
The Molecular Mechanisms of Actions, Effects, and Clinical Implications of PARP Inhibitors in Epithelial Ovarian Cancers: A Systematic Review




Abstract
:1. Introduction
2. Methods
3. Molecular Mechanisms of PARP and PARPi Actions
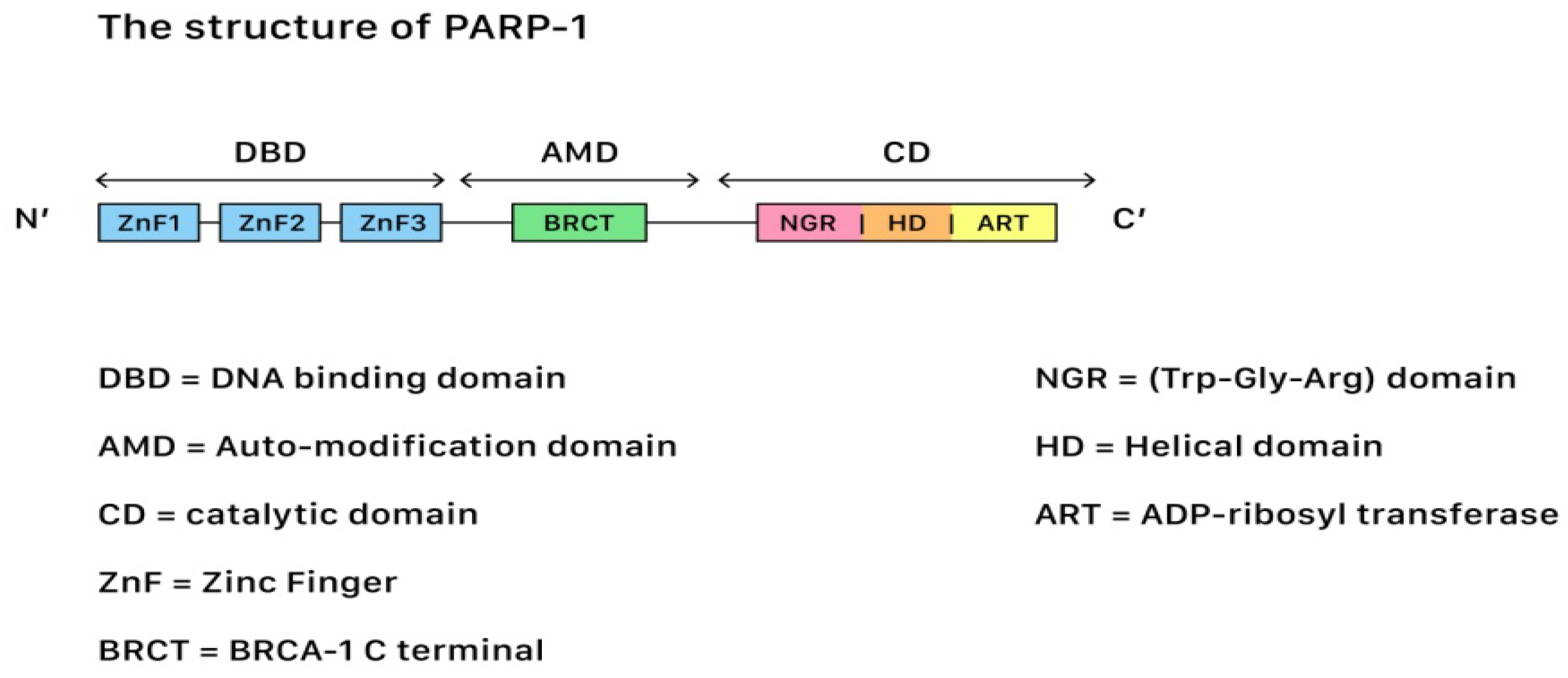
3.1. The PARP Enzymes
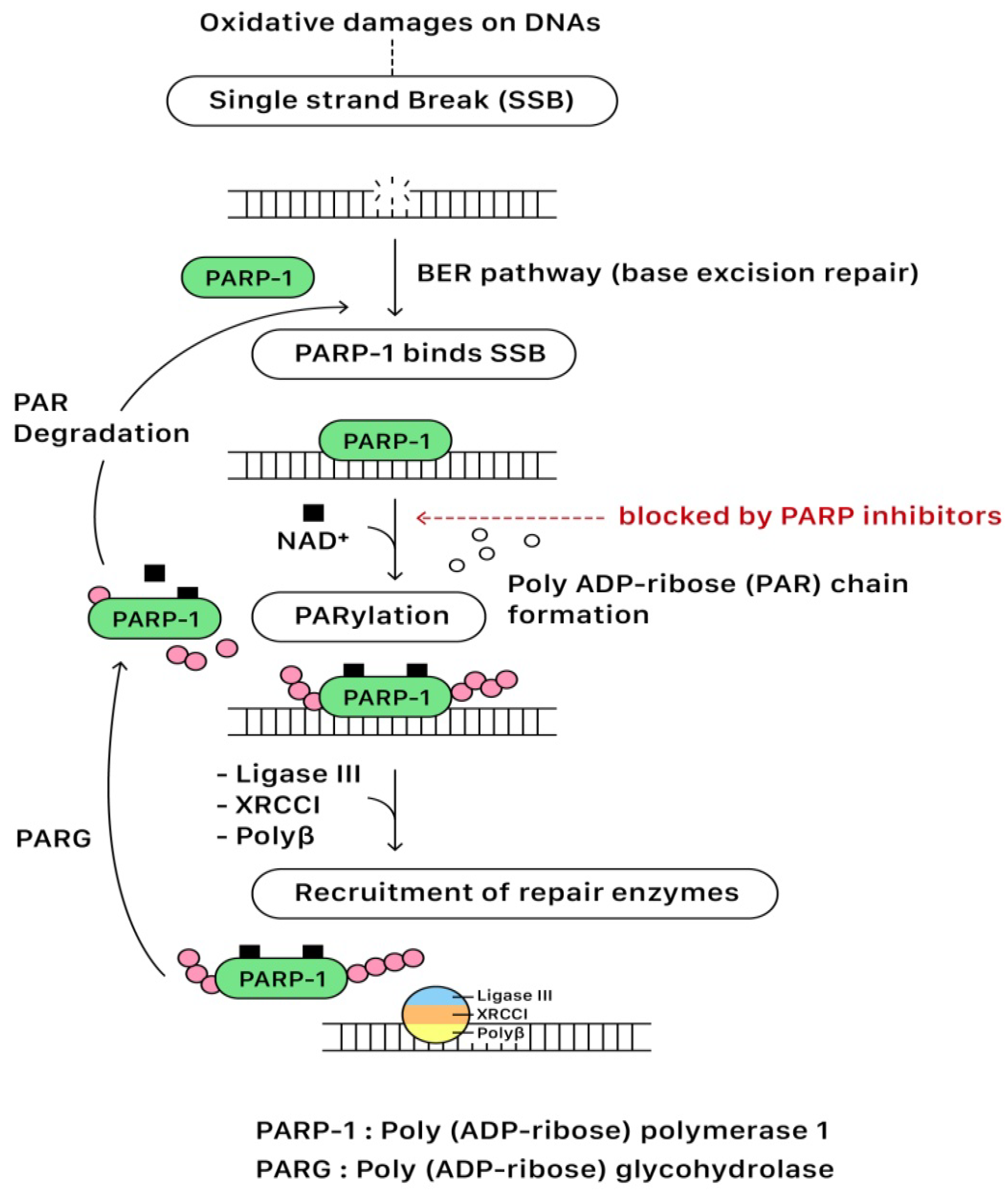
3.2. The Mechanism Underlying Single-Strand Break Repair (SSBR) of DNAs
3.3. The Mechanism Underlying Double-Strand Break Repair (DSBR) of DNAs
3.4. BRCA 1 and BRCA 2
3.5. PARP Inhibitors: The Mechanisms of Actions
4. Therapeutic Effects of PARP Inhibitors in Ovarian Cancers: Clinical Trials
4.1. Olaparib
4.2. Rucaparib
4.3. Niraparib

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PAPR Inhibitors | Study Design: RCTs Phase/Author | Patient Population | Patient Number | Intervention | Results/Outcomes |
|---|---|---|---|---|---|
| Olaparib | Study 42 Phase II/Kaufman, B. (2015) [17] | (1) Platinum-sensitive, advanced ovarian cancer (2) Received at least 3 prior lines of chemotherapy (3) Of whom 193 patients had recurrent ovarian cancer (4) BRCA 1/2 mutated | 298 | Olaparib 400 mg BID | (1)Objective response rate (ORR): 34% (46/137; 95% CI: 0.26–0.46) (2) Median duration of response (DOR): 7.9 months (95% CI: 5.6 months–9.6 months) |
| Study 19 Phase II/Ledermann, J. (2014) [19] | (1) Platinum-sensitive, relapsed, high-grade ovarian cancer (2) Received two or more platinum-based regimens and had a partial or complete response to their most recent platinum-based regimen. (3) Independent BRCA 1/2 mutational status | 265 (1:1) | Maintenance Olaparib capsule (400 mg BID) vs. Placebo | (1)Median PFS (months): 8.4 vs. 4.8 (HR: 0.35; 95% CI: 0.25–0.49; p < 0.001) (2) Median OS (months): 29.7 vs. 29.9 (HR 0.94; 95% CI: 0.63–1.39; p = 0.75) | |
| SOLO-2 Phase III/Pujade-Lauraine, E. (2017) [20] | (1) Platinum-sensitive, relapsed, high-grade ovarian cancer (2) Received two or more platinum-based regimens and had had a partial or complete response to their most recent platinum-based regimen (3) BRCA 1/2 mutated | 295 (2:1) | Maintenance Olaparib tablets (300 mg BID) vs. Placebo | (1)Median PFS (months): 19.1 vs. 5.5 (HR: 0.30; 95% CI: 0.22–0.41; p < 0.0001) (2) Median OS (months): 51.7 vs. 38.8 (HR: 0.74; 95% CI: 0.54–1.00; p = 0.0537) | |
