
Parkinson’s Disease Etiology: Insights and Associations with Phosphate Toxicity


Abstract
:1. Introduction
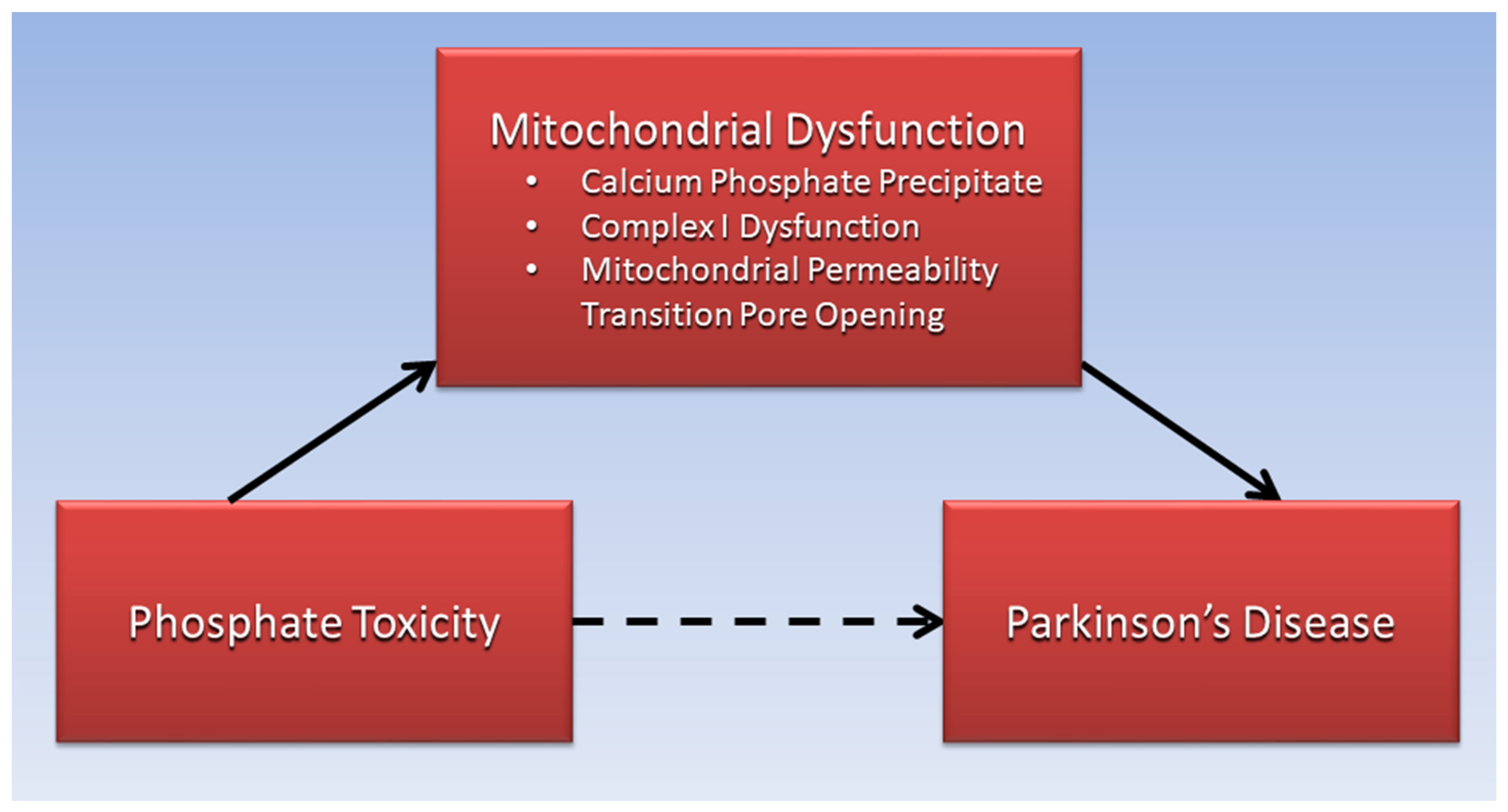
2. Calcium Phosphate and Mitochondrial Dysfunction in PD
3. Tauopathy, Diabetes, and PD
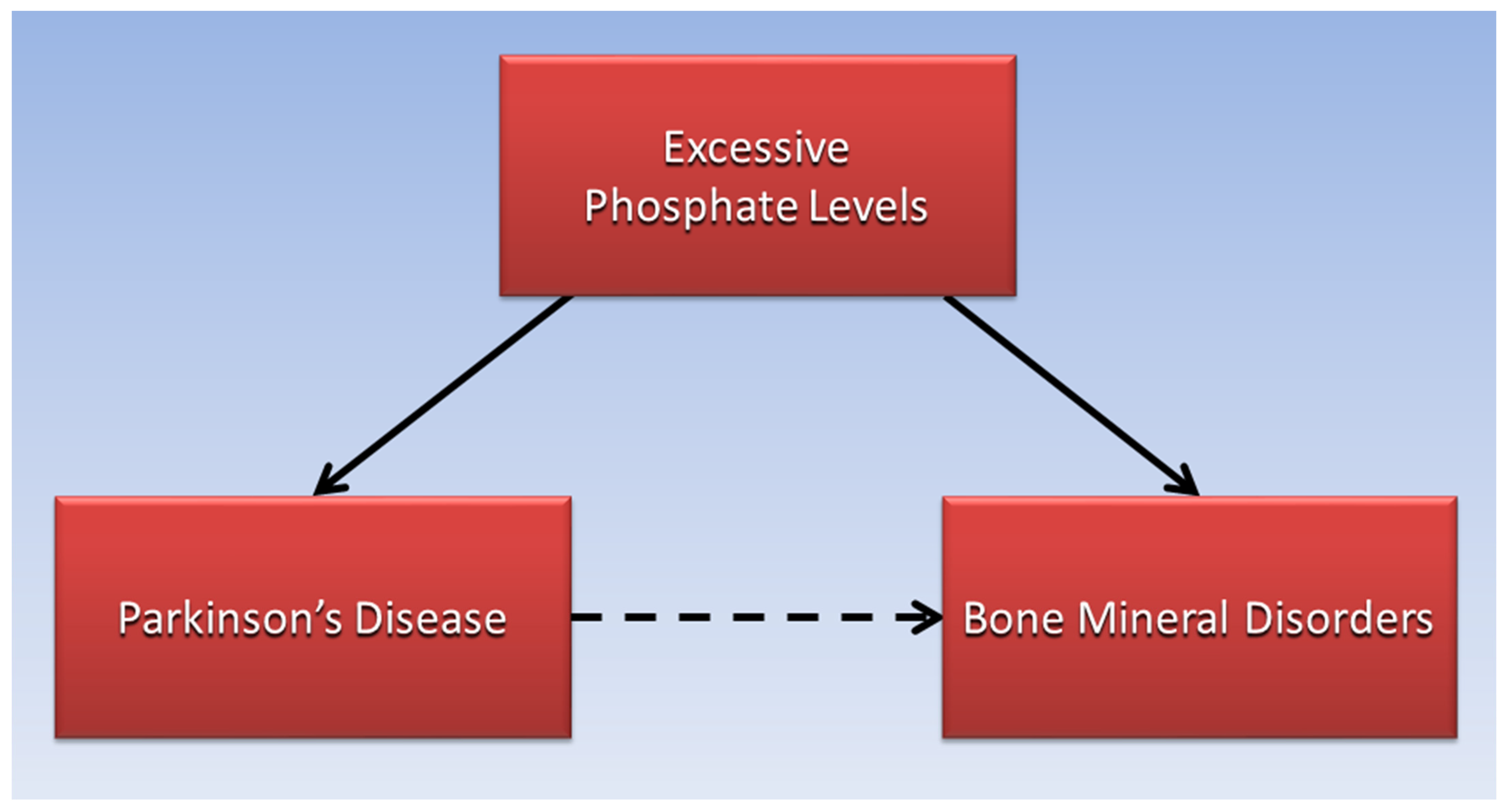
4. Ectopic Calcification and Bone Disorders in Patients with PD
5. Vitamin D, Serum Pi, and PD
6. Sarcopenia, Phosphate Toxicity, and PD
7. Cancer, Phosphate Toxicity, and PD
8. High Dietary Phosphate and PD
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Surguchov, A. Biomarkers in Parkinson’s Disease. In Neurodegenerative Diseases Biomarkers: Towards Translating Research to Clinical Practice; Peplow, P.V., Martinez, B., Gennarelli, T.A., Eds.; Springer: New York, NY, USA, 2022; pp. 155–180. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abbasi, N.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Fiandaca, M.S.; Lonser, R.R.; Elder, J.B.; Ząbek, M.; Bankiewicz, K.S. Advancing gene therapies, methods, and technologies for Parkinson’s Disease and other neurological disorders. Neurol. Neurochir. Pol. 2020, 54, 220–231. [Google Scholar] [CrossRef]
- Brown, R.B.; Razzaque, M.S. Dysregulation of phosphate metabolism and conditions associated with phosphate toxicity. BoneKEy Rep. 2015, 4, 705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfswinkel, J.F.; Furtmueller, E.; Wilderom, C.P.M. Using grounded theory as a method for rigorously reviewing literature. Eur. J. Inf. Syst. 2013, 22, 45–55. [Google Scholar] [CrossRef]
- Kristian, T.; Pivovarova, N.B.; Fiskum, G.; Andrews, S.B. Calcium-induced precipitate formation in brain mitochondria: Composition, calcium capacity, and retention. J. Neurochem. 2007, 102, 1346–1356. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.; Quan, X.; Hwang, K.-H.; Xu, S.; Das, R.; Choi, S.-K.; Wiederkehr, A.; Wollheim, C.B.; Cha, S.-K.; Park, K.-S. Mitochondrial oxidative stress mediates high-phosphate-induced secretory defects and apoptosis in insulin-secreting cells. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E933–E941. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T. Mitochondrial reactive oxygen species, endoplasmic reticulum stress and phosphate toxicity in impairment of pancreatic cells. TTU Rev. 2016, 1, 73–78. [Google Scholar]
- Hassan, A.; Sharma Kandel, R.; Mishra, R.; Gautam, J.; Alaref, A.; Jahan, N. Diabetes Mellitus and Parkinson’s Disease: Shared Pathophysiological Links and Possible Therapeutic Implications. Cureus 2020, 12, e9853. [Google Scholar] [CrossRef]
