Croghan, T.W. Depression Is a Risk Factor for Noncompliance With Medical Treatment: Meta-analysis of the Effects of Anxiety and Depression on Patient Adherence. Arch. Intern. Med. 2000, 160, 2101–2107. [Google Scholar] [CrossRef] [PubMed]
- Rathbun, A.M.; Reed, G.W.; Harrold, L.R. The temporal relationship between depression and rheumatoid arthritis disease activity, treatment persistence and response: A systematic review. Rheumatology 2013, 52, 1785–1794. [Google Scholar] [CrossRef] [PubMed]
- Vallerand, I.A.; Patten, S.B.; Barnabe, C. Depression and the risk of rheumatoid arthritis. Lippincott Williams Wilkins Open Access 2019, 31, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Sparks, J.A.; Malspeis, S.; Hahn, J.; Wang, J.; Roberts, A.L.; Kubzansky, L.D.; Costenbader, K.H. Depression and subsequent risk for incident rheumatoid arthritis among women. Arthritis Care Res. 2020, 73, 78–89. [Google Scholar] [CrossRef]
- Du, X.; Pang, T.Y. Is Dysregulation of the HPA-Axis a Core Pathophysiology Mediating Co-Morbid Depression in Neurodegenerative Diseases? Front. Psychiatry 2015, 6, 32. [Google Scholar] [CrossRef]
- Murphy, L.B.; Sacks, J.J.; Brady, T.J.; Hootman, J.M.; Chapman, D.P. Anxiety and depression among US adults with arthritis: Prevalence and correlates. Arthritis Care Res. 2012, 64, 968–976. [Google Scholar]
- Chandarana, P.C.; Eals, M.; Steingart, A.B.; Bellamy, N.; Allen, S. The Detection of Psychiatric Morbidity and Associated Factors in Patients with Rheumatoid Arthritis. Can. J. Psychiatry. Rev. Can. De Psychiatr. 1987, 32, 356–361. [Google Scholar] [CrossRef]
- Roger, H.O.; Erin, F.U.; Chua, A.; Cheak, A.; Mak, A. Clinical and psychosocial factors associated with depression and anxiety in Singaporean patients with rheumatoid arthritis. Int. J. Rheum. Dis. 2011, 14, 37–47. [Google Scholar]
- Ijaz, H.I.; Yaser, I.M. Depression in Rheumatoid Arthritis and its relation to disease activity. Pak. J. Med. Sci. 2015, 31, 393. [Google Scholar]
- Kojima, M.; Kojima, T.; Suzuki, S.; Oguchi, T.; Oba, M.; Tsuchiya, H.; Sugiura, F.; Kanayama, Y.; Furukawa, T.A.; Tokudome, S.; et al. Depression, inflammation, and pain in patients with rheumatoid arthritis. Arthritis Care Res. 2009, 61, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Mok, C.; Lok, E.; Cheung, E. Concurrent psychiatric disorders are associated with significantly poorer quality of life in patients with rheumatoid arthritis. Scand. J. Rheumatol. 2012, 41, 253–259. [Google Scholar] [CrossRef]
- Faith, M.; Sam, N.; Scott, D.L.; Sophia, S.; Matthew, H. Symptoms of depression and anxiety predict treatment response and long-term physical health outcomes in rheumatoid arthritis: Secondary analysis of a randomized controlled trial. Rheumatology 2016, 55, 268–278. [Google Scholar]
- Li, M.; Fan, Y.L.; Tang, Z.Y.; Cheng, X.S. Schizophrenia and risk of stroke: A meta-analysis of cohort studies. Int. J. Cardiol. 2014, 173, 588–590. [Google Scholar] [CrossRef]
- Chen, S.-F.; Wang, L.-Y.; Chiang, J.-H.; Hsu, C.-Y.; Shen, Y.-C. Assessing whether the association between rheumatoid arthritis and schizophrenia is bidirectional: A nationwide population-based cohort study. Sci. Rep. 2019, 9, 4493. [Google Scholar] [CrossRef]
- Feigenson, K.A.; Kusnecov, A.W.; Silverstein, S.M. Inflammation and the Two-Hit Hypothesis of Schizophrenia. Neurosci. Biobehav. Rev. 2013, 38, 72–93. [Google Scholar] [CrossRef]