| SOLO-1 Phase III/Moore, K. (2018) [22] | (1) Newly diagnosed, advanced (FIGO stage III–IV), high-grade ovarian cancer (2) Partial or complete clinical response to platinum-based chemotherapy. (3) Mutation in BRCA1, BRCA2, or both (BRCA1/2) | 391 (2:1) | Maintenance Olaparib tablets (300 mg BID) vs. Placebo | (1) Lower three-year rate of disease progression or death compared with the placebo therapy (60% vs. 27%; HR: 0.30, 95% CI: 0.23–0.41, p < 0.0001) (2) Median PFS (months): 56 (95% CI: 41.9 months not reached) vs. 14 (HR: 0.33; 95% CI: 0.25–0.43) | |
| Rucaparib | Study 10 Phase I–II/Kristeleit, R. (2017) [24] | (1) Part 1 (phase I) sought to determine the MTD, recommended phase II dose (RP2D), and pharmacokinetics of oral rucaparib administered in 21-day continuous cycles in patients with advanced solid tumors. (2) Part 2A (phase II): platinum-sensitive, high-grade serous or ovarian cancer associated with germline BRCA1/2 mutation and received between 2 and 4 prior treatment regimens (3) Part 2B of this study is currently assessing the efficacy of rucaparib in patients with relapsed HGOC associated with a germline or somatic BRCA 1/2 mutation who had received at least three prior chemotherapy regimens. (4) Part 3 is ongoing and currently assessing the pharmacokinetic (including the effect of food) and safety profile of a higher dose tablet of rucaparib in patients with a relapsed solid tumor associated with a germline or somatic BRCA1/2 mutation. | Part 1: 56 Part 2: 42 | Part 1: Rucaparib 40~500 mg QD and 240~840 mg BID Part 2A: Rucaparib 600 mg BID | (1) Part 1: No maximum tolerated dose (MTD) was identified using the protocol- specified criteria, and a dose of 600 mg BID was selected as the RP2D based on manageable toxicity and clinical activity (2) Part 2: Objective response rate (ORR): 59.5% with a median duration of response of 7.8 months (95% CI: 5.6–10.5 months) |
| ARIEL 2 Phase II/Swisher, E. M. (2017) [25] | (1) Recurrent, platinum-sensitive, high-grade ovarian carcinomas after one or more chemotherapy (part 1) and 3 or 4 cycles of prior chemotherapy (part 2) (2) BRCA 1/2 mutated | Part 1: 192 | Rucaparib at a dose of 600 mg BID | (1)Median PFS: BRCA-mutated subgroup (12.8 months; HR: 0.27, 95% CI: 0.14–0.44, p < 0.0001) (2) LOH high group (5.7 vs. 5.2 months; HR: 0.62, 95% CI: 0.42–0.90, p = 0.011) compared with the LOH low group | |
| ARIEL 3 Phase III/Coleman, R. L. (2017) [27] | (1) Recurrent, platinum-sensitive, high-grade serous or endometrioid ovarian cancer (2) Received at least two previous platinum-based chemotherapy regimens, had achieved complete or partial response to their last platinum-based regimen (3) Independent BRCA 1/2 mutational status | 564 (2:1) | Maintenance Rucaparib tablets 600 mg BID vs. placebo | Median PFS (months): Overall population: 10.8 vs. 5.4 (HR: 0.37; 95% CI: 0.30–0.45; p < 0.0001) BRCAm population: 16.6 vs. 5.4 (HR: 0.23, 95% CI: 0.16–0.34, p = 0.0001) HRD (+) population: 13.6 vs. 5.4 (HR: 0.32, 95% CI: 0.24–0.42; p < 0.0001) Intention-to-treat population: 10.8 vs. 5.4 (HR: 0.36; 95% CI: 0.30–0.45; p < 0.0001) | |
