
The mTOR Signaling Pathway in Multiple Sclerosis; from Animal Models to Human Data

 ,
,
Abstract
:1. Introduction
1.1. Multiple Sclerosis—Basic Aspects of Pathophysiology
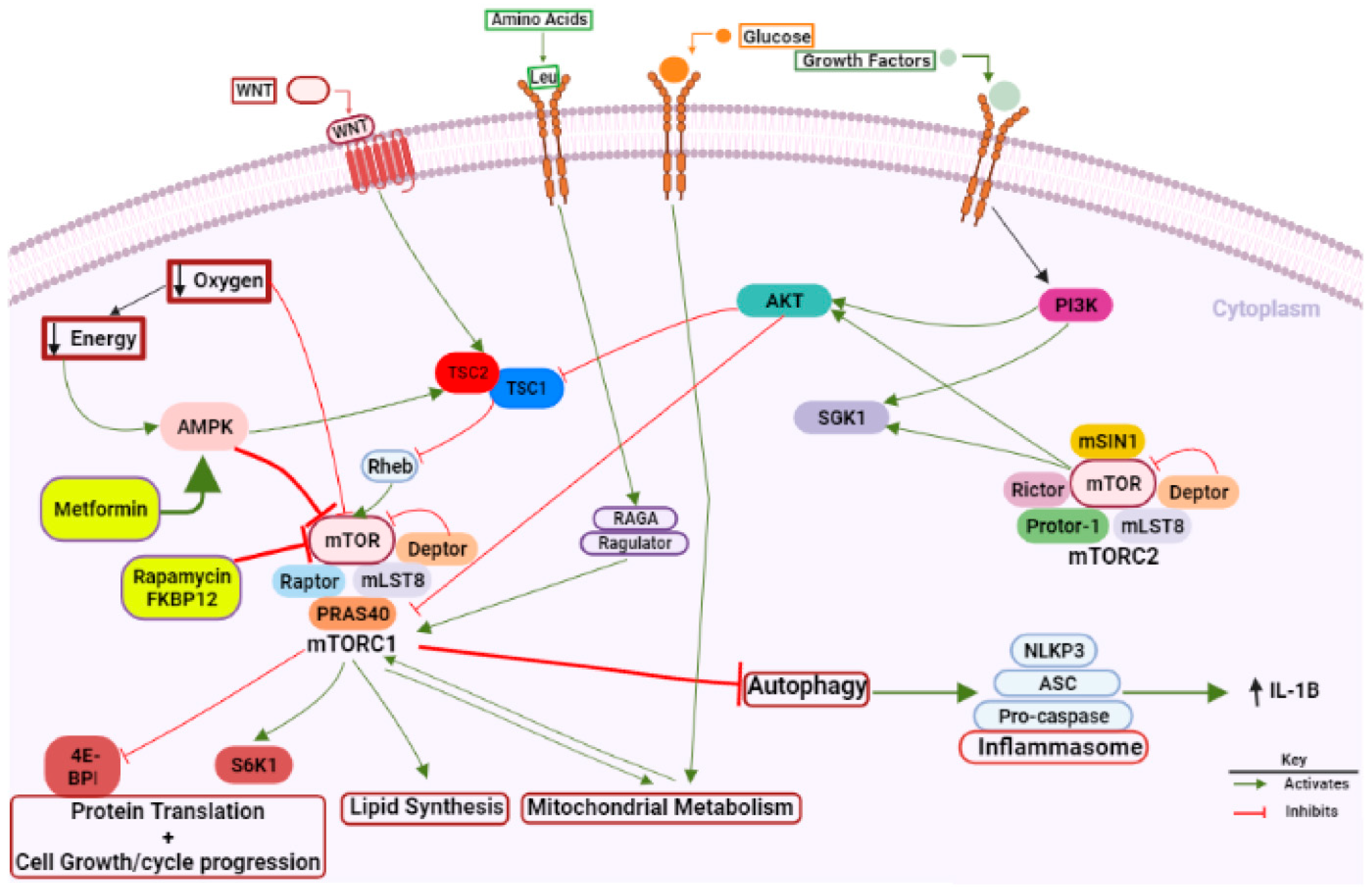
1.2. mTOR as a Master Regulator of Growth, Metabolism, and Survival
1.3. mTOR Inhibitors

{kind=link}
{kind=link}
| Animal Model | Intervention | Emerging Role of mTOR Pathway Manipulations | Change in Disease Status | Ref. |
|---|---|---|---|---|
| RR-EAE (SJL/j mice, PLP139–151 peptide) | Rapamycin | ↓ Teff, ↑ Tregs | Improvement | [43] |
| RR-EAE (SJL/j mice, PLP139–151 peptide) and adoptive transfer of encephalitogenic T cells (donor cells from SJL/j mice immunized with PLP139–151) | tNPs; polymers that encapsulate peptide Ag and rapamycin | ↑ Ag-specific Tregs and ↓ T cell-mediated autoimmunity | Improvement | [57] |
| PR-EAE (DA rats, whole DA rat spinal cord) | Rapamycin | ↓ splenic CD8 T cells, ↑ splenic Tregs | Improvement | [49] |
| M-EAE (C57BL/6 mice, MOG35–55) | Rapamycin | ↓ Th1, ↓ Th17 cells | Improvement | [45] |
| M-EAE (C57BL/6 mice, MOG35–55) | Rapamycin | ↓ Th17 cells, ↑ Tregs | Improvement | [48] |
| M-EAE (C57BL/6 mice, MOG35–55) | Rapamycin | ↓ neuronal cell death, ↑ autophagy | Improvement | [44] |
| M-EAE (C57BL/6 mice, MOG35–55) | Rapamycin ex vivo post stimulation with MOG/LPS | ↓ IL-17, TBX21, RORc, IFN-γ, TNF-α | Improvement | [58] |
| M-EAE (C57BL/6 mice, MOG35–55) | Rapamycin/MCC950 | ↓ neuronal cell death, ↑ autophagy | Improvement | [50] |
| M-EAE (C57BL/6 mice, MOG35–55) | Rapamycin/MCC950 | Restoration of gut microbiota, ↑ autophagy | Improvement | [51] |
| M-EAE (C57BL/6 mice, MOG35–55) | AZD8055 | ↓ inflammation, ↑ autophagy | Improvement | [59] |
| M-EAE (C57BL/6 mice, MOG35–55) | Rapamycin | ↑ TAM receptors, ↑ anti-inflammatory factors and ↓ TLR-3, TLR-4 | Improvement | [60] |
| M-EAE (C57BL/6 mice, MOG35–55) | Rapamycin and fingolimod | ↓ Th17 and ↑ Treg cells in the spleen and CNS | Improvement | [45] |
| C-EAE (C57BL/6 mice, MOG35–55) | HSO/EPO and/or rapamycin | ↑ regeneration of the myelin sheath | Improvement | [61] |
| C-EAE (C57BL/6 mice, MOG35–55) | HSO/EPO | ↓ STAT3 and IL-17 genes and ↑ FOXP3+ gene in spinal cord | Improvement | [62] |
| C-EAE (C57BL/6 mice, MOG35–55) | HSO/EPO and/or rapamycin | ↓ RAPTOR, IFNγ, IL-17, STAT3 and ↑ RICTOR, IL-10, FOXP3 genes | Improvement | [63] |
| Cuprizone (C56BL/6 J mice, 0.3% CPZ diet) | Rapamycin | ↓ spontaneous remyelination | Worsening | [54] |
| Cuprizone (C57BL/6 J mice 0.2% CPZ diet) | Metformin | ↑ myelin recovery in oligodendrocytes | Improvement | [53] |
| Cuprizone (Ng2-Mtor cKO of C57Bl/6 background, 0.2% CPZ diet) | OPC-specific mTOR KO | Delay in myelin production | Delayed improvement | [55] |
| Cuprizone (C57BL/6 J mice 0.2% CPZ diet) | Rapamycin | ↑ inflammation and callosal axonal damage | Worsening | [56] |
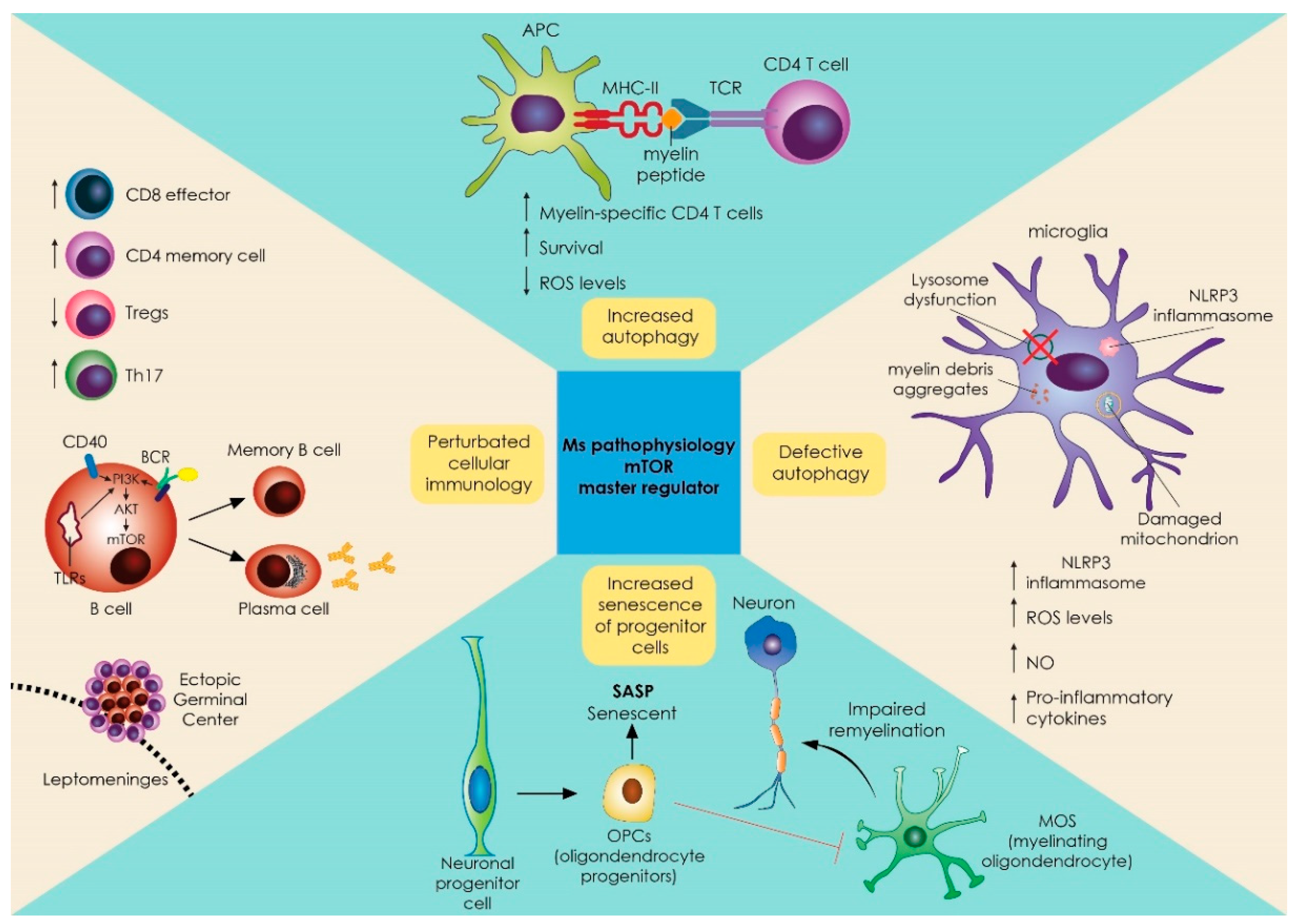
2. Specific Role of mTOR on the Biological Process of Autophagy and Its Relevance to CNS Demyelination and MS
2.1. Role of mTOR in Innate Immune Responses in Myeloid, Microglial Cells and Inflammasome
2.2. Role of mTOR in Adaptive Immune Responses of T and B Cell Development and Differentiation
3. Human Data Regarding mTOR Inhibition
| Type of Study | Patients Included | Main Results | Ref. |
|---|---|---|---|
| Double-blind, placebo-controlled phase II trial | N = 296 patients with active, relapsing MS relapses; 2, 4, 8 mg temsirolimus/d or placebo for 9 months | 8 mg group: ~50% reduction in active lesions and relapses, mouth ulcers, menstrual abnormalities | [107,108] |
| Non-randomized, prospective, controlled | N = 50 RRMS with metabolic syndrome; N = 20 metformin, N = 20 untreated | Metformin group:↓ new/expanding T2 lesions, ↓ Gd+ lesions, ↓ myelin-specific T cells, ↑ Tregs | [110] |
| Non-randomized, prospective, controlled | N = six RRMS patients on 2 mg rapamycin/d for 6 months N = six healthy controls | ↓ IFNγ, ↑ TGFβ in serum of patients after 6 months | [105] |
| Non-randomized, prospective, uncontrolled | N = eight RRMS patients on 2 mg rapamycin/d for 6 months | ↑ Tregs, ↓ in mean lesional area size after treatment | [106] |
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | AMP kinase |
| Atg5 | Autophagy-related 5 protein |