- Potvin, S.; Stip, E.; Sepehry, A.A.; Gendron, A.; Bah, R.; Kouassi, E. Inflammatory Cytokine Alterations in Schizophrenia: A Systematic Quantitative Review. Biol. Psychiatry 2008, 63, 801–808. [Google Scholar] [CrossRef]
- Lee, S.H.; Byrne, E.M.; Hultman, C.M.; Kähler, A.; Vinkhuyzen, A.A.; Ripke, S.; Andreassen, O.A.; Frisell, T.; Gusev, A.; Hu, X.; et al. New data and an old puzzle: The negative association between schizophrenia and rheumatoid arthritis. Int. J. Epidemiol. 2015, 44, 1706–1721. [Google Scholar] [CrossRef]
- Dinarello, C.A. The many worlds of reducing interleukin-1. Arthritis Rheum. 2005, 52, 1960–1967. [Google Scholar] [CrossRef] [PubMed]
- Gorwood, P.; Pouchot, J.; Vinceneux, P.; Puéchal, X.; Flipo, R.M.; Bandt, M.; Adès, J. Rheumatoid arthritis and schizophrenia: A negative association at a dimensional level. Schizophr. Res. 2004, 66, 21–29. [Google Scholar] [CrossRef]
- Wright, P.; Nimgaonkar, V.L.; Donaldson, P.T.; Murray, R.M. Schizophrenia and HLA: A review. Schizophr. Res. 2001, 47, 1–12. [Google Scholar] [CrossRef]
- Watanabe, Y.; Nunokawa, A.; Kaneko, N.; Muratake, T.; Arinami, T.; Ujike, H.; Inada, T.; Iwata, N.; Kunugi, H.; Itokawa, M. Two-stage case–control association study of polymorphisms in rheumatoid arthritis susceptibility genes with schizophrenia. J. Hum. Genet. 2009, 54, 62–65. [Google Scholar] [CrossRef][Green Version]
- Najjar, S.; Pearlman, D.M.; Alper, K. Neuroinflammation and psychiatric illness. J. Neuroinflamm. 2013, 10, 816. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Chen, S.C.; Liu, C.J.; Lu, T.; Shen, C.C.; Hu, Y.W.; Yeh, C.M.; Chen, P.M.; Chen, T.J.; Hu, L.Y. Rheumatoid Arthritis and the Risk of Bipolar Disorder: A Nationwide Population-Based Study. PLoS ONE 2014, 9, e107512. [Google Scholar] [CrossRef]
- Goldstein, B.I.; Kemp, D.E.; Soczynska, J.K.; Mcintyre, R.S. Inflammation and the Phenomenology, Pathophysiology, Comorbidity, and Treatment of Bipolar Disorder: A Systematic Review of the Literature. J. Clin. Psychiatry 2009, 70, 1078–1090. [Google Scholar] [CrossRef]
- Price, A.L.; Marzani-Nissen, G.R. Bipolar disorders: A review. Am. Fam. Physician 2012, 85, 483. [Google Scholar]
- Cutolo, M.; Sulli, A.; Straub, R.H. Estrogen metabolism and autoimmunity. Autoimmun. Rev. 2012, 11, A460–A464. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Li, X.; Li, X.; Sheng, Y.Y.; Zhuang, P.W.; Zhang, Y.J. Neuroimmune crosstalk in central nervous system injury-induced infection and pharmacological intervention. Brain Res. Bull. 2019, 153, 232–238. [Google Scholar] [CrossRef]
- Anastasia, B.; Evangelos, A.; Giatas, K.; Paraskevas, G.; Nikolaos, T.; Evangelia, K. A Systematic Review of Peripheral and Central Nervous System Involvement of Rheumatoid Arthritis, Systemic Lupus Erythematosus, Primary Sjgren’s Syndrome, and Associated Immunological Profiles. Int. J. Chronic Dis. 2015, 2015, 1–11. [Google Scholar]
- Sü, P.; Rothe, T.; Hoffmann, A.; Schlachetzki, J.; Winkler, J. The Joint-Brain Axis: Insights From Rheumatoid Arthritis on the Crosstalk Between Chronic Peripheral Inflammation and the Brain. Front. Immunol. 2020, 11, 612104. [Google Scholar]
- Fa, A.; Rt, B.; Ifm, C.; Mcg, D.; Rc, E.; Sp, F. Central nervous system involvement in rheumatoid arthritis patients and the potential implications of using biological agents. Best Pract. Res. Clin. Rheumatol. 2018, 32, 500–510. [Google Scholar]