- Chohan, H.; Senkevich, K.; Patel, R.K.; Bestwick, J.P.; Jacobs, B.M.; Bandres Ciga, S.; Gan-Or, Z.; Noyce, A.J. Type 2 Diabetes as a Determinant of Parkinson’s Disease Risk and Progression. Mov. Disord. 2021, 36, 1420–1429. [Google Scholar] [CrossRef]
- Malyala, S.; Zhang, Y.; Strubbe, J.O.; Bazil, J.N. Calcium phosphate precipitation inhibits mitochondrial energy metabolism. PLoS Comput. Biol. 2019, 15, e1006719. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Kent, A.C.; El Baradie, K.B.Y.; Hamrick, M.W. Targeting the Mitochondrial Permeability Transition Pore to Prevent Age-Associated Cell Damage and Neurodegeneration. Oxid. Med. Cell Longev. 2021, 2021, 6626484. [Google Scholar] [CrossRef] [PubMed]
- Elustondo, P.A.; Nichols, M.; Negoda, A.; Thirumaran, A.; Zakharian, E.; Robertson, G.S.; Pavlov, E.V. Mitochondrial permeability transition pore induction is linked to formation of the complex of ATPase C-subunit, polyhydroxybutyrate and inorganic polyphosphate. Cell Death Discov. 2016, 2, 16070. [Google Scholar] [CrossRef] [Green Version]
- Phadwal, K.; Vrahnas, C.; Ganley, I.G.; MacRae, V.E. Mitochondrial Dysfunction: Cause or Consequence of Vascular Calcification? Front. Cell Dev. Biol. 2021, 9, 611922. [Google Scholar] [CrossRef]
- Varanyuwatana, P.; Halestrap, A.P. The roles of phosphate and the phosphate carrier in the mitochondrial permeability transition pore. Mitochondrion 2012, 12, 120–125. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Fraley, C.; Diao, C.T.; Winkfein, R.; Colicos, M.A.; Duchen, M.R.; French, R.J.; Pavlov, E. Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proc. Natl. Acad. Sci. USA 2007, 104, 18091–18096. [Google Scholar] [CrossRef] [Green Version]
- Angelova, P.R.; Agrawalla, B.K.; Elustondo, P.A.; Gordon, J.; Shiba, T.; Abramov, A.Y.; Chang, Y.T.; Pavlov, E.V. In situ investigation of mammalian inorganic polyphosphate localization using novel selective fluorescent probes JC-D7 and JC-D8. ACS Chem. Biol. 2014, 9, 2101–2110. [Google Scholar] [CrossRef]
- Sexton, C.; Snyder, H.; Beher, D.; Boxer, A.L.; Brannelly, P.; Brion, J.P.; Buée, L.; Cacace, A.M.; Chételat, G.; Citron, M.; et al. Current directions in tau research: Highlights from Tau 2020. Alzheimers Dement. 2022, 18, 988–1007. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, F.; Wang, D.; Li, C.; Fu, Y.; He, W.; Zhang, J. Tau Pathology in Parkinson’s Disease. Front. Neurol. 2018, 9, 809. [Google Scholar] [CrossRef] [Green Version]