| BBB | Blood–brain barrier |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| EAE | Experimental autoimmune encephalomyelitis |
| DDIT4 | DNA damage-inducible transcript 4 |
| FKBP12 | FK506-binding protein of 12 kDa |
| GCs | Glucocorticoids |
| Gd | Gadolinium |
| iPSCs | Induced pluripotent stem cells |
| LPS | Lipopolysaccharide |
| MS | Multiple sclerosis |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | mTOR complex 1 |
| mTORC2 | mTOR complex 2 |
| NLRP3 | NOD-like receptor protein 3 |
| NO | Nitric oxide |
| NPCs | Neural progenitor cells |
| OLs | Mature oligodendrocytes |
| Pi3K-AKT | Phosphoinositide-3-kinase/protein kinase B |
| PPMS | Primary progressive MS |
| ROS | Reactive oxygen species |
| RRMS | Relapsing–remitting MS |
| SGK1 | Serum- and glucocorticoid-induced protein kinase 1 |
| SPMS | Secondary progressive MS |
| TCR | T cell receptor |
| Tregs | Regulatory T cells |
| TSC | Tuberous sclerosis complex |
References
- Browne, P.; Chandraratna, D.; Angood, C.; Tremlett, H.; Baker, C.; Taylor, B.V.; Thompson, A.J. Atlas of Multiple Sclerosis 2013: A Growing Global Problem with Widespread Inequity. Neurology 2014, 83, 1022–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrikios, P.; Stadelmann, C.; Kutzelnigg, A.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Brück, W.; Lucchinetti, C.; Lassmann, H. Remyelination Is Extensive in a Subset of Multiple Sclerosis Patients. Brain 2006, 129, 3165–3172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.; Staugaitis, S.M.; Dutta, R.; Batt, C.E.; Easley, K.E.; Chomyk, A.M.; Yong, V.W.; Fox, R.J.; Kidd, G.J.; Trapp, B.D. Cortical Remyelination: A New Target for Repair Therapies in Multiple Sclerosis. Ann. Neurol. 2012, 72, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Artemiadis, A.K.; Anagnostouli, M.C. Apoptosis of Oligodendrocytes and Post-Translational Modifications of Myelin Basic Protein in Multiple Sclerosis: Possible Role for the Early Stages of Multiple Sclerosis. Eur. Neurol. 2010, 63, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Brück, W.; Lassmann, H. An Updated Histological Classification System for Multiple Sclerosis Lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef]
- Dal-Bianco, A.; Grabner, G.; Kronnerwetter, C.; Weber, M.; Höftberger, R.; Berger, T.; Auff, E.; Leutmezer, F.; Trattnig, S.; Lassmann, H.; et al. Slow Expansion of Multiple Sclerosis Iron Rim Lesions: Pathology and 7 T Magnetic Resonance Imaging. Acta Neuropathol. 2017, 133, 25–42. [Google Scholar] [CrossRef] [Green Version]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-Cell Follicles in Secondary Progressive Multiple Sclerosis Associate with Early Onset of Disease and Severe Cortical Pathology. Brain 2007, 130, 1089–1104. [Google Scholar] [CrossRef]
- Barnett, M.H.; Prineas, J.W. Relapsing and Remitting Multiple Sclerosis: Pathology of the Newly Forming Lesion. Ann. Neurol. 2004, 55, 458–468. [Google Scholar] [CrossRef]
- Lucchinetti, C.; Brück, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of Multiple Sclerosis Lesions: Implications for the Pathogenesis of Demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Peterson, J.W.; Bö, L.; Mörk, S.; Chang, A.; Trapp, B.D. Transected Neurites, Apoptotic Neurons, and Reduced Inflammation in Cortical Multiple Sclerosis Lesions. Ann. Neurol. 2001, 50, 389–400. [Google Scholar] [CrossRef]
- Popescu, B.F.G.; Pirko, I.; Lucchinetti, C.F. Pathology of Multiple Sclerosis: Where Do We Stand? Contin. Lifelong Learn. Neurol. 2013, 19, 901–921. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2; Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.A.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; et al. Genetic Risk and a Primary Role for Cell-Mediated Immune Mechanisms in Multiple Sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [PubMed]