- Dhillon, N.; Liang, K. Prevention of Stroke in Rheumatoid Arthritis. Curr. Treat. Options Neurol. 2015, 17, 356. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, B.; Yuan, Y.; Zhang, L.; Hu, L.; Jin, S.; Kang, B.; Liao, X.; Sun, W.; Xu, F.; et al. Brain control of humoral immune responses amenable to behavioural modulation. Nature 2020, 581, 204–208. [Google Scholar] [CrossRef]
- Rooney, M.; Symons, J.A.; Duff, G.W. Interleukin 1 beta in synovial fluid is related to local disease activity in rheumatoid arthritis. Rheumatol. Int. 1990, 10, 217–219. [Google Scholar] [CrossRef]
- Eastgate, J.A.; Symons, J.A.; Wood, N.C.; Grinlinton, F.M.; di Giovine, F.S.; Duff, G.W. Correlation of plasma interleukin 1 levels with disease activity in rheumatoid arthritis. Lancet 1988, 2, 706–709. [Google Scholar] [CrossRef]
- Liu, X.; Nemeth, D.; McKim, D.; Zhu, L. Cell-Type-Specific Interleukin 1 Receptor 1 Signaling in the Brain Regulates Distinct Neuroimmune Activities. Immunity 2019, 50, 317–333. [Google Scholar] [CrossRef]
- Zhu, L.; Chen, P.; Sun, X.; Zhang, S. Associations between Polymorphisms in the IL-1 Gene and the Risk of Rheumatoid Arthritis and Systemic Lupus Erythematosus: Evidence from a Meta-Analysis. Int. Arch. Allergy Immunol. 2020, 182, 1–9. [Google Scholar] [CrossRef]
- Ray, M.; Curtis, J.R.; Baddley, J.W. A case report of progressive multifocal leucoencephalopathy (PML) associated with adalimumab. Ann. Rheum. Dis. 2014, 73, 1429–1430. [Google Scholar] [CrossRef]
- Pongratz, G.; Straub, R.H. Role of peripheral nerve fibres in acute and chronic inflammation in arthritis. Nat. Rev. Rheumatol. 2013, 9, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Kohm, A.P.; Tang, Y.; Sanders, V.M.; Jones, S.B. Activation of antigen-specific CD4+ Th2 cells and B cells in vivo increases norepinephrine release in the spleen and bone marrow. J. Immunol. 2000, 165, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.J. The induction of pain: An integrative review. Prog. Neurobiol. 1999, 57, 1–164. [Google Scholar] [CrossRef]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef]
- Kang, K.; Pulver, S.R.; Panzano, V.C.; Chang, E.C.; Griffith, L.C.; Theobald, D.L.; Garrity, P.A. Analysis of Drosophila TRPA1 reveals an ancient origin for human chemical nociception. Nature 2010, 464, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Karashima, Y.; Talavera, K.; Everaerts, W.; Janssens, A.; Kwan, K.Y.; Vennekens, R.; Nilius, B.; Voets, T. TRPA1 acts as a cold sensor in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Riol-Blanco, L.; Ordovas-Montanes, J.; Perro, M.; Naval, E.; Thiriot, A.; Alvarez, D.; Paust, S.; Wood, J.N.; von Andrian, U.H. Nociceptive sensory neurons drive interleukin-23-mediated psoriasiform skin inflammation. Nature 2014, 510, 157–161. [Google Scholar] [CrossRef]
- Bautista, D.M.; Siemens, J.; Glazer, J.M.; Tsuruda, P.R.; Basbaum, A.I.; Stucky, C.L.; Jordt, S.E.; Julius, D. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 2007, 448, 204–208. [Google Scholar] [CrossRef]
- Liu, T.; Gao, Y.J.; Ji, R.R. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci. Bull. 2012, 28, 131–144. [Google Scholar] [CrossRef]
- Carolan, E.J.; Casale, T.B. Effects of neuropeptides on neutrophil migration through noncellular and endothelial barriers. J. Allergy Clin. Immunol. 1993, 92, 589–598. [Google Scholar] [CrossRef]
