
Cytochrome P450 27C1 Level Dictates Lung Cancer Tumorigenicity and Sensitivity towards Multiple Anticancer Agents and Its Potential Interplay with the IGF-1R/Akt/p53 Signaling Pathway

 , , , , ,
, , , , ,  , and
, and 
Abstract
1. Introduction
2. Results
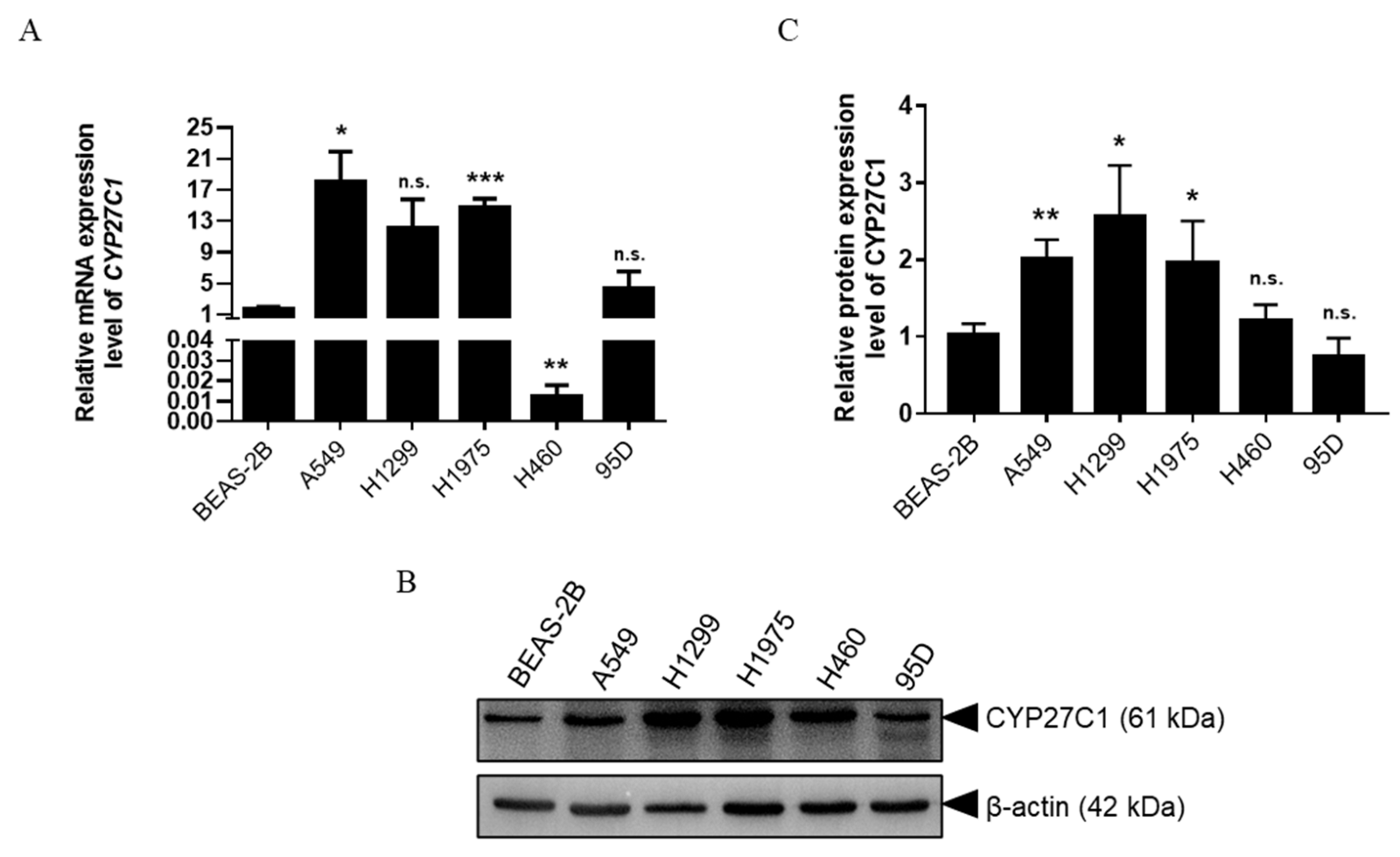
2.1. CYP27C1 Is Differentially Expressed in Human Lung Cancer Cell Lines
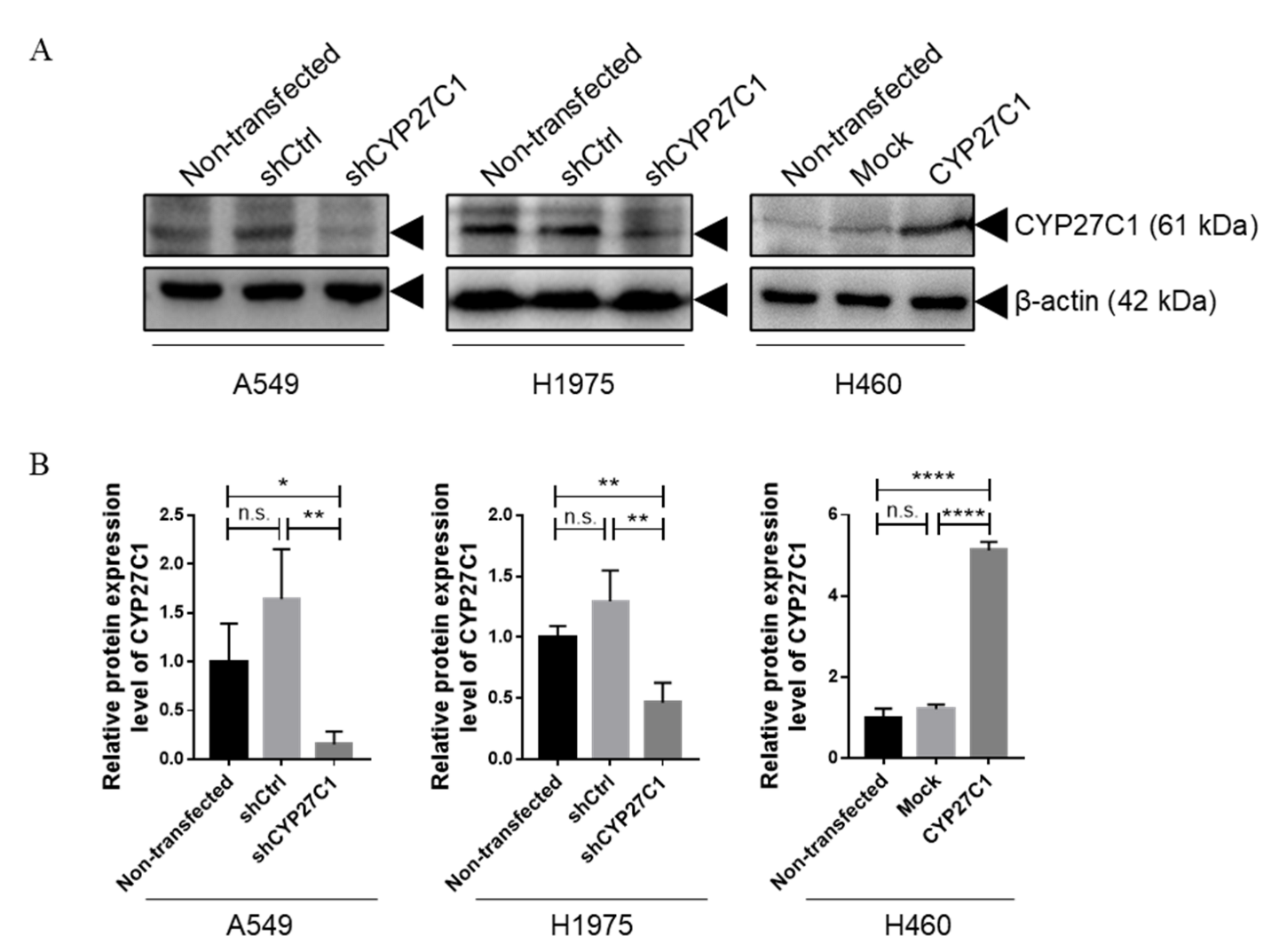
2.2. Stable CYP27C1-Knockdown Enhances Lung Cancer Progression
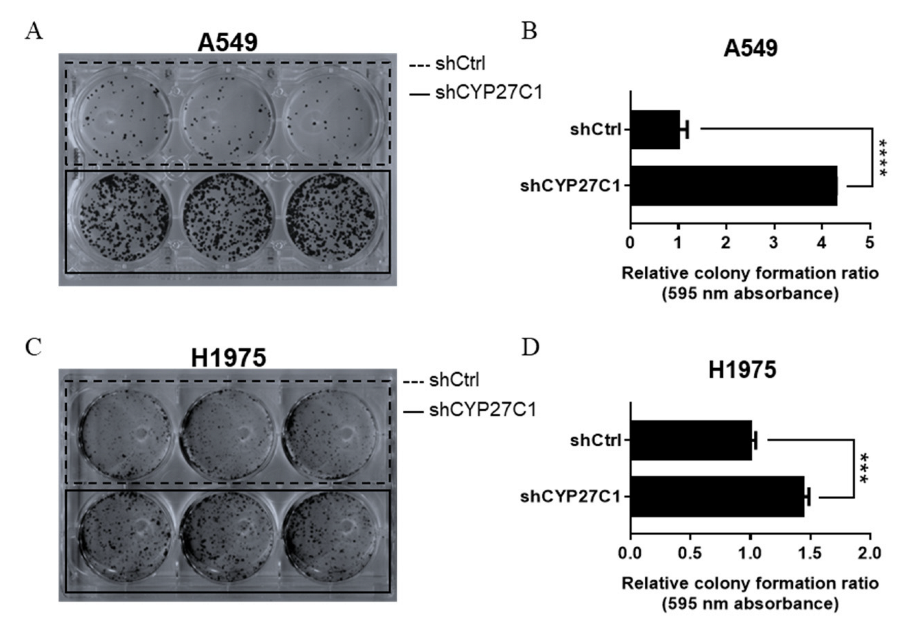
2.2.1. Stable CYP27C1-Knockdown Aggravates Colony Formation
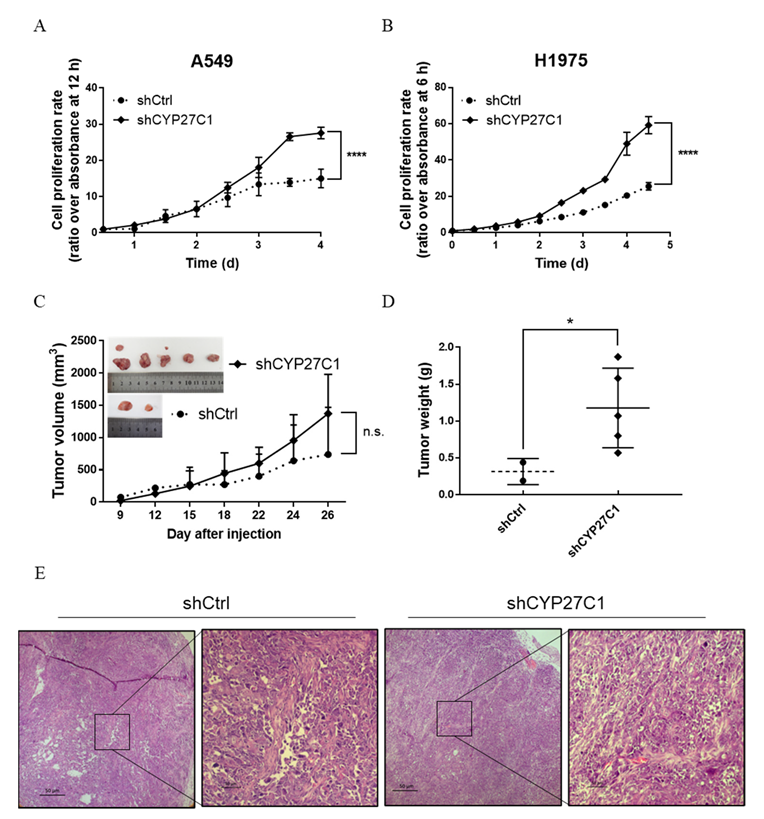
2.2.2. Stable CYP27C1-Knockdown Facilitates Cell Proliferation
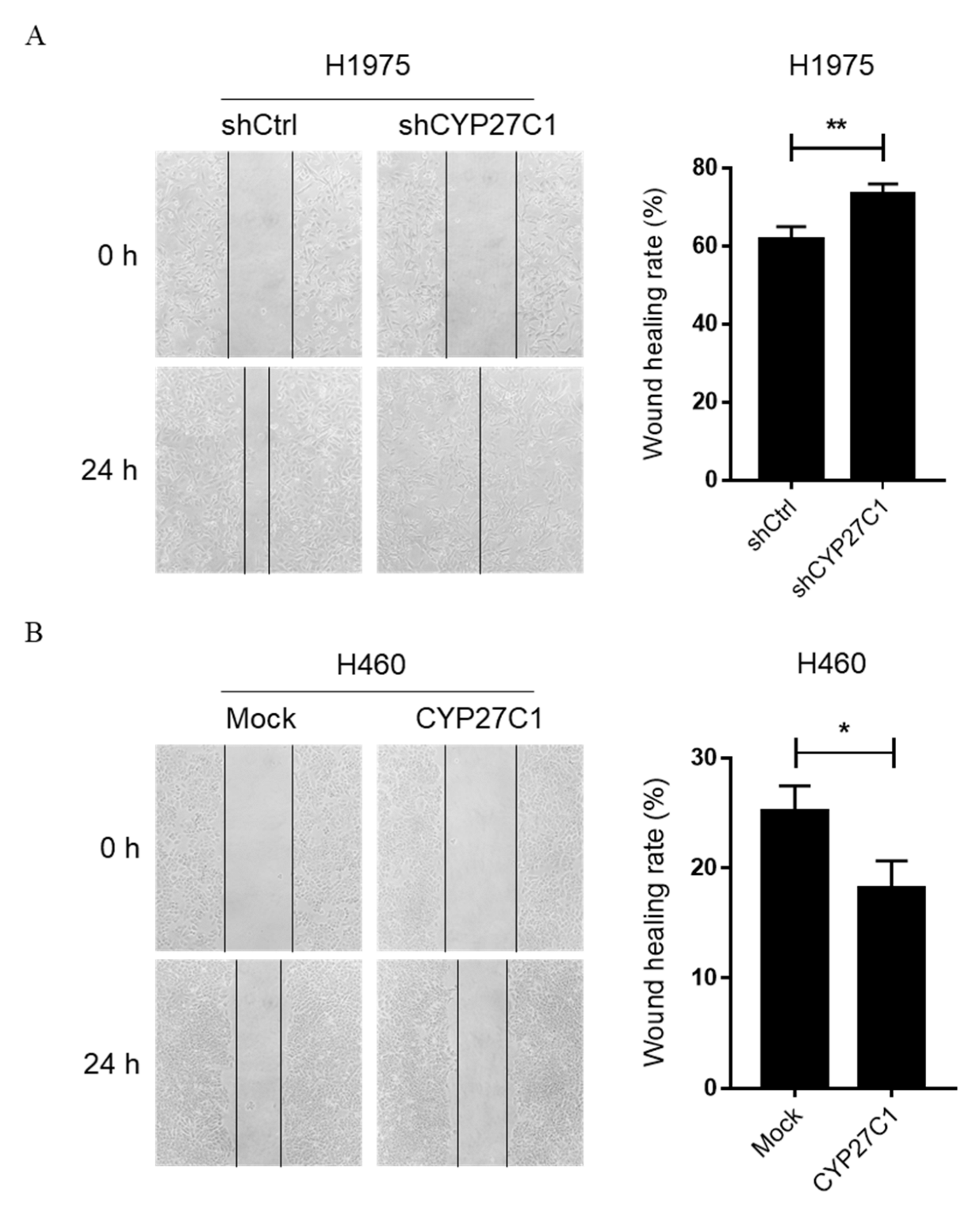
2.2.3. Stable CYP27C1-Knockdown Promotes Cell Migration
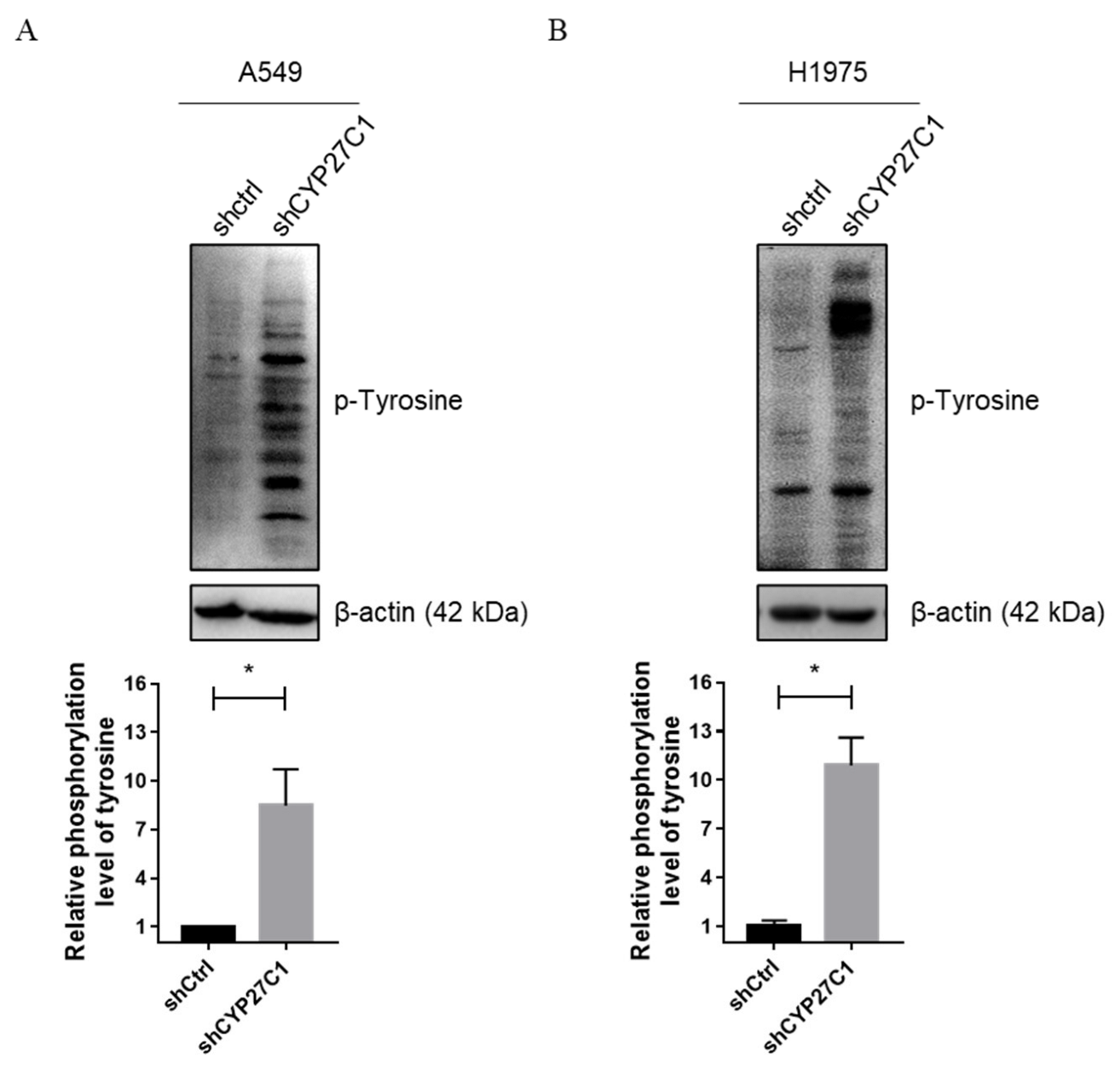
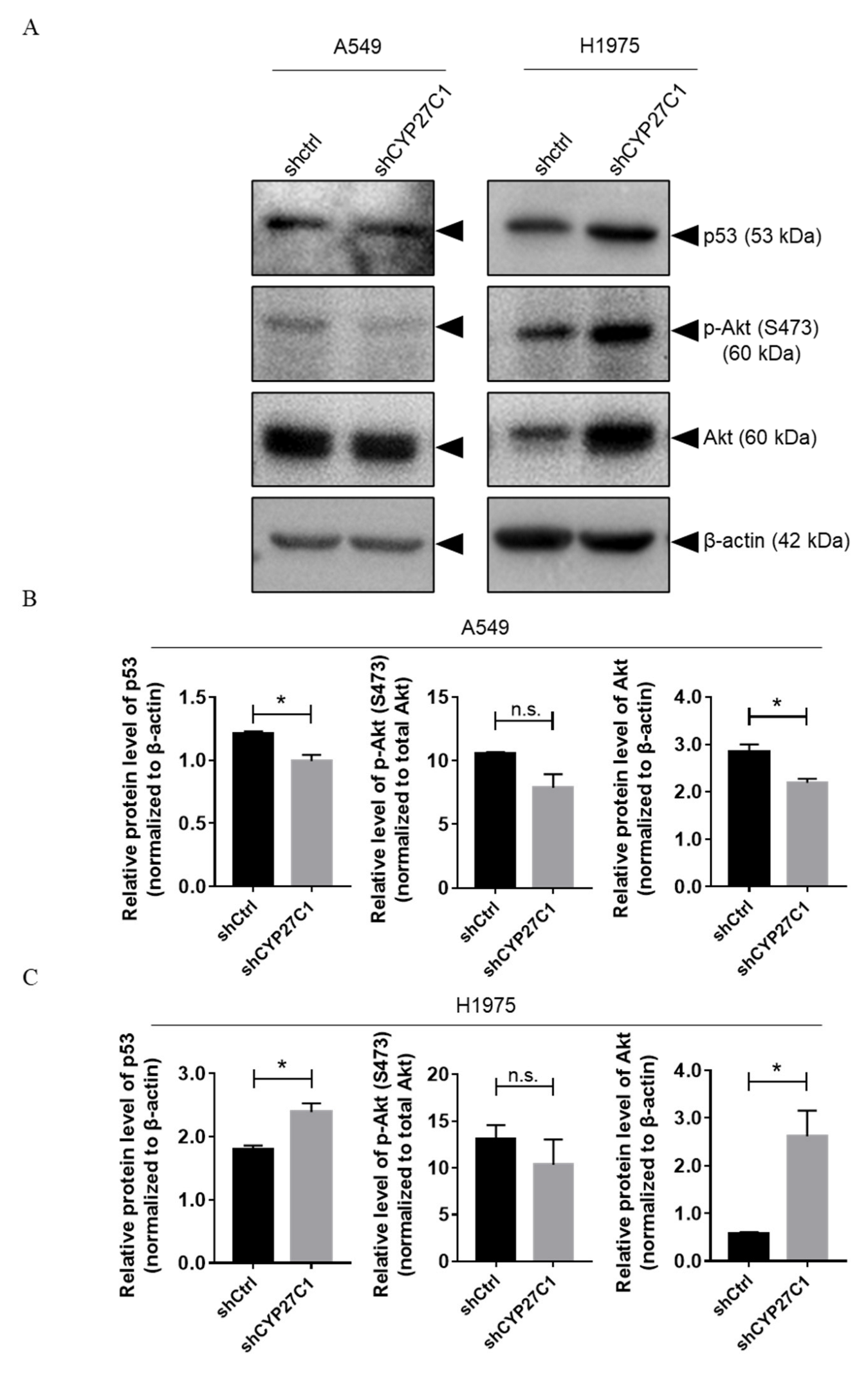
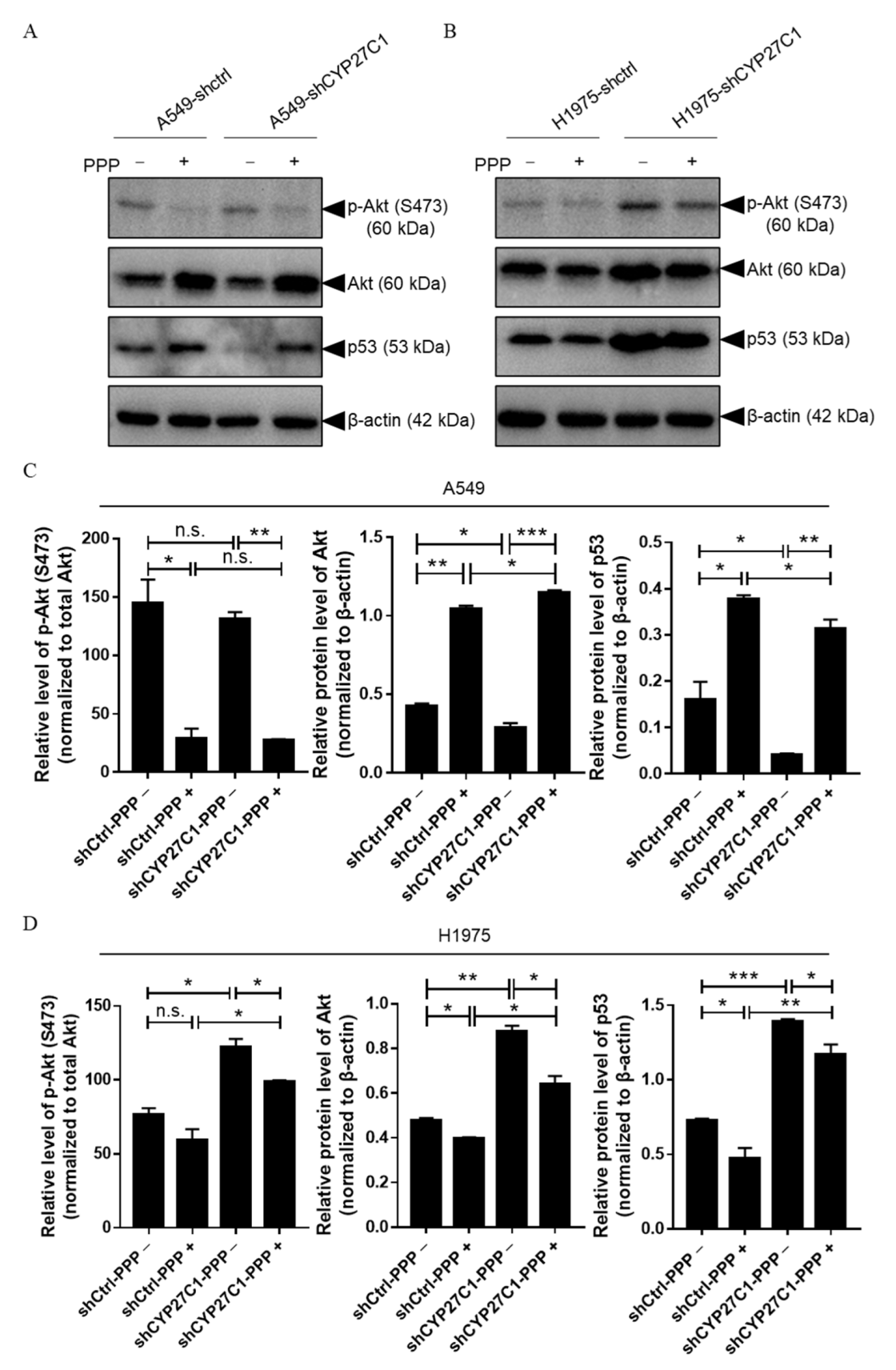
2.2.4. Stable CYP27C1-Knockdown Involves in Activation of IGF-1R/Akt/p53 Signaling Pathway
2.3. IC50 Values of Anticancer Agents in Human Lung Cancer Cells
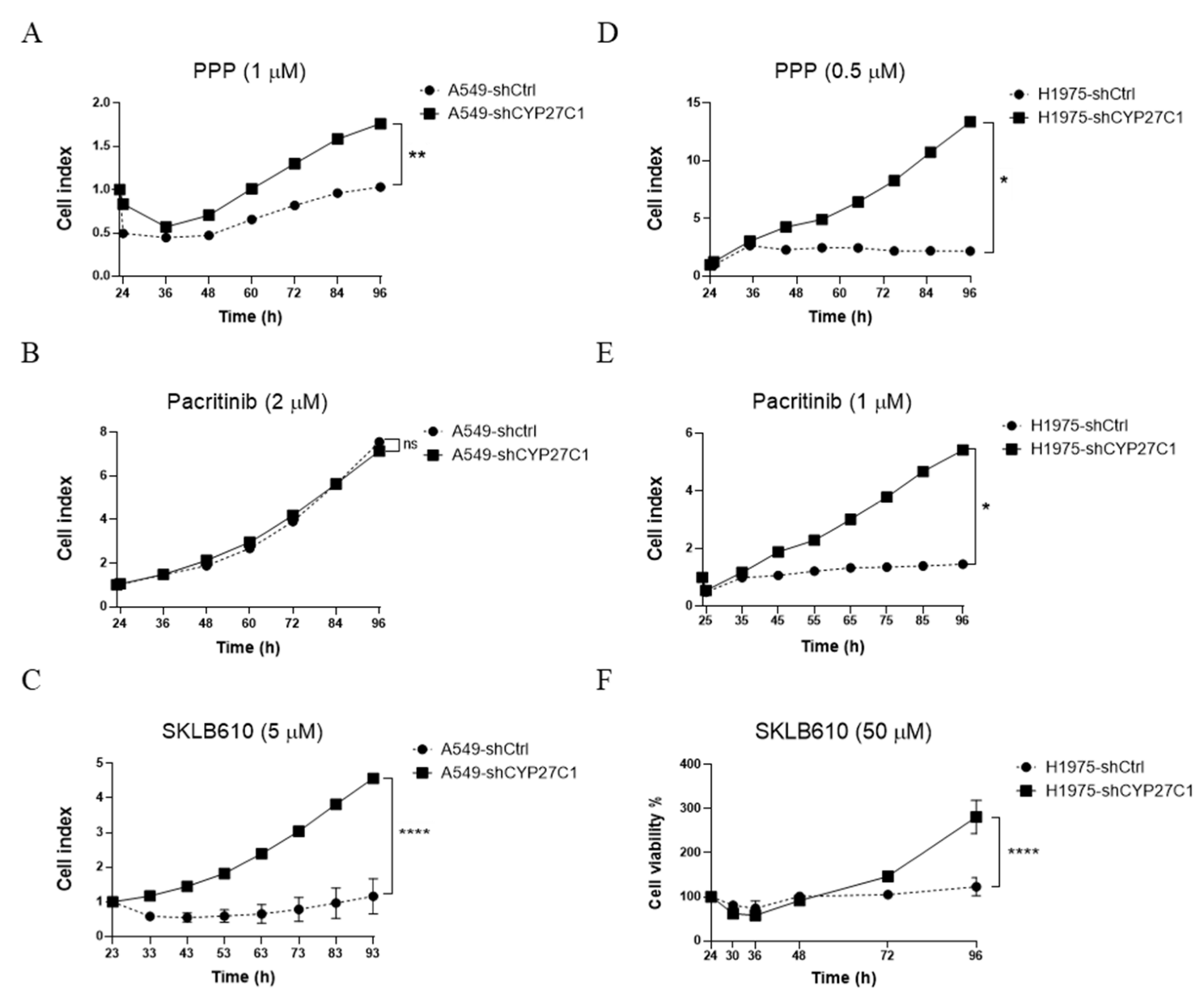
2.4. CYP27C1-Knockdown Attenuates Anticancer Potency of Protein Kinase Inhibitors
2.5. CYP27C1 Impacts Inhibitory Effect of Picropodophyllin via IGF-1R/Akt/p53 Signaling Pathway
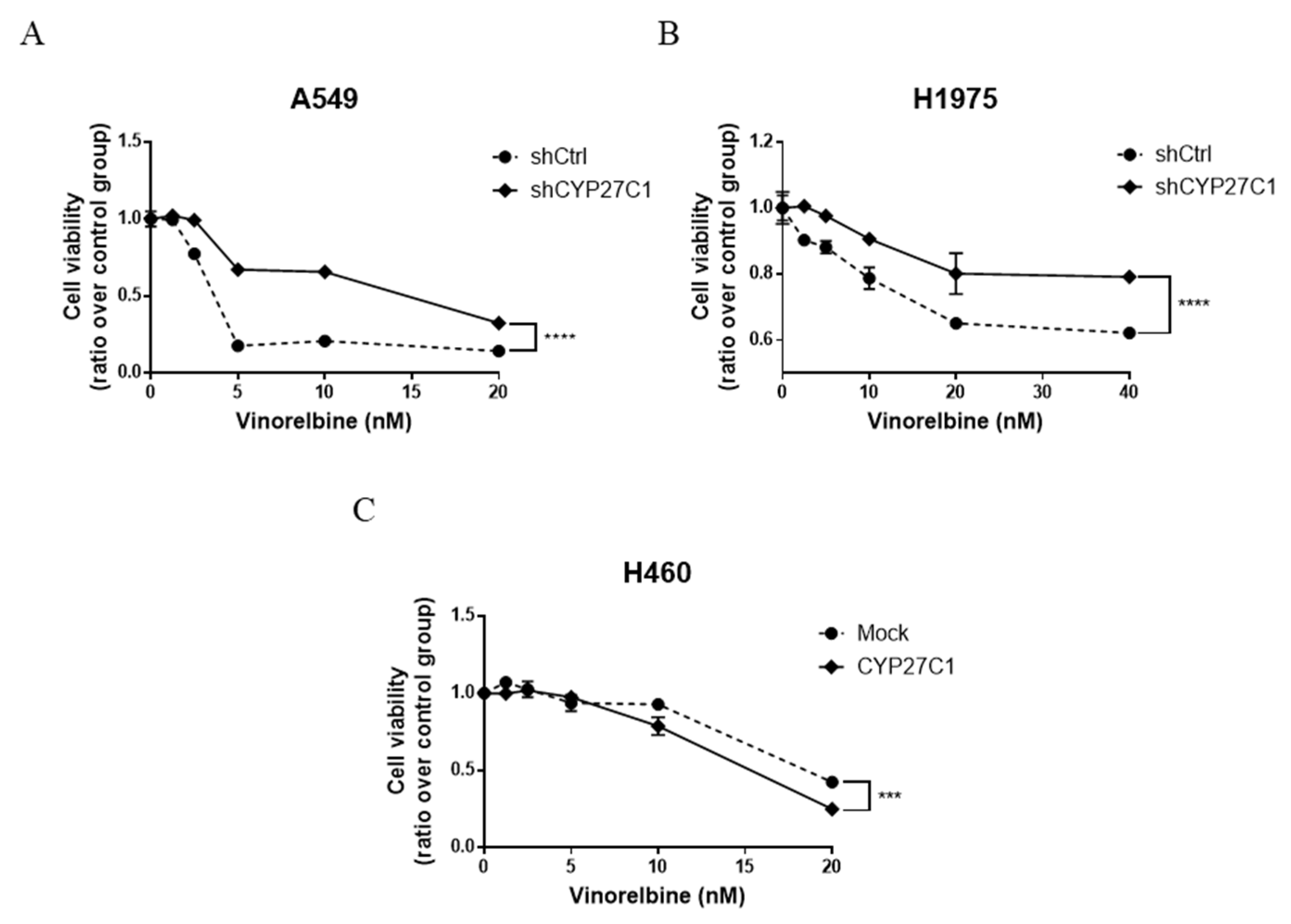
2.6. CYP27C1-Knockdown Impairs Anticancer Effect of Vinorelbine
3. Discussion
4. Materials and Methods
4.1. Chemicals and Anticancer Agents
4.2. Oligonucleotides and Plasmids
4.3. Cell Culture and Cell Lines Establishment
4.4. RNA Extraction, Reverse Transcription, and RT-qPCR
4.5. Protein Extraction and Immunoblot Analysis
4.6. Determination of the Effects of CYP27C1 on Cell Proliferation
4.7. Establishment of In Vivo Xenograft Mouse Model
4.8. Cell Colony Formation Assay
4.9. Cell Scratch (Wound Healing) Assay
4.10. IC50 Values Assessment
4.11. Vinorelbine and Protein Kinase Inhibitors Sensitivity
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nelson, D.R.; Zeldin, D.C.; Hoffman, S.M.; Maltais, L.J.; Wain, H.M.; Nebert, D.W. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics 2004, 14, 1–18. [Google Scholar] [CrossRef]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome p450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Westphal, C.; Konkel, A.; Schunck, W.H. Cytochrome p450 enzymes in the bioactivation of polyunsaturated Fatty acids and their role in cardiovascular disease. Adv. Exp. Med. Biol. 2015, 851, 151–187. [Google Scholar]