- Ganten, D.; Ruckpaul, K.; Birchmeier, W.; Epplen, J.T.; Genser, K.; Gossen, M.; Kersten, B.; Lehrach, H.; Oschkinat, H.; Ruiz, P.; et al. (Eds.) Hyperphosphorylation. In Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar] [CrossRef]
- Ma, M.R.; Hu, Z.W.; Zhao, Y.F.; Chen, Y.X.; Li, Y.M. Phosphorylation induces distinct alpha-synuclein strain formation. Sci. Rep. 2016, 6, 37130. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.; Nagy, J.D.; Elser, J.J. Biological stoichiometry of tumor dynamics: Mathematical models and analysis. Discret. Contin. Dyn. Syst. Ser. B 2004, 4, 221–240. [Google Scholar]
- Gonçalves, R.A.; Wijesekara, N.; Fraser, P.E.; De Felice, F.G. The Link between Tau and Insulin Signaling: Implications for Alzheimer’s Disease and Other Tauopathies. Front. Cell Neurosci. 2019, 13, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, C.; Beare, R.; Phan, T.G.; Bruce, D.G.; Callisaya, M.L.; Srikanth, V. Type 2 diabetes mellitus and biomarkers of neurodegeneration. Neurology 2015, 85, 1123–1130. [Google Scholar] [CrossRef] [Green Version]
- de Pablo-Fernández, E.; Courtney, R.; Rockliffe, A.; Gentleman, S.; Holton, J.L.; Warner, T.T. Faster disease progression in Parkinson’s disease with type 2 diabetes is not associated with increased α-synuclein, tau, amyloid-β or vascular pathology. Neuropathol. Appl. Neurobiol. 2021, 47, 1080–1091. [Google Scholar] [CrossRef]
- De Pablo-Fernandez, E.; Goldacre, R.; Pakpoor, J.; Noyce, A.J.; Warner, T.T. Association between diabetes and subsequent Parkinson disease: A record-linkage cohort study. Neurology 2018, 91, e139–e142. [Google Scholar] [CrossRef]
- Mullee, A.; Romaguera, D.; Pearson-Stuttard, J.; Viallon, V.; Stepien, M.; Freisling, H.; Fagherazzi, G.; Mancini, F.R.; Boutron-Ruault, M.C.; Kühn, T.; et al. Association Between Soft Drink Consumption and Mortality in 10 European Countries. JAMA Intern. Med. 2019, 179, 1479–1490. [Google Scholar] [CrossRef] [Green Version]
- Neelakantan, N.; Park, S.H.; Chen, G.C.; van Dam, R.M. Sugar-sweetened beverage consumption, weight gain, and risk of type 2 diabetes and cardiovascular diseases in Asia: A systematic review. Nutr. Rev. 2021, 80, 50–67. [Google Scholar] [CrossRef]