- International Multiple Sclerosis Genetics Consortium; Lill, C.M.; Schjeide, B.-M.M.; Graetz, C.; Ban, M.; Alcina, A.; Ortiz, M.A.; Pérez, J.; Damotte, V.; Booth, D.; et al. MANBA, CXCR5, SOX8, RPS6KB1 and ZBTB46 Are Genetic Risk Loci for Multiple Sclerosis. Brain 2013, 136, 1778–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoepner, R.; Bagnoud, M.; Pistor, M.; Salmen, A.; Briner, M.; Synn, H.; Schrewe, L.; Guse, K.; Ahmadi, F.; Demir, S.; et al. Vitamin D Increases Glucocorticoid Efficacy via Inhibition of MTORC1 in Experimental Models of Multiple Sclerosis. Acta Neuropathol. 2019, 138, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Ransohoff, R.M.; Hafler, D.A.; Lucchinetti, C.F. Multiple Sclerosis—A Quiet Revolution. Nat. Rev. Neurol. 2015, 11, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.S.; Lees, J.G.; Moalem-Taylor, G. The Contribution of Immune and Glial Cell Types in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Mult. Scler. Int. 2014, 2014, 285245. [Google Scholar] [CrossRef]
- Cao, Y.; Goods, B.A.; Raddassi, K.; Nepom, G.T.; Kwok, W.W.; Love, J.C.; Hafler, D.A. Functional Inflammatory Profiles Distinguish Myelin-Reactive T Cells from Patients with Multiple Sclerosis. Sci. Transl. Med. 2015, 7, 287ra74. [Google Scholar] [CrossRef] [Green Version]
- Bogie, J.F.J.; Stinissen, P.; Hendriks, J.J.A. Macrophage Subsets and Microglia in Multiple Sclerosis. Acta Neuropathol. 2014, 128, 191–213. [Google Scholar] [CrossRef]
- Bar-Or, A.; Li, R. Cellular Immunology of Relapsing Multiple Sclerosis: Interactions, Checks, and Balances. Lancet Neurol. 2021, 20, 470–483. [Google Scholar] [CrossRef]
- Comabella, M.; Khoury, S.J. Immunopathogenesis of Multiple Sclerosis. Clin. Immunol. 2012, 142, 2–8. [Google Scholar] [CrossRef]
- Sospedra, M. B Cells in Multiple Sclerosis. Curr. Opin. Neurol. 2018, 31, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulos, P.; Dalakas, M.C. Evolution of Anti-B Cell Therapeutics in Autoimmune Neurological Diseases. Neurotherapeutics 2022, 19, 691–710. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.; Touil, H.; Pikor, N.B.; Gommerman, J.L.; Prat, A.; Bar-Or, A. B Cells in the Multiple Sclerosis Central Nervous System: Trafficking and Contribution to CNS-Compartmentalized Inflammation. Front. Immunol. 2015, 6, 636. [Google Scholar] [CrossRef] [Green Version]
- Reali, C.; Magliozzi, R.; Roncaroli, F.; Nicholas, R.; Howell, O.W.; Reynolds, R. B Cell Rich Meningeal Inflammation Associates with Increased Spinal Cord Pathology in Multiple Sclerosis. Brain Pathol. 2020, 30, 779–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of Ectopic B-Cell Follicles with Germinal Centers in the Meninges of Patients with Secondary Progressive Multiple Sclerosis. Brain Pathol. 2004, 14, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Cepok, S.; Rosche, B.; Grummel, V.; Vogel, F.; Zhou, D.; Sayn, J.; Sommer, N.; Hartung, H.-P.; Hemmer, B. Short-Lived Plasma Blasts Are the Main B Cell Effector Subset during the Course of Multiple Sclerosis. Brain 2005, 128, 1667–1676. [Google Scholar] [CrossRef] [Green Version]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mörk, S.; Bö, L. Axonal Transection in the Lesions of Multiple Sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef]
- Lucchinetti, C.F.; Popescu, B.F.G.; Bunyan, R.F.; Moll, N.M.; Roemer, S.F.; Lassmann, H.; Brück, W.; Parisi, J.E.; Scheithauer, B.W.; Giannini, C.; et al. Inflammatory Cortical Demyelination in Early Multiple Sclerosis. N. Engl. J. Med. 2011, 365, 2188–2197. [Google Scholar] [CrossRef] [Green Version]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of “homeostatic” Microglia and Patterns of Their Activation in Active Multiple Sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef]
- Jäckle, K.; Zeis, T.; Schaeren-Wiemers, N.; Junker, A.; van der Meer, F.; Kramann, N.; Stadelmann, C.; Brück, W. Molecular Signature of Slowly Expanding Lesions in Progressive Multiple Sclerosis. Brain 2020, 143, 2073–2088. [Google Scholar] [CrossRef]
- Kamma, E.; Lasisi, W.; Libner, C.; Ng, H.S.; Plemel, J.R. Central Nervous System Macrophages in Progressive Multiple Sclerosis: Relationship to Neurodegeneration and Therapeutics. J. Neuroinflamm. 2022, 19, 45. [Google Scholar] [CrossRef] [PubMed]