- Jamieson, K.L.; Endo, T.; Darwesh, A.M.; Samokhvalov, V.; Seubert, J.M. Cytochrome P450-derived eicosanoids and heart function. Pharmacol. Ther. 2017, 179, 47–83. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef]
- Niwa, T.; Murayama, N.; Imagawa, Y.; Yamazaki, H. Regioselective hydroxylation of steroid hormones by human cytochromes P450. Drug Metab. Rev. 2015, 47, 89–110. [Google Scholar] [CrossRef]
- Fava, C.; Bonafini, S. Eicosanoids via CYP450 and cardiovascular disease: Hints from genetic and nutrition studies. Prostaglandins Other Lipid Mediat. 2018, 139, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, F.; Saba, A.; Zucchi, R. An Update on Vitamin D Metabolism. Int. J. Mol. Sci. 2020, 21, 6573. [Google Scholar] [CrossRef]
- Isoherranen, N.; Zhong, G. Biochemical and physiological importance of the CYP26 retinoic acid hydroxylases. Pharmacol. Ther. 2019, 204, 107400. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Azzi, A.; Birringer, M.; Cook-Mills, J.M.; Eggersdorfer, M.; Frank, J.; Cruciani, G.; Lorkowski, S.; Ozer, N.K. Vitamin E: Emerging aspects and new directions. Free Radic. Biol. Med. 2017, 102, 16–36. [Google Scholar] [CrossRef]
- Almazroo, O.A.; Miah, M.K.; Venkataramanan, R. Drug Metabolism in the Liver. Clin. Liver Dis. 2017, 21, 1–20. [Google Scholar] [CrossRef] [PubMed]
- De Montellano, P.R.O. Cytochrome P450-activated prodrugs. Future Med. Chem. 2013, 5, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Wikvall, K.; Miller, W.L. Human cytochromes P450 in health and disease. Philos. Trans. R Soc. Lond. B Biol. Sci. 2013, 368, 20120431. [Google Scholar] [CrossRef] [PubMed]
- Mittal, B.; Tulsyan, S.; Kumar, S.; Mittal, R.D.; Agarwal, G. Cytochrome P450 in Cancer Susceptibility and Treatment. Adv. Clin. Chem. 2015, 71, 77–139. [Google Scholar] [PubMed]
- Megaraj, V.; Zhou, X.; Xie, F.; Liu, Z.; Yang, W.; Ding, X. Role of CYP2A13 in the bioactivation and lung tumorigenicity of the tobacco-specific lung procarcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone: In Vivo studies using a CYP2A13-humanized mouse model. Carcinogenesis 2014, 35, 131–137. [Google Scholar] [CrossRef]