- Harder, S.; Hopp, K.; Ward, H.; Neglio, H.; Gitlin, J.; Kido, D. Mineralization of the deep gray matter with age: A retrospective review with susceptibility-weighted MR imaging. Am. J. Neuroradiol. 2008, 29, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Vermersch, P.; Leys, D.; Pruvo, J.P.; Clarisse, J.; Petit, H. Parkinson’s disease and basal ganglia calcifications: Prevalence and clinico-radiological correlations. Clin. Neurol. Neurosurg. 1992, 94, 213–217. [Google Scholar] [CrossRef]
- Vorland, C.J.; Stremke, E.R.; Moorthi, R.N.; Hill Gallant, K.M. Effects of Excessive Dietary Phosphorus Intake on Bone Health. Curr. Osteoporos. Rep. 2017, 15, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Torsney, K.M.; Noyce, A.J.; Doherty, K.M.; Bestwick, J.P.; Dobson, R.; Lees, A.J. Bone health in Parkinson’s disease: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2014, 85, 1159–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakşi, E.; Yaşar, M.F. Bone mineral density and vitamin D levels in parkinson’s disease: A retrospective controlled study. Northwest Med. J. 2022, 2, 51–58. [Google Scholar] [CrossRef]
- Zhang, H.-J.; Zhang, J.-R.; Mao, C.-J.; Li, K.; Wang, F.; Chen, J.; Liu, C.-F. Relationship between 25-Hydroxyvitamin D, bone density, and Parkinson’s disease symptoms. Acta Neurol. Scand. 2019, 140, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, E.A.; Gundogdu, I.; Tonuk, B.; Umay, E.; Kocer, B.G.; Cakci, A. Bone mineral density and serum vitamin D status in Parkinson’s disease: Are the stage and clinical features of the disease important? Neurol. India 2020, 68, 394–400. [Google Scholar] [CrossRef]
- Chowdhury, A.; Balogh, E.; Ababneh, H.; Tóth, A.; Jeney, V. Activation of Nrf2/HO-1 Antioxidant Pathway by Heme Attenuates Calcification of Human Lens Epithelial Cells. Pharmaceuticals 2022, 15, 493. [Google Scholar] [CrossRef]
- Lai, S.-W.; Lin, C.-L.; Liao, K.-F.; Chang-Ou, K.-C. Increased risk of Parkinson’s disease in cataract patients: A population-based cohort study. Parkinsonism Relat. Disord. 2015, 21, 68–71. [Google Scholar] [CrossRef]
- Meléndez-Flores, J.D.; Estrada-Bellmann, I. Linking chronic kidney disease and Parkinson’s disease: A literature review. Metab Brain Dis. 2021, 36, 1–12. [Google Scholar] [CrossRef]
- Bover, J.; Aguilar, A.; Arana, C.; Molina, P.; Lloret, M.J.; Ochoa, J.; Berná, G.; Gutiérrez-Maza, Y.G.; Rodrigues, N.; D’Marco, L.; et al. Clinical Approach to Vascular Calcification in Patients With Non-dialysis Dependent Chronic Kidney Disease: Mineral-Bone Disorder-Related Aspects. Front. Med. 2021, 8, 642718. [Google Scholar] [CrossRef]