| Ariel 4 Phase III/Kristeleit, R. (2022) [28] | (1) Relapsed, ovarian cancers who have received at least two cycles of previous chemotherapy regimens. (2) BRCA1/2 mutated | 394 (2:1) | Rucaparib 600 mg BID vs. chemotherapy (weekly paclitaxel or platinum-based) | Median PFS (months): Overall population: 7.4 vs. 5.7 (HR: 0.64, 95% CI: 0.49–0.84; p = 0.0010) Intention-to-treat population: 7.4 vs. 5.7 (HR: 0.67; 95% CI: 0.52–0.86; p < 0.0017) | |
| Niraparib | NOVA study Phase III/Mirza, M.R (2016) [29] | (1) Recurrent, platinum-sensitive ovarian cancers with predominantly high-grade serous histologic features. (2) gBRCA cohort and non-gBRCA cohort | 553 (2:) | Maintenance Niraparib 300 mg QD vs. Placebo | Median PFS (months): gBRCAm cohort: 21.0 vs. 5.5 (HR: 0.27; 95% CI: 0.17–0.41; p < 0.001) HRD-positive and non-gBRCAm: 12.9 vs. 3.8 (HR: 0.38; 95% CI: 0.24–0.59; p < 0.001) HRD-negative and non-gBRCAm: 6.9 vs. 3.8 (HR: 0.58; 95% CI: 0.36–0.92; p = 0.02) Median OS (months): gBRCAm cohort: 45.9 vs. 43.2 (restricted mean) non-gBRCAm cohort: 38.5 vs. 39.1(restricted mean) |
| QUADRA Phase II/Moore, K. N. (2019) [30] | (1) Metastatic, relapsed, high-grade serous epithelial ovarian cancer (2) Received treatment with three or more previous chemotherapy regimens. (3) Homologous recombination deficiency (HRD)-positive tumors (including patients with/without BRCA mutations) | 463 | Niraparib 300 mg QD continuously, beginning on day 1 and every cycle (28 days) thereafter until disease progression. | Objective response rate (ORR): 27.5% (95% CI: 15.9%–41.7%) Disease control rate (DCR): 68.6% Duration of response (DOR): 9.2 months | |
| PRIMA study Phase III/González-Martín, A. (2019) [31] | (1) Newly diagnosed advanced ovarian cancers (2) Complete or partial response to platinum-based chemotherapy (3) Independent BRCA 1/2 mutational status | 733 (2:1) | Maintenance Niraparib tablets (300 mg BID) vs. Placebo | Median PFS (months): Overall population: 13.8 vs. 8.2 (HR: 0.62; 95% CI: 0.50–0.76; p < 0.0001) HRD (+) population: 21.9 vs. 10.4 (HR: 0.43; 95% CI: 0.31–0.59; p < 0.0001) HRD (+), BRCAm population: 22.1 vs. 10.9 (HR: 0.40; 95% CI: 0.27–0.62) HRD (+), non-BRCAm population: 19.6 vs. 8.2 (HR: 0.50; 95% CI: 0.31–0.83) HRD (-) population: 8.1 vs. 5.4 (HR: 0.68; 95% CI: 0.49–0.94) HRD-unknown population: median not reported (HR: 0.59; 95% CI: 0.51–1.43) |