- Sulc, M.; Indra, R.; Moserova, M.; Schmeiser, H.H.; Frei, E.; Arlt, V.M.; Stiborova, M. The impact of individual cytochrome P450 enzymes on oxidative metabolism of benzo[a]pyrene in human livers. Environ. Mol. Mutagen. 2016, 57, 229–235. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Tang, Z.; Salamanca-Pinzón, S.G.; Cheng, Q. Characterizing proteins of unknown function: Orphan cytochrome p450 enzymes as a paradigm. Mol. Interv. 2010, 10, 153–163. [Google Scholar] [CrossRef]
- Guengerich, F.P.; Cheng, Q. Orphans in the human cytochrome P450 superfamily: Approaches to discovering functions and relevance in pharmacology. Pharmacol. Rev. 2011, 63, 684–699. [Google Scholar] [CrossRef]
- Wu, Z.L.; Bartleson, C.J.; Ham, A.J.; Guengerich, F.P. Heterologous expression, purification, and properties of human cytochrome P450 27C1. Arch. Biochem. Biophys 2006, 445, 138–146. [Google Scholar] [CrossRef]
- Johnson, K.M.; Phan, T.T.N.; Albertolle, M.E.; Guengerich, F.P. Human mitochondrial cytochrome P450 27C1 is localized in skin and preferentially desaturates trans-retinol to 3,4-dehydroretinol. J. Biol. Chem. 2017, 292, 13672–13687. [Google Scholar] [CrossRef]
- Jiang, J.G.; Ning, Y.G.; Chen, C.; Ma, D.; Liu, Z.J.; Yang, S.; Zhou, J.; Xiao, X.; Zhang, X.A.; Edin, M.L.; et al. Cytochrome p450 epoxygenase promotes human cancer metastasis. Cancer Res. 2007, 67, 6665–6674. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.T.; Chan, T.C.; Li, C.F.; Huan, S.K.H.; Tian, Y.F.; Liang, P.I.; Pan, C.T.; Shiue, Y.L. Downregulation of the cytochrome P450 4B1 protein confers a poor prognostic factor in patients with urothelial carcinomas of upper urinary tracts and urinary bladder. APMIS 2019, 127, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Maki, C.G. The IGF-1R/AKT pathway determines cell fate in response to p53. Transl. Cancer Res. 2016, 5, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Denduluri, S.K.; Idowu, O.; Wang, Z.; Liao, Z.; Yan, Z.; Mohammed, M.K.; Ye, J.; Wei, Q.; Wang, J.; Zhao, L.; et al. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015, 2, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Capasso, A. Vinorelbine in cancer therapy. Curr. Drug Targets 2012, 13, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Nobili, S.; Lavacchi, D.; Perrone, G.; Vicini, G.; Tassi, R.; Landini, I.; Grosso, A.; Roviello, G.; Mazzanti, R.; Santomaggio, C.; et al. Vinorelbine in Non-Small Cell Lung Cancer: Real-World Data from a Single-Institution Experience. Oncol. Res. 2020, 28, 237–248. [Google Scholar] [CrossRef]
