
Preclinical Assessment of the Combination of PSMA-Targeting Radionuclide Therapy with PARP Inhibitors for Prostate Cancer Treatment

 ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. PARPi Did Not Radiosensitize PCa Cells to PSMA-TRT
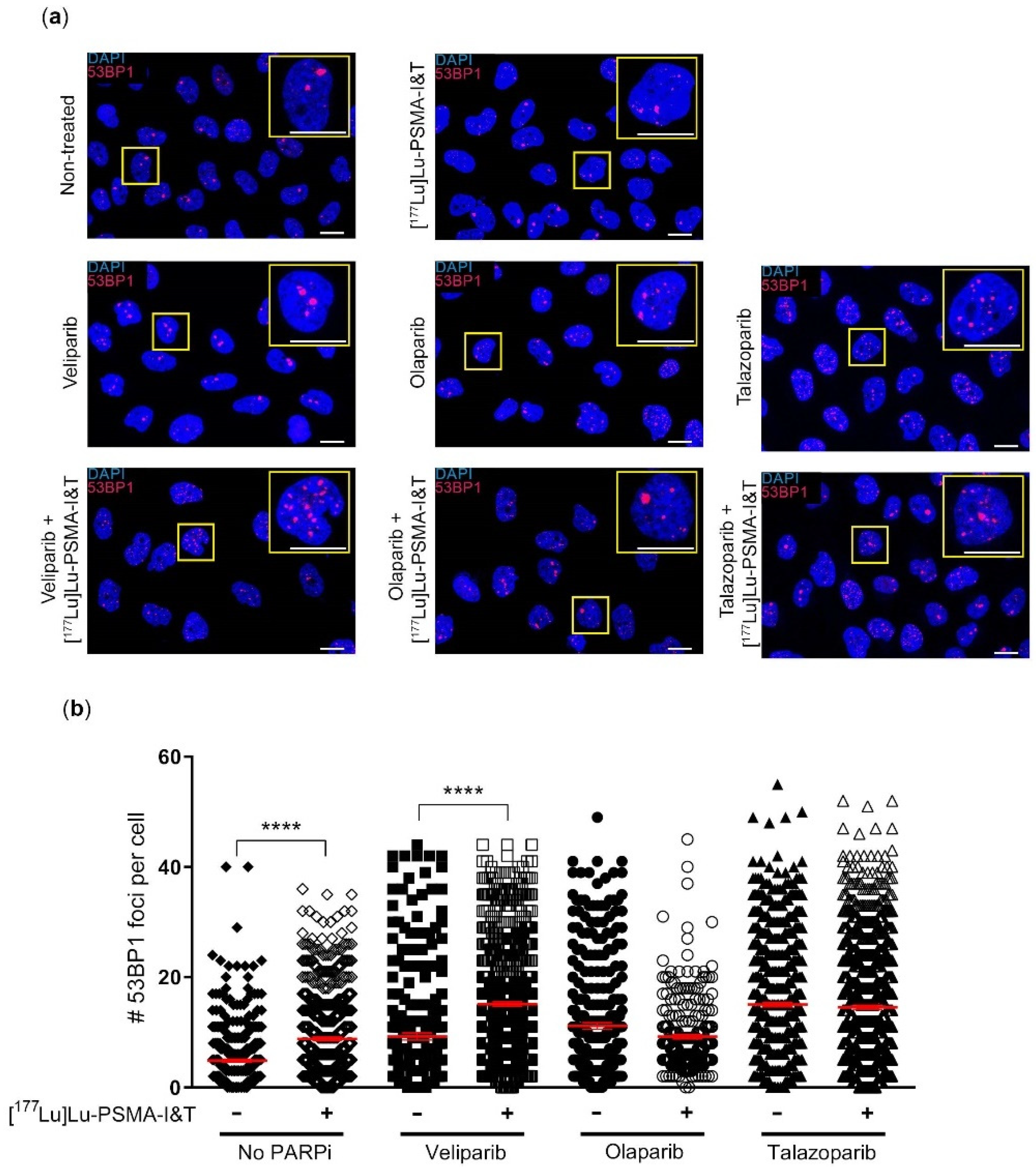
2.2. DNA Damage Induction Was Not Significantly Enhanced by the Combination Treatment
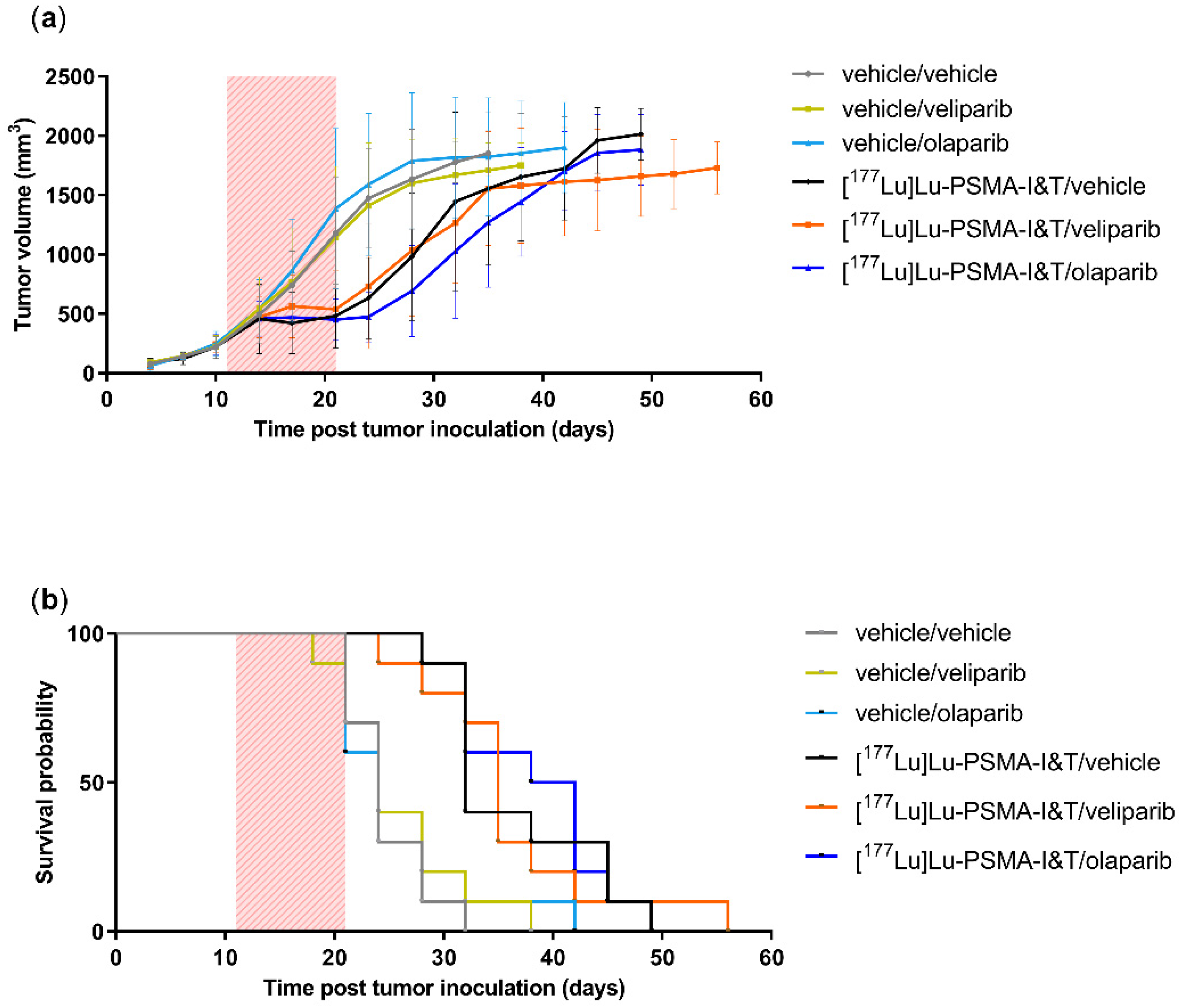
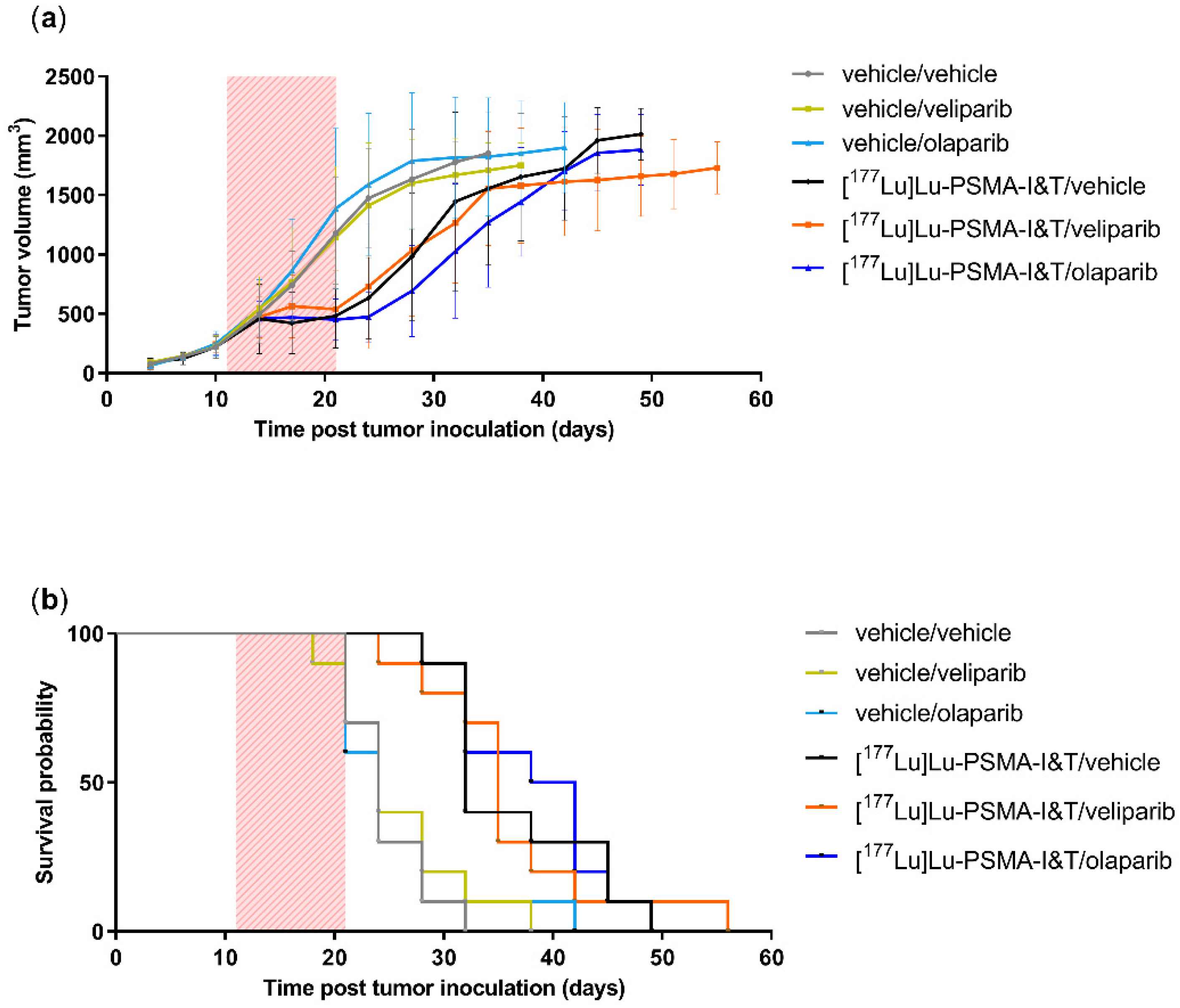
2.3. PARPi Treatment Started Simultaneously with PSMA-TRT Did Not Enhance In Vivo Treatment Efficacy
3. Discussion
4. Materials and Methods
4.1. Reagents and Chemicals
4.2. Radiolabeling
4.3. PARP-Inhibitors
4.4. Cell Culture
4.5. Clonogenic Survival Assay
4.6. DNA Damage Assay
4.7. In Vivo Therapy Study
4.8. Olaparib Measurements
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Czerwińska, M.; Bilewicz, A.; Kruszewski, M.; Wegierek-Ciuk, A.; Lankoff, A. Targeted Radionuclide Therapy of Prostate Cancer—From Basic Research to Clinical Perspectives. Molecules 2020, 25, 1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Niaz, M.O.; Nelson, A.; Skafida, M.; Niaz, M.J. Review of 177Lu-PSMA-617 in Patients with Metastatic Castration-Resistant Prostate Cancer. Cureus 2020, 12, e8921. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.; Griffiths, K.; Barata, P.C.; Paller, C.J. PSMA Theranostics: Review of the Current Status of PSMA-Targeted Imaging and Radioligand Therapy. Cancers 2020, 12, 1367. [Google Scholar] [CrossRef] [PubMed]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177–PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Luining, W.I.; Cysouw, M.C.F.; Meijer, D.; Hendrikse, N.H.; Boellaard, R.; Vis, A.N.; Oprea-Lager, D.E. Targeting PSMA Revolutionizes the Role of Nuclear Medicine in Diagnosis and Treatment of Prostate Cancer. Cancers 2022, 14, 1169. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Bruchertseifer, F.; Rathke, H.; Hohenfellner, M.; Giesel, F.L.; Haberkorn, U.; Morgenstern, A. Targeted α-Therapy of Metastatic Castration-Resistant Prostate Cancer with 225Ac-PSMA-617: Swimmer-Plot Analysis Suggests Efficacy Regarding Duration of Tumor Control. J. Nucl. Med. 2018, 59, 795–802. [Google Scholar] [CrossRef] [Green Version]
- Awang, Z.H.; Essler, M.; Ahmadzadehfar, H. Radioligand therapy of metastatic castration-resistant prostate cancer: Current approaches. Radiat. Oncol. 2018, 13, 98. [Google Scholar] [CrossRef]
- Shibata, A.; Jeggo, P. DNA Double-strand Break Repair in a Cellular Context. Clin. Oncol. 2014, 26, 243–249. [Google Scholar] [CrossRef]
- Jagtap, P.; Szabó, C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar] [CrossRef]
- Shen, Y.; Aoyagi-Scharber, M.; Wang, B. Trapping Poly(ADP-Ribose) Polymerase. J. Pharmacol. Exp. Ther. 2015, 353, 446–457. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Wang, J.; Mishail, D.; Wang, C.-Y. Recent advancements in PARP inhibitors-based targeted cancer therapy. Precis. Clin. Med. 2020, 3, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Zandarashvili, L.; Langelier, M.-F.; Velagapudi, U.K.; Hancock, M.A.; Steffen, J.D.; Billur, R.; Hannan, Z.M.; Wicks, A.J.; Krastev, D.B.; Pettitt, S.J.; et al. Structural basis for allosteric PARP-1 retention on DNA breaks. Science 2020, 368, eaax6367. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullinane, C.; Waldeck, K.; Kirby, L.; Rogers, B.E.; Eu, P.; Tothill, R.W.; Hicks, R.J. Enhancing the anti-tumour activity of 177Lu-DOTA-octreotate radionuclide therapy in somatostatin receptor-2 expressing tumour models by targeting PARP. Sci. Rep. 2020, 10, 10196. [Google Scholar] [CrossRef] [PubMed]
- Purohit, N.K.; Shah, R.G.; Adant, S.; Hoepfner, M.; Shah, G.M.; Beauregard, J.-M. Potentiation of 177Lu-octreotate peptide receptor radionuclide therapy of human neuroendocrine tumor cells by PARP inhibitor. Oncotarget 2018, 9, 24693–24706. [Google Scholar] [CrossRef] [Green Version]
- Nonnekens, J.; van Kranenburg, M.; Beerens, C.E.; Suker, M.; Doukas, M.; van Eijck, C.H.; de Jong, M.; van Gent, D.C. Potentiation of Peptide Receptor Radionuclide Therapy by the PARP Inhibitor Olaparib. Theranostics 2016, 6, 1821–1832. [Google Scholar] [CrossRef] [Green Version]
- Peter MacCallum Cancer Centre, Australia. 177Lu-PSMA-617 Therapy and Olaparib in Patients with Metastatic Castration Resistant Prostate Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03874884 (accessed on 1 May 2022).