5. PARP Inhibitors and Antiangiogenic Agents
6. Results and Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Prat, J. New Insights into Ovarian Cancer Pathology. Ann. Oncol. 2012, 23 (Suppl. S10), x111–x117. [Google Scholar] [CrossRef] [PubMed]
- Jelovac, D.; Armstrong, D.K. Recent Progress in The Diagnosis and Treatment of Ovarian Cancer. CA Cancer J. Clin. 2011, 61, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Gadducci, A.; Guerrieri, M.E. PARP Inhibitors Alone and In Combination with Other Biological Agents in Homologous Recombination Deficient Epithelial Ovarian Cancer. Crit. Rev. Oncol. Hematol. 2017, 114, 153–165. [Google Scholar] [CrossRef]
- Shigeaki, S.; Akira, N.; Yoshio, M. Crosstalk of DNA Double-Strand Break Repair Pathways in Poly(ADP-ribose) Polymerase Inhibitor Treatment of Breast Cancer Susceptibility Gene 1/2-Mutated Cancer. Cancer Sci. 2018, 109, 893–899. [Google Scholar] [CrossRef]
- Manchana, T.; Phoolcharoen, N.; Tantbirojn, P. BRCA Mutation in High Grade Epithelial Ovarian Cancers. Gynecol. Oncol. Rep. 2019, 29, 102–105. [Google Scholar] [CrossRef]
- Dziadkowiec, K.N.; Gasiorowska, E.; Nowak-Markwitz, E.; Jankowska, A. PARP inhibitors: Review of Mechanisms of Action and BRCA1/2 Mutation Targeting. Prz. Menopauzalny 2016, 15, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP Inhibitors: Synthetic Lethality in The Clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Kelly, E.M. Poly-ADP-Ribosyl-Polymerase Inhibitor Resistance Mechanisms and Their Therapeutic Implications. Curr. Opin. Obstet. Gynecol. 2019, 31, 12–17. [Google Scholar] [CrossRef]
- Kubalanza, K.; Konecny, G.E. Mechanisms of PARP Inhibitor Resistance in Ovarian Cancer. Curr. Opin. Obstet. Gynecol. 2020, 32, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Zhang, Y.; Chen, S.; Wang, X.; Rao, Y.; Fang, H. Mechanism and Current Progress of Poly ADP-Ribose Polymerase (PARP) Inhibitors in The Treatment of Ovarian Cancer. Biomed. Pharmacother. 2020, 123, 109661. [Google Scholar] [CrossRef] [PubMed]
- Loizzi, V.; Del Vecchio, V.; Cicinelli, E.; Cormio, G.; Ranieri, G.; Laforgia, M.; Gadaleta, C.D.; Gargano, G.; Kardhashi, A.; De Liso, M.; et al. PARP Inhibitors and Epithelial Ovarian Cancer: Molecular Mechanisms, Clinical Development and Future Prospective (Review). Oncol. Lett. 2020, 20, 90. [Google Scholar] [CrossRef]
- Li, L.Y.; Guan, Y.D.; Chen, X.S.; Yang, J.M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2021, 11, 629266. [Google Scholar] [CrossRef]