- Wingerchuk, D.M.; Carter, J.L. Multiple Sclerosis: Current and Emerging Disease-Modifying Therapies and Treatment Strategies. Mayo Clin. Proc. 2014, 89, 225–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bross, M.; Hackett, M.; Bernitsas, E. Approved and Emerging Disease Modifying Therapies on Neurodegeneration in Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 4312. [Google Scholar] [CrossRef]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. MTOR Controls Cell Cycle Progression through Its Cell Growth Effectors S6K1 and 4E-BP1/Eukaryotic Translation Initiation Factor 4E. Mol. Cell. Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Zhang, Q.; Tan, L.; Xu, Y.; Xie, X.; Zhao, Y. The Regulatory Effects of MTOR Complexes in the Differentiation and Function of CD4+ T Cell Subsets. J. Immunol. Res. 2020, 2020, 3406032. [Google Scholar] [CrossRef] [Green Version]
- Nicaise, A.M.; Wagstaff, L.J.; Willis, C.M.; Paisie, C.; Chandok, H.; Robson, P.; Fossati, V.; Williams, A.; Crocker, S.J. Cellular Senescence in Progenitor Cells Contributes to Diminished Remyelination Potential in Progressive Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 9030–9039. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. MTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, K.G.; Fingar, D.C. Mammalian Target of Rapamycin (MTOR): Conducting the Cellular Signaling Symphony. J. Biol. Chem. 2010, 285, 14071–14077. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Dibble, C.C.; Matsuzaki, M.; Manning, B.D. The TSC1-TSC2 Complex Is Required for Proper Activation of MTOR Complex 2. Mol. Cell. Biol. 2008, 28, 4104–4115. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-MTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, A.S.; RAPAMUNE Global Study Group. A Worldwide, Phase III, Randomized, Controlled, Safety and Efficacy Study of a Sirolimus/Cyclosporine Regimen for Prevention of Acute Rejection in Recipients of Primary Mismatched Renal Allografts. Transplantation 2001, 71, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One Drug, Many Effects. Cell. Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, M.; Ruffini, F.; Bellone, M.; Gagliani, N.; Battaglia, M.; Martino, G.; Furlan, R. Rapamycin Inhibits Relapsing Experimental Autoimmune Encephalomyelitis by Both Effector and Regulatory T Cells Modulation. J. Neuroimmunol. 2010, 220, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Hou, H.; Zou, Y.; Guo, L. Defective Autophagy Is Associated with Neuronal Injury in a Mouse Model of Multiple Sclerosis. Bosn. J. Basic Med. Sci. 2017, 17, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, H.; Miao, J.; Cao, R.; Han, M.; Sun, Y.; Liu, X.; Guo, L. Rapamycin Ameliorates Experimental Autoimmune Encephalomyelitis by Suppressing the MTOR-STAT3 Pathway. Neurochem. Res. 2017, 42, 2831–2840. [Google Scholar] [CrossRef] [PubMed]
- DiToro, D.; Harbour, S.N.; Bando, J.K.; Benavides, G.; Witte, S.; Laufer, V.A.; Moseley, C.; Singer, J.R.; Frey, B.; Turner, H.; et al. Insulin-Like Growth Factors Are Key Regulators of T Helper 17 Regulatory T Cell Balance in Autoimmunity. Immunity 2020, 52, 650–667.e10. [Google Scholar] [CrossRef] [PubMed]
- Petrou, P.; Kassis, I.; Levin, N.; Paul, F.; Backner, Y.; Benoliel, T.; Oertel, F.C.; Scheel, M.; Hallimi, M.; Yaghmour, N.; et al. Beneficial Effects of Autologous Mesenchymal Stem Cell Transplantation in Active Progressive Multiple Sclerosis. Brain 2020, 143, 3574–3588. [Google Scholar] [CrossRef]
- Li, Z.; Nie, L.; Chen, L.; Sun, Y.; Li, G. Rapamycin Relieves Inflammation of Experimental Autoimmune Encephalomyelitis by Altering the Balance of Treg/Th17 in a Mouse Model. Neurosci. Lett. 2019, 705, 39–45. [Google Scholar] [CrossRef]
- Donia, M.; Mangano, K.; Amoroso, A.; Mazzarino, M.C.; Imbesi, R.; Castrogiovanni, P.; Coco, M.; Meroni, P.; Nicoletti, F. Treatment with Rapamycin Ameliorates Clinical and Histological Signs of Protracted Relapsing Experimental Allergic Encephalomyelitis in Dark Agouti Rats and Induces Expansion of Peripheral CD4+CD25+Foxp3+ Regulatory T Cells. J. Autoimmun. 2009, 33, 135–140. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, C.; Jiang, N.; He, D.; Bai, Y.; Xin, Y. Rapamycin Combined with MCC950 to Treat Multiple Sclerosis in Experimental Autoimmune Encephalomyelitis. J. Cell. Biochem. 2019, 120, 5160–5168. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, C.; He, D.; Jiang, N.; Bai, Y.; Xin, Y. Rapamycin and MCC950 Modified Gut Microbiota in Experimental Autoimmune Encephalomyelitis Mouse by Brain Gut Axis. Life Sci. 2020, 253, 117747. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, G.K.; Morell, P. The Neurotoxicant, Cuprizone, as a Model to Study Demyelination and Remyelination in the Central Nervous System. Brain Pathol. 2001, 11, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Sanadgol, N.; Barati, M.; Houshmand, F.; Hassani, S.; Clarner, T.; Shahlaei, M.; Golab, F. Metformin Accelerates Myelin Recovery and Ameliorates Behavioral Deficits in the Animal Model of Multiple Sclerosis via Adjustment of AMPK/Nrf2/MTOR Signaling and Maintenance of Endogenous Oligodendrogenesis during Brain Self-Repairing Period. Pharmacol. Rep. 2020, 72, 641–658. [Google Scholar] [CrossRef] [PubMed]