- Banerejee, S.R.; Kumar, V.; Lisok, A.; Plyku, D.; Nováková, Z.; Brummet, M.; Wharram, B.; Barinka, C.; Hobbs, R.F.; Pomper, M.G. Evaluation of 111In-DOTA-5D3, a Surrogate SPECT Imaging Agent for Radioimmunotherapy of Prostate-Specific Membrane Antigen. J. Nucl. Med. 2018, 60, 400–406. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, T.A.; Ainsworth, W.B.; Ellis, P.A.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Abraham, V.C.; Algire, M.A.; Shi, Y.; Olson, A.M.; et al. PARP1 Trapping by PARP Inhibitors Drives Cytotoxicity in Both Cancer Cells and Healthy Bone Marrow. Mol. Cancer Res. 2019, 17, 409–419. [Google Scholar] [CrossRef] [Green Version]
- LaFargue, C.J.; Molin, G.Z.D.; Sood, A.K.; Coleman, R.L. Exploring and comparing adverse events between PARP inhibitors. Lancet Oncol. 2019, 20, e15–e28. [Google Scholar] [CrossRef]
- Smith, M.A.; Reynolds, C.P.; Kang, M.H.; Kolb, E.A.; Gorlick, R.; Carol, H.; Lock, R.B.; Keir, S.T.; Maris, J.M.; Billups, C.A.; et al. Synergistic Activity of PARP Inhibition by Talazoparib (BMN 673) with Temozolomide in Pediatric Cancer Models in the Pediatric Preclinical Testing Program. Clin. Cancer Res. 2015, 21, 819–832. [Google Scholar] [CrossRef] [Green Version]
- Baldwin, P.; Likhotvorik, R.; Baig, N.; Cropper, J.; Carlson, R.; Kurmasheva, R.; Sridhar, S. Nanoformulation of Talazoparib Increases Maximum Tolerated Doses in Combination With Temozolomide for Treatment of Ewing Sarcoma. Front. Oncol. 2019, 9, 1416. [Google Scholar] [CrossRef] [PubMed]
- Gani, C.; Coackley, C.; Kumareswaran, R.; Schütze, C.; Krause, M.; Zafarana, G.; Bristow, R.G. In vivo studies of the PARP inhibitor, AZD-2281, in combination with fractionated radiotherapy: An exploration of the therapeutic ratio. Radiother. Oncol. 2015, 116, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Aerts, A.; Eberlein, U.; Holm, S.; Hustinx, R.; Konijnenberg, M.; Strigari, L.; van Leeuwen, F.W.; Glatting, G.; Lassmann, M. EANM position paper on the role of radiobiology in nuclear medicine. Eur. J. Pediatr. 