- Beulz-Riche, D.; Grude, P.; Puozzo, C.; Sautel, F.; Filaquier, C.; Riche, C.; Ratanasavanh, D. Characterization of human cytochrome P450 isoenzymes involved in the metabolism of vinorelbine. Fundam. Clin. Pharmacol. 2005, 19, 545–553. [Google Scholar] [CrossRef]
- Girnita, A.; Girnita, L.; del Prete, F.; Bartolazzi, A.; Larsson, O.; Axelson, M. Cyclolignans as inhibitors of the insulin-like growth factor-1 receptor and malignant cell growth. Cancer. Res. 2004, 64, 236–242. [Google Scholar] [CrossRef]
- Hart, S.; Goh, K.C.; Novotny-Diermayr, V.; Hu, C.Y.; Hentze, H.; Tan, Y.C.; Madan, B.; Amalini, C.; Loh, Y.K.; Ong, L.C.; et al. SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia 2011, 25, 1751–1759. [Google Scholar] [CrossRef]
- Cao, Z.X.; Zheng, R.L.; Lin, H.J.; Luo, S.D.; Zhou, Y.; Xu, Y.Z.; Zeng, X.X.; Wang, Z.; Zhou, L.N.; Mao, Y.Q.; et al. SKLB610: A novel potential inhibitor of vascular endothelial growth factor receptor tyrosine kinases inhibits angiogenesis and tumor growth in vivo. Cell Physiol. Biochem. 2011, 27, 565–574. [Google Scholar] [CrossRef]
- Shimada, T. Inhibition of Carcinogen-Activating Cytochrome P450 Enzymes by Xenobiotic Chemicals in Relation to Antimutagenicity and Anticarcinogenicity. Toxicol. Res. 2017, 33, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Kramlinger, V.M.; Nagy, L.D.; Fujiwara, R.; Johnson, K.M.; Phan, T.T.; Xiao, Y.; Enright, J.M.; Toomey, M.B.; Corbo, J.C.; Guengerich, F.P. Human cytochrome P450 27C1 catalyzes 3,4-desaturation of retinoids. FEBS Lett. 2016, 590, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Buters, J.T.; Sakai, S.; Richter, T.; Pineau, T.; Alexander, D.L.; Savas, U.; Doehmer, J.; Ward, J.M.; Jefcoate, C.R.; Gonzalez, F.J. Cytochrome P450 CYP1B1 determines susceptibility to 7, 12-dimethylbenz[a]anthracene-induced lymphomas. Proc. Natl. Acad. Sci. USA 1999, 96, 1977–1982. [Google Scholar] [CrossRef] [PubMed]