- Walsh, C.S. Two Decades Beyond BRCA1/2: Homologous Recombination, Hereditary Cancer Risk and A Target for Ovarian Cancer Therapy. Gynecol. Oncol. 2015, 137, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Mitchell, J.B.G.; Fried, G.; Stemmer, S.M.; Hubert, A.; Rosengarten, O.; et al. Olaparib Monotherapy in Patients with Advanced Cancer and A Germline BRCA1/2 Mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Domchek, S.M.; Aghajanian, C.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; et al. Efficacy and Safety of Olaparib Monotherapy in Germline BRCA1/2 Mutation Carriers with Advanced Ovarian Cancer and Three or More Lines of Prior Therapy. Gynecol. Oncol. 2016, 140, 199–203. [Google Scholar] [CrossRef] [Green Version]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib Maintenance Therapy in Patients with Platinum-Sensitive Relapsed Serous Ovarian Cancer: A Preplanned Retrospective Analysis of Outcomes by BRCA Status in A Randomised Phase 2 Trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib Tablets as Maintenance Therapy in Patients with Platinum-Sensitive, Relapsed Ovarian Cancer and A BRCA1/2 Mutation (SOLO2/ENGOT-Ov21): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Poveda, A.; Ledermann, J.A.; Selle, F.; Gebski, V.; Floquet, A.; Asher, R.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; et al. Olaparib Tablets as Maintenance Therapy in Patients with Platinum-Sensitive Relapsed Ovarian Cancer and A BRCA1/2 Mutation (SOLO2/ENGOT-Ov21): A Final Analysis of a Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2021, 22, 620–631. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 27, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Moore, K.N.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; et al. Maintenance Olaparib for Patients with Newly Diagnosed Advanced Ovarian Cancer and A BRCA Mutation (SOLO1/GOG 3004): 5-Year Follow-Up of A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2021, 22, 1721–1731. [Google Scholar] [CrossRef]
- Kristeleit, R.; Shapiro, G.I.; Burris, H.A.; Oza, A.M.; LoRusso, P.; Patel, M.R.; Domchek, S.M.; Balmaña, J.; Drew, Y.; Chen, L.; et al. A Phase I-II Study of The Oral PARP Inhibitor Rucaparib in Patients with Germline BRCA1/2-Mutated Ovarian Carcinoma or Other Solid Tumors. Clin. Cancer Res. 2017, 23, 4095–4106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; James, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in Relapsed, Platinum-Sensitive High-Grade Ovarian Carcinoma (ARIEL2 Part 