2021, 48, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Hanna, C.; Kurian, K.M.; Williams, K.; Watts, C.; Jackson, A.; Carruthers, R.; Strathdee, K.; Cruickshank, G.; Dunn, L.; Erridge, S.; et al. Pharmacokinetics, safety, and tolerability of olaparib and temozolomide for recurrent glioblastoma: Results of the phase I OPARATIC trial. Neuro-Oncology 2020, 22, 1840–1850. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liao, C.-Y.; Chtatou, H.; Incrocci, L.; van Gent, D.C.; van Weerden, W.M.; Nonnekens, J. Apalutamide Sensitizes Prostate Cancer to Ionizing Radiation via Inhibition of Non-Homologous End-Joining DNA Repair. Cancers 2019, 11, 1593. [Google Scholar] [CrossRef] [Green Version]
- Nonnekens, J.; Schottelius, M. “Luke! Luke! Don’t! It’s a trap!”—Spotlight on bias in animal experiments in nuclear oncology. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1024–1026. [Google Scholar] [CrossRef] [Green Version]
- Chatalic, K.L.; Heskamp, S.; Konijnenberg, M.; Molkenboer-Kuenen, J.D.; Franssen, G.M.; Groningen, M.C.C.-V.; Schottelius, M.; Wester, H.-J.; van Weerden, W.M.; Boerman, O.C.; et al. Towards Personalized Treatment of Prostate Cancer: PSMA I&T, a Promising Prostate-Specific Membrane Antigen-Targeted Theranostic Agent. Theranostics 2016, 6, 849–861. [Google Scholar] [CrossRef]
- Müller, C.; Umbricht, C.A.; Gracheva, N.; Tschan, V.J.; Pellegrini, G.; Bernhardt, P.; Zeevaart, J.R.; Köster, U.; Schibli, R.; van der Meulen, N.P. Terbium-161 for PSMA-targeted radionuclide therapy of prostate cancer. Eur. J. Pediatr. 2019, 46, 1919–1930. [Google Scholar] [CrossRef] [Green Version]
- To, C.; Kim, E.-H.; Royce, D.B.; Williams, C.R.; Collins, R.M.; Risingsong, R.; Sporn, M.B.; Liby, K.T. The PARP Inhibitors, Veliparib and Olaparib, Are Effective Chemopreventive Agents for Delaying Mammary Tumor Development in BRCA1-deficient Mice. Cancer Prev. Res. 2014, 7, 698–707. [Google Scholar] [CrossRef] [Green Version]