- Karkhanis, A.; Hong, Y.; Chan, E.C.Y. Inhibition and inactivation of human CYP2J2: Implications in cardiac pathophysiology and opportunities in cancer therapy. Biochem. Pharmacol. 2017, 135, 12–21. [Google Scholar] [CrossRef]
- Chen, C.; Li, G.; Liao, W.; Wu, J.; Liu, L.; Ma, D.; Zhou, J.; Elbekai, R.H.; Edin, M.L.; Zeldin, D.C.; et al. Selective inhibitors of CYP2J2 related to terfenadine exhibit strong activity against human cancers in vitro and in vivo. J. Pharmacol. Exp. Ther. 2009, 329, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Chen, G.G.; Liu, Y.; Su, X.; Hu, B.; Leung, B.C.; Wang, Y.; Ho, R.L.; Yang, S.; Lu, G.; et al. Cytochrome P450 1A2 Metabolizes 17beta-Estradiol to Suppress Hepatocellular Carcinoma. PLoS ONE 2016, 11, e0153863. [Google Scholar] [CrossRef]
- Feng, Z. p53 regulation of the IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring Harb. Perspect. Biol. 2010, 2, a001057. [Google Scholar] [CrossRef]
- Powell, E.; Piwnica-Worms, D.; Piwnica-Worms, H. Contribution of p53 to metastasis. Cancer Discov. 2014, 4, 405–414. [Google Scholar] [CrossRef]
- Vahlquist, A.; Andersson, E.; Coble, B.I.; Rollman, O.; Torma, H. Increased concentrations of 3,4-didehydroretinol and retinoic acid-binding protein (CRABPII) in human squamous cell carcinoma and keratoacanthoma but not in basal cell carcinoma of the skin. J. Investig. Dermatol. 1996, 106, 1070–1074. [Google Scholar] [CrossRef][Green Version]
- Kajita, J.; Kuwabara, T.; Kobayashi, H.; Kobayashi, S. CYP3A4 is mainly responsibile for the metabolism of a new vinca alkaloid, vinorelbine, in human liver microsomes. Drug Metab. Dispos. 2000, 28, 1121–1127. [Google Scholar]
- Kannaiyan, R.; Mahadevan, D. A comprehensive review of protein kinase inhibitors for cancer therapy. Expert. Rev. Anticancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J. Hematol. Oncol. 2020, 13, 143. [Google Scholar] [CrossRef] [PubMed]
- Hussaarts, K.; Veerman, G.D.M.; Jansman, F.G.A.; van Gelder, T.; Mathijssen, R.H.J.; van Leeuwen, R.W.F. Clinically relevant drug interactions with multikinase inhibitors: A review. Ther. Adv. Med. Oncol. 2019, 11, 1758835918818347. [Google Scholar] [CrossRef] [PubMed]
- Van Erp, N.P.; Gelderblom, H.; Guchelaar, H.J. Clinical pharmacokinetics of tyrosine kinase inhibitors. Cancer Treat. Rev. 2009, 35, 692–706. [Google Scholar] [CrossRef]