- Sachs, H.H.; Bercury, K.K.; Popescu, D.C.; Narayanan, S.P.; Macklin, W.B. A New Model of Cuprizone-Mediated Demyelination/Remyelination. ASN Neuro 2014, 6, 1759091414551955. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, M.A.; McLane, L.E.; Khandker, L.; Mather, M.L.; Evangelou, A.V.; Kantak, D.; Bourne, J.N.; Macklin, W.B.; Wood, T.L. MTOR Signaling Regulates Metabolic Function in Oligodendrocyte Precursor Cells and Promotes Efficient Brain Remyelination in the Cuprizone Model. J. Neurosci. 2021, 41, 8321–8337. [Google Scholar] [CrossRef] [PubMed]
- Yamate-Morgan, H.; Lauderdale, K.; Horeczko, J.; Merchant, U.; Tiwari-Woodruff, S.K. Functional Effects of Cuprizone-Induced Demyelination in the Presence of the MTOR-Inhibitor Rapamycin. Neuroscience 2019, 406, 667–683. [Google Scholar] [CrossRef] [PubMed]
- LaMothe, R.A.; Kolte, P.N.; Vo, T.; Ferrari, J.D.; Gelsinger, T.C.; Wong, J.; Chan, V.T.; Ahmed, S.; Srinivasan, A.; Deitemeyer, P.; et al. Tolerogenic Nanoparticles Induce Antigen-Specific Regulatory T Cells and Provide Therapeutic Efficacy and Transferrable Tolerance against Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2018, 9, 281. [Google Scholar] [CrossRef] [Green Version]
- Borim, P.A.; Mimura, L.A.N.; Zorzella-Pezavento, S.F.G.; Polonio, C.M.; Peron, J.P.S.; Sartori, A.; Fraga-Silva, T.F.d.C. Effect of Rapamycin on MOG-Reactive Immune Cells and Lipopolysaccharide-Activated Microglia: An In Vitro Approach for Screening New Therapies for Multiple Sclerosis. J. Interferon Cytokine Res. 2022, 42, 153–160. [Google Scholar] [CrossRef]
- He, M.; Wu, D.-M.; Zhao, Y.-Y.; Yu, Y.; Deng, S.-H.; Liu, T.; Zhang, T.; Li, J.; Wang, F.; Xu, Y. AZD8055 Ameliorates Experimental Autoimmune Encephalomyelitis via the MTOR/ROS/NLRP3 Pathway. Biochem. Biophys. Res. Commun. 2021, 573, 27–34. [Google Scholar] [CrossRef]
- Li, X.-L.; Zhang, B.; Liu, W.; Sun, M.-J.; Zhang, Y.-L.; Liu, H.; Wang, M.-X. Rapamycin Alleviates the Symptoms of Multiple Sclerosis in Experimental Autoimmune Encephalomyelitis (EAE) Through Mediating the TAM-TLRs-SOCS Pathway. Front. Neurol. 2020, 11, 590884. [Google Scholar] [CrossRef]
- Rezapour-Firouzi, S.; Shahabi, S.; Mohammadzadeh, A.; Tehrani, A.A.; Kheradmand, F.; Mazloomi, E. The Potential Effects of Hemp Seed/Evening Primrose Oils on the Mammalian Target of Rapamycin Complex 1 and Interferon-Gamma Genes Expression in Experimental Autoimmune Encephalomyelitis. Res. Pharm. Sci. 2018, 13, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Rezapour-Firouzi, S.; Kheradmand, F.; Shahabi, S.; Tehrani, A.A.; Mazloomi, E.; Mohammadzadeh, A. Regulatory Effects of Hemp Seed/Evening Primrose Oil Supplement in Comparison with Rapamycin on the Expression of the Mammalian Target of Rapamycin-Complex 2 and Interleukin-10 Genes in Experimental Autoimmune Encephalomyelitis. Res. Pharm. Sci. 2019, 14, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Rezapour-Firouzi, S.; Mohammadian, M.; Sadeghzadeh, M.; Mazloomi, E. Effects of Co-Administration of Rapamycin and Evening Primrose/Hemp Seed Oil Supplement on Immunologic Factors and Cell Membrane Fatty Acids in Experimental Autoimmune Encephalomyelitis. Gene 2020, 759, 144987. [Google Scholar] [CrossRef] [PubMed]
- Alirezaei, M.; Fox, H.S.; Flynn, C.T.; Moore, C.S.; Hebb, A.L.O.; Frausto, R.F.; Bhan, V.; Kiosses, W.B.; Whitton, J.L.; Robertson, G.S.; et al. Elevated ATG5 Expression in Autoimmune Demyelination and Multiple Sclerosis. Autophagy 2009, 5, 152–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and Molecular Mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- New, J.; Thomas, S.M. Autophagy-Dependent Secretion: Mechanism, Factors Secreted, and Disease Implications. Autophagy 2019, 15, 1682–1693. [Google Scholar] [CrossRef]