- Lorton, D.; Lubahn, C.; Engan, C.; Schaller, J.; Felten, D.L.; Bellinger, D.L. Local application of capsaicin into the draining lymph nodes attenuates expression of adjuvant-induced arthritis. Neuroimmunomodulation 2000, 7, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, T.; Sakai, A.; Ito, H.; Suzuki, H. Intra-articular administration of tachykinin NK₁ receptor antagonists reduces hyperalgesia and cartilage destruction in the inflammatory joint in rats with adjuvant-induced arthritis. Eur. J. Pharmacol. 2011, 668, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Leroy, V.; Mauser, P.; Gao, Z.; Peet, N.P. Neurokinin receptor antagonists. Expert Opin. Investig. Drugs 2000, 9, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, Y.; Zheng, X.; Gao, J.; Wang, L.; Li, Y. Investigation of the Potential Use of Sialic Acid as a Biomarker for Rheumatoid Arthritis. Ann. Clin. Lab. Sci. 2019, 49, 224–231. [Google Scholar]
- Fonseca, J.E.; Santos, M.J.; Canhão, H.; Choy, E. Interleukin-6 as a key player in systemic inflammation and joint destruction. Autoimmun. Rev. 2009, 8, 538–542. [Google Scholar] [CrossRef]
- Nowell, M.A.; Richards, P.J.; Horiuchi, S.; Yamamoto, N.; Rose-John, S.; Topley, N.; Williams, A.S.; Jones, S.A. Soluble IL-6 receptor governs IL-6 activity in experimental arthritis: Blockade of arthritis severity by soluble glycoprotein 130. J. Immunol. 2003, 171, 3202–3209. [Google Scholar] [CrossRef]
- Axmann, R.; Böhm, C.; Krönke, G.; Zwerina, J.; Smolen, J.; Schett, G. Inhibition of interleukin-6 receptor directly blocks osteoclast formation in vitro and in vivo. Arthritis Rheum. 2009, 60, 2747–2756. [Google Scholar] [CrossRef]
- Jones, S.A.; Richards, P.J.; Scheller, J.; Rose-John, S. IL-6 transsignaling: The in vivo consequences. J. Interferon Cytokine Res. 2005, 25, 241–253. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Bondeson, J.; Blom, A.B.; Wainwright, S.; Hughes, C.; Caterson, B.; van den Berg, W.B. The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis. Arthritis Rheum. 2010, 62, 647–657. [Google Scholar] [CrossRef]
- Ebbinghaus, M.; Uhlig, B.; Richter, F.; von Banchet, G.S.; Gajda, M.; Bräuer, R.; Schaible, H.G. The role of interleukin-1β in arthritic pain: Main involvement in thermal, but not mechanical, hyperalgesia in rat antigen-induced arthritis. Arthritis Rheum. 2012, 64, 3897–3907. [Google Scholar] [CrossRef] [PubMed]
- Copray, J.C.; Mantingh, I.; Brouwer, N.; Biber, K.; Küst, B.M.; Liem, R.S.; Huitinga, I.; Tilders, F.J.; Van Dam, A.M.; Boddeke, H.W. Expression of interleukin-1 beta in rat dorsal root ganglia. J. Neuroimmunol. 2001, 118, 203–211. [Google Scholar] [CrossRef]
- Obreja, O.; Rathee, P.K.; Lips, K.S.; Distler, C.; Kress, M. IL-1 beta potentiates heat-activated currents in rat sensory neurons: Involvement of IL-1RI, tyrosine kinase, and protein kinase C. FASEB J. 2002, 16, 1497–1503. [Google Scholar] [CrossRef] [PubMed]
- Schäfers, M.; Sorkin, L. Effect of cytokines on neuronal excitability. Neurosci. Lett. 2008, 437, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, H.; Kawatani, M.; Hisamitsu, T.; Takeshige, C. Cutaneous hyperalgesia induced by peripheral injection of interleukin-1 beta in the rat. Brain Res. 1994, 657, 133–140. [Google Scholar] [CrossRef]
- Pappu, R.; Ramirez-Carrozzi, V.; Ota, N.; Ouyang, W.; Hu, Y. The IL-17 family cytokines in immunity and disease. J. Clin. Immunol. 2010, 30, 185–195. [Google Scholar] [CrossRef]