- Brown, R.B. Diabetes, diabetic complications, and phosphate toxicity: A scoping review. Curr. Diabetes Rev. 2020, 16, 674–689. [Google Scholar] [CrossRef]
- Kummer, B.R.; Diaz, I.; Wu, X.; Aaroe, A.E.; Chen, M.L.; Iadecola, C.; Kamel, H.; Navi, B.B. Associations between cerebrovascular risk factors and parkinson disease. Ann. Neurol. 2019, 86, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Acar, S.; Özkan, B. Vitamin D Metabolism. In Vitamin D; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Erben, R.G. Physiological Actions of Fibroblast Growth Factor-23. Front. Endocrinol. 2018, 9, 267. [Google Scholar] [CrossRef] [PubMed]
- Koduah, P.; Paul, F.; Dörr, J.M. Vitamin D in the prevention, prediction and treatment of neurodegenerative and neuroinflammatory diseases. Epma J. 2017, 8, 313–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samavarchi Tehrani, S.; Sarfi, M.; Yousefi, T.; Ahmadi Ahangar, A.; Gholinia, H.; Mohseni Ahangar, R.; Maniati, M.; Saadat, P. Comparison of the calcium-related factors in Parkinson’s disease patients with healthy individuals. Casp. J. Intern. Med. 2020, 11, 28–33. [Google Scholar] [CrossRef]
- Amanzadeh, J.; Reilly, R.F. Hypophosphatemia: An evidence-based approach to its clinical consequences and management. Nat. Clin. Pract. Nephrol. 2006, 2, 136–148. [Google Scholar] [CrossRef]
- Osuka, S.; Razzaque, M.S. Can features of phosphate toxicity appear in normophosphatemia? J. Bone Miner. Metab. 2012, 30, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Hiller, A.L.; Murchison, C.F.; Lobb, B.M.; O’Connor, S.; O’Connor, M.; Quinn, J.F. A randomized, controlled pilot study of the effects of vitamin D supplementation on balance in Parkinson’s disease: Does age matter? PLoS ONE 2018, 13, e0203637. [Google Scholar] [CrossRef]
- Luthra, N.S.; Kim, S.; Zhang, Y.; Christine, C.W.; On behalf of the NINDS NET-PD Investigators. Characterization of vitamin D supplementation and clinical outcomes in a large cohort of early Parkinson’s disease. J. Clin. Mov. Disord. 2018, 5, 7. [Google Scholar] [CrossRef] [Green Version]
- Tsekoura, M.; Kastrinis, A.; Katsoulaki, M.; Billis, E.; Gliatis, J. Sarcopenia and Its Impact on Quality of Life. Adv. Exp. Med. Biol. 2017, 987, 213–218. [Google Scholar] [CrossRef]