- Misrielal, C.; Mauthe, M.; Reggiori, F.; Eggen, B.J.L. Autophagy in Multiple Sclerosis: Two Sides of the Same Coin. Front. Cell. Neurosci. 2020, 14, 603710. [Google Scholar] [CrossRef]
- Bankston, A.N.; Forston, M.D.; Howard, R.M.; Andres, K.R.; Smith, A.E.; Ohri, S.S.; Bates, M.L.; Bunge, M.B.; Whittemore, S.R. Autophagy Is Essential for Oligodendrocyte Differentiation, Survival, and Proper Myelination. Glia 2019, 67, 1745–1759. [Google Scholar] [CrossRef]
- Bercury, K.K.; Dai, J.X.; Sachs, H.H.; Ahrendsen, J.T.; Wood, T.L.; Macklin, W.B. Conditional Ablation of Raptor or Rictor Has Differential Impact on Oligodendrocyte Differentiation and CNS Myelination. J. Neurosci. 2014, 34, 4466–4480. [Google Scholar] [CrossRef] [Green Version]
- Wahl, S.E.; McLane, L.E.; Bercury, K.K.; Macklin, W.B.; Wood, T.L. Mammalian Target of Rapamycin Promotes Oligodendrocyte Differentiation, Initiation and Extent of CNS Myelination. J. Neurosci. 2014, 34, 4453–4465. [Google Scholar] [CrossRef]
- Choi, I.Y.; Piccio, L.; Childress, P.; Bollman, B.; Ghosh, A.; Brandhorst, S.; Suarez, J.; Michalsen, A.; Cross, A.H.; Morgan, T.E.; et al. A Diet Mimicking Fasting Promotes Regeneration and Reduces Autoimmunity and Multiple Sclerosis Symptoms. Cell Rep. 2016, 15, 2136–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, A.; Parillon, X.; Zeng, S.; Han, S.; Eissa, N.T. Deficiency of Autophagy in Dendritic Cells Protects against Experimental Autoimmune Encephalomyelitis. J. Biol. Chem. 2014, 289, 26525–26532. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Song, W.; Xu, J.; Postoak, J.L.; Cheng, F.; Martinez, J.; Zhang, J.; Wu, L.; Van Kaer, L. Pik3c3 Deficiency in Myeloid Cells Imparts Partial Resistance to Experimental Autoimmune Encephalomyelitis Associated with Reduced IL-1β Production. Cell. Mol. Immunol. 2021, 18, 2024–2039. [Google Scholar] [CrossRef]
- Albert, M.; Barrantes-Freer, A.; Lohrberg, M.; Antel, J.P.; Prineas, J.W.; Palkovits, M.; Wolff, J.R.; Brück, W.; Stadelmann, C. Synaptic Pathology in the Cerebellar Dentate Nucleus in Chronic Multiple Sclerosis. Brain Pathol. 2017, 27, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Castellazzi, M.; Bonora, M.; Marchi, S.; Casetta, I.; Pugliatti, M.; Giorgi, C.; Granieri, E.; Pinton, P. Autophagy and Mitophagy Elements Are Increased in Body Fluids of Multiple Sclerosis-Affected Individuals. J. Neurol. Neurosurg. Psychiatry 2018, 89, 439–441. [Google Scholar] [CrossRef] [PubMed]
- Igci, M.; Baysan, M.; Yigiter, R.; Ulasli, M.; Geyik, S.; Bayraktar, R.; Bozgeyik, İ.; Bozgeyik, E.; Bayram, A.; Cakmak, E.A. Gene Expression Profiles of Autophagy-Related Genes in Multiple Sclerosis. Gene 2016, 588, 38–46. [Google Scholar] [CrossRef]
- Parnell, G.P.; Gatt, P.N.; McKay, F.C.; Schibeci, S.; Krupa, M.; Powell, J.E.; Visscher, P.M.; Montgomery, G.W.; Lechner-Scott, J.; Broadley, S.; et al. Ribosomal Protein S6 MRNA Is a Biomarker Upregulated in Multiple Sclerosis, Downregulated by Interferon Treatment, and Affected by Season. Mult. Scler. 2014, 20, 675–685. [Google Scholar] [CrossRef] [Green Version]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules That Regulate Macrophage Polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Wang, T.; Tait, S. Chapter 20–Metabolic Regulation of Immunity. In Kelley and Firestein’s Textbook of Rheumatology, 10th ed.; Firestein, G.S., Budd, R.C., Gabriel, S.E., McInnes, I.B., O’Dell, J.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 318–326. ISBN 978-0-323-31696-5. [Google Scholar]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of Innate Immune Cell Function by MTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef]
- Kimura, T.; Nada, S.; Takegahara, N.; Okuno, T.; Nojima, S.; Kang, S.; Ito, D.; Morimoto, K.; Hosokawa, T.; Hayama, Y.; et al. Polarization of M2 Macrophages Requires Lamtor1 That Integrates Cytokine and Amino-Acid Signals. Nat. Commun. 2016, 7, 13130. [Google Scholar] [CrossRef]