- Lubberts, E. IL-17/Th17 targeting: On the road to prevent chronic destructive arthritis? Cytokine 2008, 41, 84–91. [Google Scholar] [CrossRef]
- Hueber, W.; Patel, D.D.; Dryja, T.; Wright, A.M.; Koroleva, I.; Bruin, G.; Antoni, C.; Draelos, Z.; Gold, M.H.; Durez, P.; et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci. Transl. Med. 2010, 2, 52ra72. [Google Scholar] [CrossRef]
- Inglis, J.J.; Notley, C.A.; Essex, D.; Wilson, A.W.; Feldmann, M.; Anand, P.; Williams, R. Collagen-induced arthritis as a model of hyperalgesia: Functional and cellular analysis of the analgesic actions of tumor necrosis factor blockade. Arthritis Rheum. 2007, 56, 4015–4023. [Google Scholar] [CrossRef]
- Boettger, M.K.; Hensellek, S.; Richter, F.; Gajda, M.; Stöckigt, R.; von Banchet, G.S.; Bräuer, R.; Schaible, H.G. Antinociceptive effects of tumor necrosis factor alpha neutralization in a rat model of antigen-induced arthritis: Evidence of a neuronal target. Arthritis Rheum. 2008, 58, 2368–2378. [Google Scholar] [CrossRef]
- Christianson, C.A.; Corr, M.; Firestein, G.S.; Mobargha, A.; Yaksh, T.L.; Svensson, C.I. Characterization of the acute and persistent pain state present in K/BxN serum transfer ar.rthritis. Pain 2010, 151, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Inglis, J.J.; Nissim, A.; Lees, D.M.; Hunt, S.P.; Chernajovsky, Y.; Kidd, B.L. The differential contribution of tumour necrosis factor to thermal and mechanical hyperalgesia during chronic inflammation. Arthritis Res. Ther. 2005, 7, R807–R816. [Google Scholar] [CrossRef] [PubMed]
- Hess, A.; Axmann, R.; Rech, J.; Finzel, S.; Heindl, C.; Kreitz, S.; Sergeeva, M.; Saake, M.; Garcia, M.; Kollias, G.; et al. Blockade of TNF-α rapidly inhibits pain responses in the central nervous system. Proc. Natl. Acad. Sci. USA 2011, 108, 3731–3736. [Google Scholar] [CrossRef]
- Straub, R.H.; Cutolo, M.; Buttgereit, F.; Pongratz, G. Energy regulation and neuroendocrine-immune control in chronic inflammatory diseases. J. Intern. Med. 2010, 267, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Straub, R.H. Evolutionary medicine and chronic inflammatory state--known and new concepts in pathophysiology. J. Mol. Med. 2012, 90, 523–534. [Google Scholar] [CrossRef]
- Besedovsky, H.O.; del Rey, A. Immune-neuro-endocrine interactions: Facts and hypotheses. Endocr. Rev. 1996, 17, 64–102. [Google Scholar] [CrossRef]
- Dhabhar, F.S.; Miller, A.H.; Stein, M.; McEwen, B.S.; Spencer, R.L. Diurnal and acute stress-induced changes in distribution of peripheral blood leukocyte subpopulations. Brain Behav. Immun. 1994, 8, 66–79. [Google Scholar] [CrossRef]
- Dhabhar, F.S.; McEwen, B.S. Acute stress enhances while chronic stress suppresses cell-mediated immunity in vivo: A potential role for leukocyte trafficking. Brain Behav. Immun. 1997, 11, 286–306. [Google Scholar] [CrossRef]
- Benschop, R.J.; Rodriguez-Feuerhahn, M.; Schedlowski, M. Catecholamine-induced leukocytosis: Early observations, current research, and future directions. Brain Behav. Immun. 1996, 10, 77–91. [Google Scholar] [CrossRef]
- Levine, J.D.; Coderre, T.J.; Helms, C.; Basbaum, A.I. Beta 2-adrenergic mechanisms in experimental arthritis. Proc. Natl. Acad. Sci. USA 1988, 85, 4553–4556. [Google Scholar] [CrossRef]