- Wu, Y.N.; Chen, M.H.; Chiang, P.L.; Lu, C.H.; Chen, H.L.; Yu, C.C.; Chen, Y.S.; Chang, Y.Y.; Lin, W.C. Associations between Brain Structural Damage and Core Muscle Loss in Patients with Parkinson’s Disease. J. Clin. Med. 2020, 9, 239. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Feng, F.; Wei, Q.; Jiang, Z.; Ou, R.; Shang, H. Sarcopenia in Patients With Parkinson’s Disease: A Systematic Review and Meta-Analysis. Front. Neurol. 2021, 12, 598035. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.M.; Moon, J.S.; Chang, M.C. Prevalence of Sarcopenia and Its Association with Diabetes: A Meta-Analysis of Community-Dwelling Asian Population. Front. Med. 2021, 8, 681232. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Tokumoto, M.; Tatsumoto, N.; Taniguchi, M.; Noguchi, H.; Nakano, T.; Masutani, K.; Ooboshi, H.; Tsuruya, K.; Kitazono, T. Phosphate overload directly induces systemic inflammation and malnutrition as well as vascular calcification in uremia. Am. J. Physiol. Ren. Physiol. 2014, 306, F1418–F1428. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.; Nunes, J.M.; Page, M.J.; Roberts, T.; Carr, J.; Nell, T.A.; Kell, D.B.; Pretorius, E. Parkinson’s Disease: A Systemic Inflammatory Disease Accompanied by Bacterial Inflammagens. Front. Aging Neurosci. 2019, 11, 210. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-Y.; Yang, M.; Bao, J.-F.; Gu, L.-J.; Yu, H.-L.; Yuan, W.-J. Phosphate stimulates myotube atrophy through autophagy activation: Evidence of hyperphosphatemia contributing to skeletal muscle wasting in chronic kidney disease. BMC Nephrol. 2018, 19, 45. [Google Scholar] [CrossRef]
- Brown, R.B.; Razzaque, M.S. Phosphate toxicity and tumorigenesis. Biochim. et Biophys. Acta Rev. Cancer 2018, 1869, 303–309. [Google Scholar] [CrossRef] [Green Version]
- Feng, D.D.; Cai, W.; Chen, X. The associations between Parkinson’s disease and cancer: The plot thickens. Transl. Neurodegener. 2015, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Raatz, S.K.; Jahns, L.; Johnson, L.K.; Scheett, A.; Carriquiry, A.; Lemieux, A.; Nakajima, M.; al’Absi, M. Smokers report lower intake of key nutrients than nonsmokers, yet both fall short of meeting recommended intakes. Nutr. Res. 2017, 45, 30–37. [Google Scholar] [CrossRef]
- Phosphorus Fact Sheet for Health Professionals. Available online: https://ods.od.nih.gov/factsheets/Phosphorus-HealthProfessional/#:~:text=Phosphorus%20Intakes%20and%20Status,-Most%20Americans%20consume&text=In%20adults%20aged%2020%20and,mg%20for%20men%20%5B30%5D. (accessed on 3 July 2022).