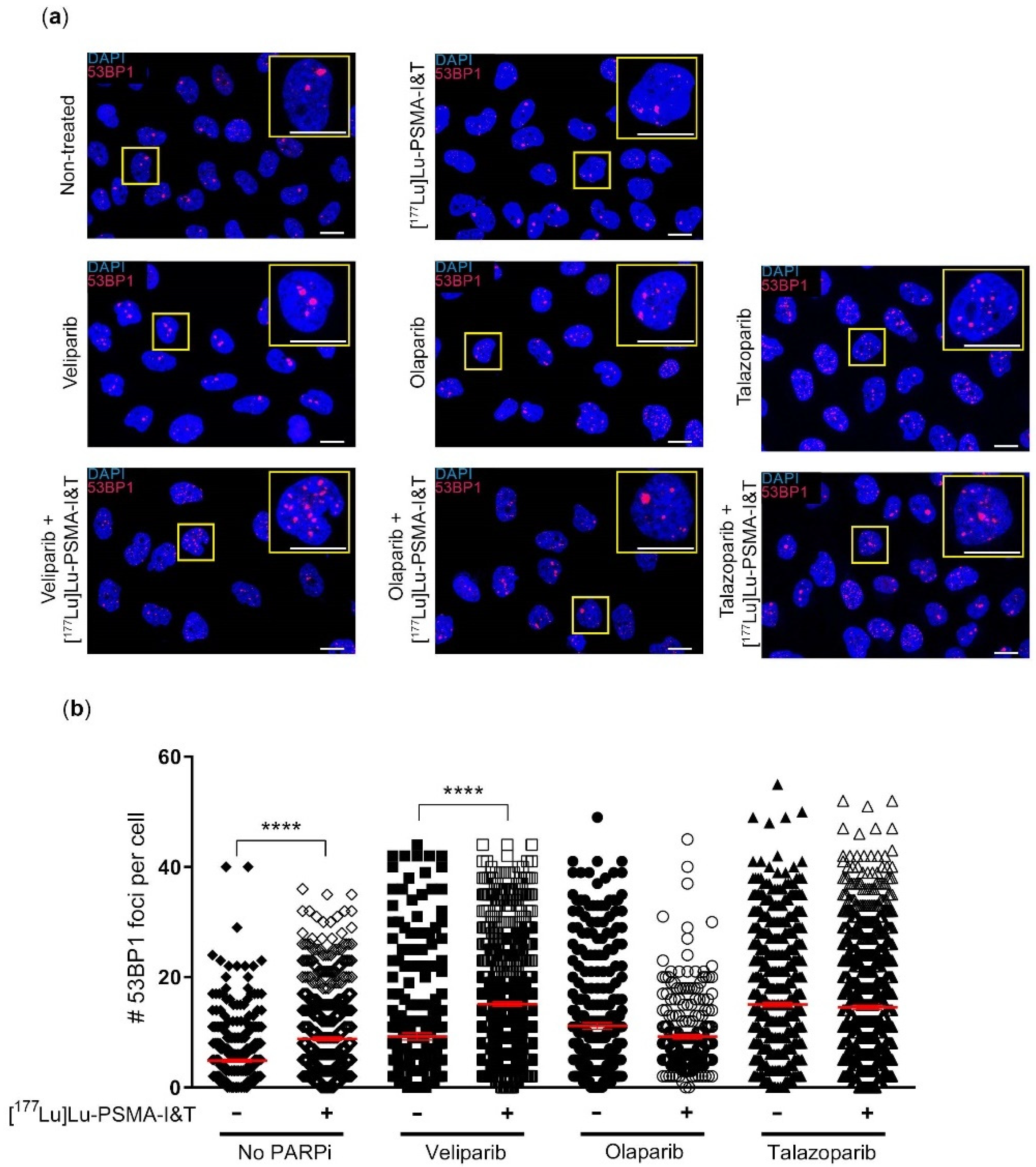

{kind=link}
{kind=link}
{kind=link}
| Animal Number | Olaparib Level in Tumor Tissue (ng/mg) | Olaparib Level in Plasma (nM) |
|---|---|---|
| 9 | 2.17 | 13,234 |
| 10 | 0.144 | - |
| 11 | 0.747 | 2864 |
| 12 | 1.35 | 8047 |
| 40 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruigrok, E.A.M.; Verkaik, N.S.; de Blois, E.; de Ridder, C.; Stuurman, D.; Roobol, S.J.; Van Gent, D.C.; de Jong, M.; Van Weerden, W.M.; Nonnekens, J. Preclinical Assessment of the Combination of PSMA-Targeting Radionuclide Therapy with PARP Inhibitors for Prostate Cancer Treatment. Int. J. Mol. Sci. 2022, 23, 8037. https://doi.org/10.3390/ijms23148037
Ruigrok EAM, Verkaik NS, de Blois E, de Ridder C, Stuurman D, Roobol SJ, Van Gent DC, de Jong M, Van Weerden WM, Nonnekens J. Preclinical Assessment of the Combination of PSMA-Targeting Radionuclide Therapy with PARP Inhibitors for Prostate Cancer Treatment. International Journal of Molecular Sciences. 2022; 23(14):8037. https://doi.org/10.3390/ijms23148037
Chicago/Turabian StyleRuigrok, Eline A. M., Nicole S. Verkaik, Erik de Blois, Corrina de Ridder, Debra Stuurman, Stefan J. Roobol, Dik C. Van Gent, Marion de Jong, Wytske M. Van Weerden, and Julie Nonnekens. 2022. "Preclinical Assessment of the Combination of PSMA-Targeting Radionuclide Therapy with PARP Inhibitors for Prostate Cancer Treatment" International Journal of Molecular Sciences 23, no. 14: 8037. https://doi.org/10.3390/ijms23148037
APA StyleRuigrok, E. A. M., Verkaik, N. S., de Blois, E., de Ridder, C., Stuurman, D., Roobol, S. J., Van Gent, D. C., de Jong, M., Van Weerden, W. M., & Nonnekens, J. (2022). Preclinical Assessment of the Combination of PSMA-Targeting Radionuclide Therapy with PARP Inhibitors for Prostate Cancer Treatment. International Journal of Molecular Sciences, 23(14), 8037. https://doi.org/10.3390/ijms23148037