- Weichhart, T.; Costantino, G.; Poglitsch, M.; Rosner, M.; Zeyda, M.; Stuhlmeier, K.M.; Kolbe, T.; Stulnig, T.M.; Hörl, W.H.; Hengstschläger, M.; et al. The TSC-MTOR Signaling Pathway Regulates the Innate Inflammatory Response. Immunity 2008, 29, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, A.C.P.; Candelario-Jalil, E.; Langbein, J.; Wendeburg, L.; Bhatia, H.S.; Schlachetzki, J.C.M.; Biber, K.; Fiebich, B.L. Pharmacological Inhibition of Akt and Downstream Pathways Modulates the Expression of COX-2 and MPGES-1 in Activated Microglia. J. Neuroinflamm. 2012, 9, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, R.; Turnquist, H.R.; Taner, T.; Thomson, A.W. Use of Rapamycin in the Induction of Tolerogenic Dendritic Cells. In Dendritic Cells; Lombardi, G., Riffo-Vasquez, Y., Eds.; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 215–232. ISBN 978-3-540-71029-5. [Google Scholar]
- Chong, Z.Z.; Li, F.; Maiese, K. The Pro-Survival Pathways of MTOR and Protein Kinase B Target Glycogen Synthase Kinase-3β and Nuclear Factor-ΚB to Foster Endogenous Microglial Cell Protection. Int. J. Mol. Med. 2007, 19, 263–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, N.M.; Chi, H. MTOR Links Environmental Signals to T Cell Fate Decisions. Front. Immunol. 2014, 5, 686. [Google Scholar] [CrossRef] [Green Version]
- Chi, H. Regulation and Function of MTOR Signalling in T Cell Fate Decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. The MTOR Kinase Differentially Regulates Effector and Regulatory T Cell Lineage Commitment. Immunity 2009, 30, 832–844. [Google Scholar] [CrossRef] [Green Version]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The Kinase MTOR Regulates the Differentiation of Helper T Cells through the Selective Activation of Signaling by MTORC1 and MTORC2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, T.; Kimura, T.; Nada, S.; Okuno, T.; Ito, D.; Kang, S.; Nojima, S.; Yamashita, K.; Nakatani, T.; Hayama, Y.; et al. Lamtor1 Is Critically Required for CD4+ T Cell Proliferation and Regulatory T Cell Suppressive Function. J. Immunol. 2017, 199, 2008–2019. [Google Scholar] [CrossRef] [Green Version]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic Regulation of T Lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef] [Green Version]
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory Functions of MTOR Inhibition. Nat. Rev. Immunol. 2009, 9, 324–337. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Readinger, J.A.; DuBois, W.; Janka-Junttila, M.; Robinson, R.; Pruitt, M.; Bliskovsky, V.; Wu, J.Z.; Sakakibara, K.; Patel, J.; et al. Constitutive Reductions in MTOR Alter Cell Size, Immune Cell Development, and Antibody Production. Blood 2011, 117, 1228–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stathopoulou, C.; Nikoleri, D.; Bertsias, G. Immunometabolism: An Overview and Therapeutic Prospects in Autoimmune Diseases. Immunotherapy 2019, 11, 813–829. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.; Limon, J. Akt and MTOR in B Cell Activation and Differentiation. Front. Immunol. 2012, 3, 228. [Google Scholar]
- Omori, S.A.; Cato, M.H.; Anzelon-Mills, A.; Puri, K.D.; Shapiro-Shelef, M.; Calame, K.; Rickert, R.C. Regulation of Class-Switch Recombination and Plasma Cell Differentiation by Phosphatidylinositol 3-Kinase Signaling. Immunity 2006, 25, 545–557. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Li, Z.; Wang, P.; Huang, Q.; Xu, L.; He, R.; Ye, L.; Bai, Q. Mammalian Target of Rapamycin Complex 1 Signalling Is Essential for Germinal Centre Reaction. Immunology 2017, 152, 276–286. [Google Scholar] [CrossRef]
- Ersching, J.; Efeyan, A.; Mesin, L.; Jacobsen, J.T.; Pasqual, G.; Grabiner, B.C.; Dominguez-Sola, D.; Sabatini, D.M.; Victora, G.D. Germinal Center Selection and Affinity Maturation Require Dynamic Regulation of MTORC1 Kinase. Immunity 2017, 46, 1045–1058.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, Q.; Zhang, H.; Qin, J.; Xu, Z.; Gui, L.; Liu, B.; Liu, C.; Xu, C.; Liu, W.; Zhang, S.; et al. Rapamycin Inhibits BAFF-Stimulated Cell Proliferation and Survival by Suppressing MTOR-Mediated PP2A-Erk1/2 Signaling Pathway in Normal and Neoplastic B-Lymphoid Cells. Cell. Mol. Life Sci. 2015, 72, 4867–4884. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Liang, D.; Zeng, Q.; Ren, Q.; Ma, H.; Gui, L.; Chen, S.; Guo, M.; Xu, Y.; Gao, W.; et al. HsBAFF Promotes Proliferation and Survival in Cultured B Lymphocytes via Calcium Signaling Activation of MTOR Pathway. Cytokine 2013, 62, 310–321. [Google Scholar] [CrossRef]