- Jiang, F.; Chen, L.; Yang, Y.C.; Wang, X.M.; Wang, R.Y.; Li, L.; Wen, W.; Chang, Y.X.; Chen, C.Y.; Tang, J.; et al. CYP3A5 Functions as a Tumor Suppressor in Hepatocellular Carcinoma by Regulating mTORC2/Akt Signaling. Cancer Res. 2015, 75, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chai, H.; Li, Y.; Zhao, H.; Xie, X.; Zheng, H.; Wang, C.; Wang, X.; Yang, G.; Cai, X.; et al. Increased expression of CYP4Z1 promotes tumor angiogenesis and growth in human breast cancer. Toxicol. Appl. Pharmacol. 2012, 264, 73–83. [Google Scholar] [CrossRef]
- Zhang, Q.; Kanterewicz, B.; Buch, S.; Petkovich, M.; Parise, R.; Beumer, J.; Lin, Y.; Diergaarde, B.; Hershberger, P.A. CYP24 inhibition preserves 1alpha,25-dihydroxyvitamin D(3) anti-proliferative signaling in lung cancer cells. Mol. Cell Endocrinol. 2012, 355, 153–161. [Google Scholar] [CrossRef]
- Yu, W.; Chen, L.; Yang, Y.Q.; Falck, J.R.; Guo, A.M.; Li, Y.; Yang, J. Cytochrome P450 omega-hydroxylase promotes angiogenesis and metastasis by upregulation of VEGF and MMP-9 in non-small cell lung cancer. Cancer Chemother. Pharmacol. 2011, 68, 619–629. [Google Scholar] [CrossRef]
- Jiang, J.G.; Chen, C.L.; Card, J.W.; Yang, S.; Chen, J.X.; Fu, X.N.; Ning, Y.G.; Xiao, X.; Zeldin, D.C.; Wang, D.W. Cytochrome P450 2J2 promotes the neoplastic phenotype of carcinoma cells and is up-regulated in human tumors. Cancer Res. 2005, 65, 4707–4715. [Google Scholar] [CrossRef]
- Hart, S.; Goh, K.C.; Novotny-Diermayr, V.; Tan, Y.C.; Madan, B.; Amalini, C.; Ong, L.C.; Kheng, B.; Cheong, A.; Zhou, J.; et al. Pacritinib (SB1518), a JAK2/FLT3 inhibitor for the treatment of acute myeloid leukemia. Blood Cancer J. 2011, 1, e44. [Google Scholar] [CrossRef]
- Ayele, T.M.; Muche, Z.T.; Teklemariam, A.B.; Kassie, A.B.; Abebe, E.C. Role of JAK2/STAT3 Signaling Pathway in the Tumorigenesis, Chemotherapy Resistance, and Treatment of Solid Tumors: A Systemic Review. J. Inflamm. Res. 2022, 15, 1349–1364. [Google Scholar] [CrossRef]
- Lin, J.; Tang, H.; Jin, X.; Jia, G.; Hsieh, J.T. p53 regulates Stat3 phosphorylation and DNA binding activity in human prostate cancer cells expressing constitutively active Stat3. Oncogene 2002, 21, 3082–3088. [Google Scholar] [CrossRef] [PubMed]