1): An International, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Oza, A.M.; Tinker, A.V.; Oaknin, A.; Shapira-Frommer, R.; McNeish, I.A.; Swisher, E.M.; Ray-Coquard, I.; Bell-McGuinn, K.; Coleman, R.L.; O’Malley, D.M.; et al. Antitumor Activity and Safety of The PARP Inhibitor Rucaparib in Patients with High-Grade Ovarian Carcinoma and A Germline or Somatic BRCA1 or BRCA2 Mutation: Integrated Analysis of Data from Study 10 and ARIEL2. Gynecol. Oncol. 2017, 147, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib Maintenance Treatment for Recurrent Ovarian Carcinoma After Response to Platinum Therapy (ARIEL3): A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Kristeleit, R.; Lisyanskaya, A.; Fedenko, A.; Dvorkin, M.; de Melo, A.C.; Shparyk, Y.; Rakhmatullina, I.; Bondarenko, I.; Colombo, N.; Svintsitskiy, V.; et al. Rucaparib versus Standard-Of-Care Chemotherapy in Patients with Relapsed Ovarian Cancer and A Deleterious BRCA1 or BRCA2 Mutation (ARIEL4): An International, Open-Label, Randomized, Phase 3 Trial. Lancet Oncol. 2022, 23, 465–478. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Hendrickson, A.E.W.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib Monotherapy for Late-Line Treatment of Ovarian Cancer (QUADRA): A Multicentre, Open-Label, Single-Arm, Phase 2 Trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monk, B.J.; Minion, L.E.; Coleman, R.L. Anti-Angiogenic Agents in Ovarian Cancer: Past, Present, and Future. Ann. Oncol. 2016, 27 (Suppl. S1), i33–i39. [Google Scholar] [CrossRef] [PubMed]
- Sopo, M.; Anttila, M.; Hämäläinen, K.; Kivelä, A.; Ylä-Herttuala, S.; Kosma, V.-M.; Keski-Nisula, L.; Sallinen, H. Expression Profiles of VEGF-A, VEGF-D and VEGFR1 Are Higher in Distant Metastases Than in Matched Primary High Grade Epithelial Ovarian Cancer. BMC Cancer 2019, 19, 584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindra, R.S.; Gibson, S.L.; Meng, A.; Westermark, U. Hypoxia-Induced Down-Regulation of BRCA1 Expression by E2Fs. Cancer Res. 2005, 65, 11597–11604. [Google Scholar] [CrossRef] [Green Version]
- Brave, S.R.; Ratcliffe, K.; Wilson, Z.; James, N.H.; Ashton, S.; Wainwright, A.; Kendrew, J.; Dudley, P.; Broadbent, N.; Sproat, G.; et al. Assessing the Activity of Cediranib, A VEGFR-2/3 Tyrosine Kinase Inhibitor, Against VEGFR-1 and Members of The Structurally Related PDGFR Family. Mol. Cancer Ther. 2011, 10, 861–873. [Google Scholar] [CrossRef] [Green Version]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of Bevacizumab in The Primary Treatment of Ovarian Cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef] [Green Version]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A Phase 3 Trial of Bevacizumab in Ovarian Cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [Green Version]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib Plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Moore, K.N.; Mirza, M.R.; Matulonis, U.A. The Poly (ADP Ribose) Polymerase Inhibitor Niraparib: Management of Toxicities. Gynecol. Oncol. 2018, 149, 214–220. [Google Scholar] [CrossRef]
- Berek, J.S.; Matulonis, U.A.; Peen, U.; Ghatage, P.; Mahner, S.; Redondo, A.; Lesoin, A.; Colombo, N.; Vergote, I.; Rosengarten, O.; et al. Safety and Dose Modification for Patients Receiving Niraparib. Ann. Oncol. 2018, 29, 1784–1792. [Google Scholar] [CrossRef]
- Richards, J.S.; Candelaria, N.R.; Lanz, R.B. Polyploid giant cancer cells and ovarian cancer: New insights into mitotic regulators and polyploidy. Biol. Reprod. 2021, 105, 305–316. [Google Scholar] [CrossRef] [PubMed]


| Conditions | HR | BER | Outcomes |
|---|---|---|---|
| Cancer cells (no BRCA mutation) without treatment | + | + | Cell survival |
| Cancer cells (no BRCA mutation) treated with PARPi | + | − | Cell survival |
| BRCA-mutated (HRD) cancer cells without treatment | − | + | Cell survival |
| BRCA-mutated (HRD) cancer cells treated with PARPi | − | − | Cell death |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lau, C.-H.; Seow, K.-M.; Chen, K.-H. The Molecular Mechanisms of Actions, Effects, and Clinical Implications of PARP Inhibitors in Epithelial Ovarian Cancers: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 8125. https://doi.org/10.3390/ijms23158125
Lau C-H, Seow K-M, Chen K-H. The Molecular Mechanisms of Actions, Effects, and Clinical Implications of PARP Inhibitors in Epithelial Ovarian Cancers: A Systematic Review. International Journal of Molecular Sciences. 2022; 23(15):8125. https://doi.org/10.3390/ijms23158125
Chicago/Turabian StyleLau, Chien-Hui, Kok-Min Seow, and Kuo-Hu Chen. 2022. "The Molecular Mechanisms of Actions, Effects, and Clinical Implications of PARP Inhibitors in Epithelial Ovarian Cancers: A Systematic Review" International Journal of Molecular Sciences 23, no. 15: 8125. https://doi.org/10.3390/ijms23158125
APA StyleLau, C.-H., Seow, K.-M., & Chen, K.-H. (2022). The Molecular Mechanisms of Actions, Effects, and Clinical Implications of PARP Inhibitors in Epithelial Ovarian Cancers: A Systematic Review. International Journal of Molecular Sciences, 23(15), 8125. https://doi.org/10.3390/ijms23158125