- Wu, T.; Qin, X.; Kurepa, Z.; Kumar, K.R.; Liu, K.; Kanta, H.; Zhou, X.J.; Satterthwaite, A.B.; Davis, L.S.; Mohan, C. Shared Signaling Networks Active in B Cells Isolated from Genetically Distinct Mouse Models of Lupus. J. Clin. Investig. 2007, 117, 2186–2196. [Google Scholar] [CrossRef] [Green Version]
- Raza, I.G.A.; Clarke, A.J. B Cell Metabolism and Autophagy in Autoimmunity. Front. Immunol. 2021, 12, 2157. [Google Scholar] [CrossRef]
- Ireland, J.M.; Unanue, E.R. Autophagy in Antigen-Presenting Cells Results in Presentation of Citrullinated Peptides to CD4 T Cells. J. Exp. Med. 2011, 208, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.J.; Ellinghaus, U.; Cortini, A.; Stranks, A.; Simon, A.K.; Botto, M.; Vyse, T.J. Autophagy Is Activated in Systemic Lupus Erythematosus and Required for Plasmablast Development. Ann. Rheum. Dis. 2015, 74, 912–920. [Google Scholar] [CrossRef] [Green Version]
- Salehi, M.; Bagherpour, B.; Shayghannejad, V.; Mohebi, F.; Jafari, R. Th1, Th2 and Th17 Cytokine Profile in Patients with Multiple Sclerosis Following Treatment with Rapamycin. Iran. J. Immunol. 2016, 13, 141–147. [Google Scholar] [PubMed]
- Bagherpour, B.; Salehi, M.; Jafari, R.; Bagheri, A.; Kiani-Esfahani, A.; Edalati, M.; Kardi, M.T.; Shaygannejad, V. Promising Effect of Rapamycin on Multiple Sclerosis. Mult. Scler. Relat. Disord. 2018, 26, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Barkhof, F.; Desmet, A. The Effect of Oral Temsirolimus on New Magnetic Resonance Imaging Scan Lesions, Brain Atrophy, and the Number of Relapses in Multiple Sclerosis: Results from a Randomised, Controlled Clinical Trial. J. Neurol. 2005, 252, 46. [Google Scholar]
- Moraal, B.; van den Elskamp, I.J.; Knol, D.L.; Uitdehaag, B.M.J.; Geurts, J.J.G.; Vrenken, H.; Pouwels, P.J.W.; van Schijndel, R.A.; Meier, D.S.; Guttmann, C.R.G.; et al. Long-Interval T2-Weighted Subtraction Magnetic Resonance Imaging A Powerful New Outcome Measure in Multiple Sclerosis Trials. Ann. Neurol. 2010, 67, 667–675. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Meuth, S.G.; Stüve, O.; Kieseier, B.; Wiendl, H. Multiple Sclerosis Therapy: An Update on Recently Finished Trials. J. Neurol. 2007, 254, 1473–1490. [Google Scholar] [CrossRef]
- Negrotto, L.; Farez, M.F.; Correale, J. Immunologic Effects of Metformin and Pioglitazone Treatment on Metabolic Syndrome and Multiple Sclerosis. JAMA Neurol. 2016, 73, 520–528. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in Immunity and Inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef]
| Inhibitor | Category of mTOR Inhibitor | Inhibition Effect |
|---|---|---|
| Rapamycin (AY-22989) | Rapamycin | Direct mTOR inhibition |
| Temsirolimus (CCI-779) | Rapalogs | |
| Everolimus (RAD-001) | ||
| Ridaforolimus (AP23573) | ||
| OSI-027 | TORC1/TORC2 inhibitors | |
| Vistusertib (AZD2014) | ||
| AZD8055 | ||
| Dactolisib (BEZ235) | PI3K/mTOR inhibitors | Indirect mTOR inhibition |
| Apitolisib (GDC-0980) | ||
| Gedatolisib (PF05212384) | ||
| Omipalisib (GSK2126458) | ||
| Metformin (A10BA02) | AMPK activation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vakrakou, A.G.; Alexaki, A.; Brinia, M.-E.; Anagnostouli, M.; Stefanis, L.; Stathopoulos, P. The mTOR Signaling Pathway in Multiple Sclerosis; from Animal Models to Human Data. Int. J. Mol. Sci. 2022, 23, 8077. https://doi.org/10.3390/ijms23158077
Vakrakou AG, Alexaki A, Brinia M-E, Anagnostouli M, Stefanis L, Stathopoulos P. The mTOR Signaling Pathway in Multiple Sclerosis; from Animal Models to Human Data. International Journal of Molecular Sciences. 2022; 23(15):8077. https://doi.org/10.3390/ijms23158077
Chicago/Turabian StyleVakrakou, Aigli G., Anastasia Alexaki, Maria-Evgenia Brinia, Maria Anagnostouli, Leonidas Stefanis, and Panos Stathopoulos. 2022. "The mTOR Signaling Pathway in Multiple Sclerosis; from Animal Models to Human Data" International Journal of Molecular Sciences 23, no. 15: 8077. https://doi.org/10.3390/ijms23158077
APA StyleVakrakou, A. G., Alexaki, A., Brinia, M.-E., Anagnostouli, M., Stefanis, L., & Stathopoulos, P. (2022). The mTOR Signaling Pathway in Multiple Sclerosis; from Animal Models to Human Data. International Journal of Molecular Sciences, 23(15), 8077. https://doi.org/10.3390/ijms23158077