- Mappin-Kasirer, B.; Pan, H.; Lewington, S.; Kizza, J.; Gray, R.; Clarke, R.; Peto, R. Tobacco smoking and the risk of Parkinson disease. Neurology 2020, 94, e2132–e2138. [Google Scholar] [CrossRef]
- Tran, J.; Anastacio, H.; Bardy, C. Genetic predispositions of Parkinson’s disease revealed in patient-derived brain cells. NPJ Parkinson’s Dis. 2020, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.R.; Lazo, M.; Appel, L.J.; Gutiérrez, O.M.; Grams, M.E. High dietary phosphorus intake is associated with all-cause mortality: Results from NHANES III. Am. J. Clin. Nutr. 2013, 99, 320–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erem, S.; Razzaque, M.S. Dietary phosphate toxicity: An emerging global health concern. Histochem. Cell Biol. 2018, 150, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Kyrozis, A.; Ghika, A.; Stathopoulos, P.; Vassilopoulos, D.; Trichopoulos, D.; Trichopoulou, A. Dietary and lifestyle variables in relation to incidence of Parkinson’s disease in Greece. Eur. J. Epidemiol. 2013, 28, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Olsson, E.; Byberg, L.; Höijer, J.; Kilander, L.; Larsson, S.C. Milk and Fermented Milk Intake and Parkinson’s Disease: Cohort Study. Nutrients 2020, 12, 2763. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, C.; Piccoli, G.B.; Cupisti, A. The “phosphorus pyramid”: A visual tool for dietary phosphate management in dialysis and CKD patients. BMC Nephrol. 2015, 16, 9. [Google Scholar] [CrossRef]
- Hughes, K.C.; Gao, X.; Kim, I.Y.; Wang, M.; Weisskopf, M.G.; Schwarzschild, M.A.; Ascherio, A. Intake of dairy foods and risk of Parkinson disease. Neurology 2017, 89, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Gough, S.M.; Casella, A.; Ortega, K.J.; Hackam, A.S. Neuroprotection by the Ketogenic Diet: Evidence and Controversies. Front. Nutr. 2021, 8, 782657. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.; Hallett, M.; Ehrlich, D. Nutritional Ketosis in Parkinson’s Disease—A Review of Remaining Questions and Insights. Neurotherapeutics 2021, 18, 1637–1649. [Google Scholar] [CrossRef]
- Metcalfe-Roach, A.; Yu, A.C.; Golz, E.; Cirstea, M.; Sundvick, K.; Kliger, D.; Foulger, L.H.; Mackenzie, M.; Finlay, B.B.; Appel-Cresswell, S. MIND and Mediterranean Diets Associated with Later Onset of Parkinson’s Disease. Mov. Disord. 2021, 36, 977–984. [Google Scholar] [CrossRef]
- Strikwerda, A.J.; Dommershuijsen, L.J.; Ikram, M.K.; Voortman, T. Diet Quality and Risk of Parkinson’s Disease: The Rotterdam Study. Nutrients 2021, 13, 3970. [Google Scholar] [CrossRef]
- USDA. National Nutrient Database for Standard Reference, Legacy. 2018. Available online: https://fdc.nal.usda.gov/index.html (accessed on 16 July 2022).