- Lorton, D.; Lubahn, C.; Klein, N.; Schaller, J.; Bellinger, D.L. Dual role for noradrenergic innervation of lymphoid tissue and arthritic joints in adjuvant-induced arthritis. Brain Behav. Immun. 1999, 13, 315–334. [Google Scholar] [CrossRef] [PubMed]
- Härle, P.; Möbius, D.; Carr, D.J.; Schölmerich, J.; Straub, R.H. An opposing time-dependent immune-modulating effect of the sympathetic nervous system conferred by altering the cytokine profile in the local lymph nodes and spleen of mice with type II collagen-induced arthritis. Arthritis Rheum. 2005, 52, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Kin, N.W.; Sanders, V.M. It takes nerve to tell T and B cells what to do. J. Leukoc. Biol. 2006, 79, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K.; Marshall, G.D., Jr. Beta-adrenergic modulation of human type-1/type-2 cytokine balance. J. Allergy Clin. Immunol. 2000, 105, 91–98. [Google Scholar] [CrossRef]
- Sitkovsky, M.V. Use of the A(2A) adenosine receptor as a physiological immunosuppressor and to engineer inflammation in vivo. Biochem. Pharmacol. 2003, 65, 493–501. [Google Scholar] [CrossRef]
- Sanders, V.M.; Straub, R.H. Norepinephrine, the beta-adrenergic receptor, and immunity. Brain Behav. Immun. 2002, 16, 290–332. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Vida, G.; Peña, G.; Kanashiro, A.; Thompson-Bonilla Mdel, R.; Palange, D.; Deitch, E.A.; Ulloa, L. β2-Adrenoreceptors of regulatory lymphocytes are essential for vagal neuromodulation of the innate immune system. FASEB J. 2011, 25, 4476–4485. [Google Scholar] [CrossRef]
- Goldstein, R.S.; Bruchfeld, A.; Yang, L.; Qureshi, A.R.; Gallowitsch-Puerta, M.; Patel, N.B.; Huston, B.J.; Chavan, S.; Rosas-Ballina, M.; Gregersen, P.K.; et al. Cholinergic anti-inflammatory pathway activity and High Mobility Group Box-1 (HMGB1) serum levels in patients with rheumatoid arthritis. Mol. Med. 2007, 13, 210–215. [Google Scholar] [CrossRef]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef]
- Rosas-Ballina, M.; Ochani, M.; Parrish, W.R.; Ochani, K.; Harris, Y.T.; Huston, J.M.; Chavan, S.; Tracey, K.J. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc. Natl. Acad. Sci. USA 2008, 105, 11008–11013. [Google Scholar] [CrossRef] [PubMed]
- Saeed, R.W.; Varma, S.; Peng-Nemeroff, T.; Sherry, B.; Balakhaneh, D.; Huston, J.; Tracey, K.J.; Al-Abed, Y.; Metz, C.N. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J. Exp. Med. 2005, 201, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- van Maanen, M.A.; Lebre, M.C.; van der Poll, T.; LaRosa, G.J.; Elbaum, D.; Vervoordeldonk, M.J.; Tak, P.P. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum. 2009, 60, 114–122. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, D.; Gao, X.; Yang, F.; Wang, Q. Neuroimmune Crosstalk in Rheumatoid Arthritis. Int. J. Mol. Sci. 2022, 23, 8158. https://doi.org/10.3390/ijms23158158
Gao D, Gao X, Yang F, Wang Q. Neuroimmune Crosstalk in Rheumatoid Arthritis. International Journal of Molecular Sciences. 2022; 23(15):8158. https://doi.org/10.3390/ijms23158158
Chicago/Turabian StyleGao, Dashuang, Xu Gao, Fan Yang, and Qingwen Wang. 2022. "Neuroimmune Crosstalk in Rheumatoid Arthritis" International Journal of Molecular Sciences 23, no. 15: 8158. https://doi.org/10.3390/ijms23158158
APA StyleGao, D., Gao, X., Yang, F., & Wang, Q. (2022). Neuroimmune Crosstalk in Rheumatoid Arthritis. International Journal of Molecular Sciences, 23(15), 8158. https://doi.org/10.3390/ijms23158158