- Wörmann, S.M.; Song, L.; Ai, J.; Diakopoulos, K.N.; Kurkowski, M.U.; Görgülü, K.; Ruess, D.; Campbell, A.; Doglioni, C.; Jodrell, D.; et al. Loss of P53 Function Activates JAK2-STAT3 Signaling to Promote Pancreatic Tumor Growth, Stroma Modification, and Gemcitabine Resistance in Mice and Is Associated With Patient Survival. Gastroenterology 2016, 151, 180–193.e112. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D.; Roberts, C.T. The insulin-like growth factor system and cancer. Cancer Lett. 2003, 195, 127–137. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef]
- Roszkowska, K.A.; Gizinski, S.; Sady, M.; Gajewski, Z.; Olszewski, M.B. Gain-of-Function Mutations in p53 in Cancer Invasiveness and Metastasis. Int. J. Mol. Sci. 2020, 21, 1334. [Google Scholar] [CrossRef]
- Lv, T.; Lv, H.; Fei, J.; Xie, Y.; Lian, D.; Hu, J.; Tang, L.; Shi, X.; Wang, J.; Zhang, S.; et al. p53-R273H promotes cancer cell migration via upregulation of neuraminidase-1. J. Cancer 2020, 11, 6874–6882. [Google Scholar] [CrossRef]
- Jung, S.; Kim, D.H.; Choi, Y.J.; Kim, S.Y.; Park, H.; Lee, H.; Choi, C.M.; Sung, Y.H.; Lee, J.C.; Rho, J.K. Contribution of p53 in sensitivity to EGFR tyrosine kinase inhibitors in non-small cell lung cancer. Sci. Rep. 2021, 11, 19667. [Google Scholar] [CrossRef]
- Liang, Z.L.; Wu, D.D.; Yao, Y.; Yu, F.Y.; Yang, L.; Tan, H.W.; Hylkema, M.N.; Rots, M.G.; Xu, Y.M.; Lau, A.T.Y. Epiproteome profiling of cadmium-transformed human bronchial epithelial cells by quantitative histone post-translational modification-enzyme-linked immunosorbent assay. J. Appl. Toxicol. 2018, 38, 888–895. [Google Scholar] [CrossRef]
- Qin, S.H.; Lau, A.T.Y.; Liang, Z.L.; Tan, H.W.; Ji, Y.C.; Zhong, Q.H.; Zhao, X.Y.; Xu, Y.M. Resveratrol Promotes Tumor Microvessel Growth via Endoglin and Extracellular Signal-Regulated Kinase Signaling Pathway and Enhances the Anticancer Efficacy of Gemcitabine against Lung Cancer. Cancers 2020, 12, 974. [Google Scholar] [CrossRef]
- Tan, H.W.; Liang, Z.L.; Yao, Y.; Wu, D.D.; Mo, H.Y.; Gu, J.; Chiu, J.F.; Xu, Y.M.; Lau, A.T.Y. Lasting DNA Damage and Aberrant DNA Repair Gene Expression Profile Are Associated with Post-Chronic Cadmium Exposure in Human Bronchial Epithelial Cells. Cells 2019, 8, 842. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Time | IC50 Values (Mean ± SD) | |||
|---|---|---|---|---|---|
| Vinorelbine (nM) | Picropodophyllin (μM) | Pacritinib (μM) | SKLB610 (μM) | ||