- Jenkins, D.J.; Wolever, T.M.; Taylor, R.H.; Barker, H.; Fielden, H.; Baldwin, J.M.; Bowling, A.C.; Newman, H.C.; Jenkins, A.L.; Goff, D.V. Glycemic index of foods: A physiological basis for carbohydrate exchange. Am. J. Clin. Nutr. 1981, 34, 362–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Li, L.; Bennett, D.; Guo, Y.; Turnbull, I.; Yang, L.; Bragg, F.; Bian, Z.; Chen, Y.; Chen, J.; et al. Fresh fruit consumption in relation to incident diabetes and diabetic vascular complications: A 7-y prospective study of 0.5 million Chinese adults. PLoS Med. 2017, 14, e1002279. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Miao, S.; Huang, Y.; Liu, Z.; Tian, H.; Yin, X.; Tang, W.; Steffen, L.M.; Xi, B. Fruit intake decreases risk of incident type 2 diabetes: An updated meta-analysis. Endocrine 2015, 48, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Massy, Z.A.; Drüeke, T.B. Strategies for Phosphate Control in Patients with CKD. Kidney Int. Rep. 2019, 4, 1043–1056. [Google Scholar] [CrossRef] [Green Version]
- Steitz, S.A.; Speer, M.Y.; McKee, M.D.; Liaw, L.; Almeida, M.; Yang, H.; Giachelli, C.M. Osteopontin inhibits mineral deposition and promotes regression of ectopic calcification. Am. J. Pathol. 2002, 161, 2035–2046. [Google Scholar] [CrossRef] [Green Version]
- Finch, J.L.; Lee, D.H.; Liapis, H.; Ritter, C.; Zhang, S.; Suarez, E.; Ferder, L.; Slatopolsky, E. Phosphate restriction significantly reduces mortality in uremic rats with established vascular calcification. Kidney Int. 2013, 84, 1145–1153. [Google Scholar] [CrossRef]
- Jono, S.; Peinado, C.; Giachelli, C.M. Phosphorylation of osteopontin is required for inhibition of vascular smooth muscle cell calcification. J. Biol. Chem. 2000, 275, 20197–20203. [Google Scholar] [CrossRef] [Green Version]
- Hunter, G.K. Role of Osteopontin in Modulation of Hydroxyapatite Formation. Calcif. Tissue Int. 2013, 93, 348–354. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Food Item | Phosphorus mg/kcal | Food Item | Phosphorus mg/kcal |
|---|---|---|---|
| Pineapple | 0.16 | Potato, white | 0.90 |
| Pear | 0.21 | Corn | 1.03 |
| Apple | 0.21 | Wheat flour, wholegrain | 1.05 |
| Date, Medjool | 0.22 | Brazil nut | 1.10 |
| Macadamia nut | 0.26 | Beef, lean | 1.54 |
| Coconut | 0.32 | Fish, Tilapia | 1.77 |
| Avocado | 0.33 | Chicken breast, skinless | 1.78 |
| Rice, brown | 0.73 | Cow milk, non-fat | 2.89 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, R.B. Parkinson’s Disease Etiology: Insights and Associations with Phosphate Toxicity. Int. J. Mol. Sci. 2022, 23, 8060. https://doi.org/10.3390/ijms23158060
Brown RB. Parkinson’s Disease Etiology: Insights and Associations with Phosphate Toxicity. International Journal of Molecular Sciences. 2022; 23(15):8060. https://doi.org/10.3390/ijms23158060
Chicago/Turabian StyleBrown, Ronald B. 2022. "Parkinson’s Disease Etiology: Insights and Associations with Phosphate Toxicity" International Journal of Molecular Sciences 23, no. 15: 8060. https://doi.org/10.3390/ijms23158060
APA StyleBrown, R. B. (2022). Parkinson’s Disease Etiology: Insights and Associations with Phosphate Toxicity. International Journal of Molecular Sciences, 23(15), 8060. https://doi.org/10.3390/ijms23158060