| A549 | 48 h | 1.89 ± 0.66 | 0.29 ± 0.14 | 2.73 ± 1.02 | 5.71 ± 0.73 |
| 72 h | 1.76 ± 0.60 | 0.31 ± 0.11 | 2.61 ± 0.54 | 5.68 ± 0.75 | |
| H1975 | 48 h | 33.87 ± 0.12 | 0.45 ± 0.05 | 1.34 ± 0.45 | N/A |
| 72 h | 10.06 ± 0.70 | 0.44 ± 0.10 | 1.21 ± 0.30 | N/A | |
| H460 | 48 h | 2.27 ± 0.50 | 0.40 ± 0.02 | 2.73 ± 0.44 | 18.35 ± 1.26 |
| 72 h | 1.60 ± 0.54 | 0.44 ± 0.07 | 3.70 ± 0.43 | 13.09 ± 1.12 | |
| H1299 | 48 h | N/A | 0.38 ± 0.03 | 3.42 ± 0.53 | 8.78 ± 0.94 |
| 72 h | N/A | 0.29 ± 0.09 | 2.70 ± 0.43 | 6.41 ± 0.81 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mo, H.-Y.; Wei, Q.-Y.; Zhong, Q.-H.; Zhao, X.-Y.; Guo, D.; Han, J.; Noracharttiyapot, W.; Visser, L.; van den Berg, A.; Xu, Y.-M.; et al. Cytochrome P450 27C1 Level Dictates Lung Cancer Tumorigenicity and Sensitivity towards Multiple Anticancer Agents and Its Potential Interplay with the IGF-1R/Akt/p53 Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 7853. https://doi.org/10.3390/ijms23147853
Mo H-Y, Wei Q-Y, Zhong Q-H, Zhao X-Y, Guo D, Han J, Noracharttiyapot W, Visser L, van den Berg A, Xu Y-M, et al. Cytochrome P450 27C1 Level Dictates Lung Cancer Tumorigenicity and Sensitivity towards Multiple Anticancer Agents and Its Potential Interplay with the IGF-1R/Akt/p53 Signaling Pathway. International Journal of Molecular Sciences. 2022; 23(14):7853. https://doi.org/10.3390/ijms23147853
Chicago/Turabian StyleMo, Hai-Ying, Qi-Yao Wei, Qiu-Hua Zhong, Xiao-Yun Zhao, Dan Guo, Jin Han, Wachiraporn Noracharttiyapot, Lydia Visser, Anke van den Berg, Yan-Ming Xu, and et al. 2022. "Cytochrome P450 27C1 Level Dictates Lung Cancer Tumorigenicity and Sensitivity towards Multiple Anticancer Agents and Its Potential Interplay with the IGF-1R/Akt/p53 Signaling Pathway" International Journal of Molecular Sciences 23, no. 14: 7853. https://doi.org/10.3390/ijms23147853
APA StyleMo, H.-Y., Wei, Q.-Y., Zhong, Q.-H., Zhao, X.-Y., Guo, D., Han, J., Noracharttiyapot, W., Visser, L., van den Berg, A., Xu, Y.-M., & Lau, A. T. Y. (2022). Cytochrome P450 27C1 Level Dictates Lung Cancer Tumorigenicity and Sensitivity towards Multiple Anticancer Agents and Its Potential Interplay with the IGF-1R/Akt/p53 Signaling Pathway. International Journal of Molecular Sciences, 23(14), 7853. https://doi.org/10.3390/ijms23147853