
A Splice Switch in SIGIRR Causes a Defect of IL-37-Dependent Anti-Inflammatory Activity in Cystic Fibrosis Airway Epithelial Cells



Abstract
:
1. Introduction
2. Results
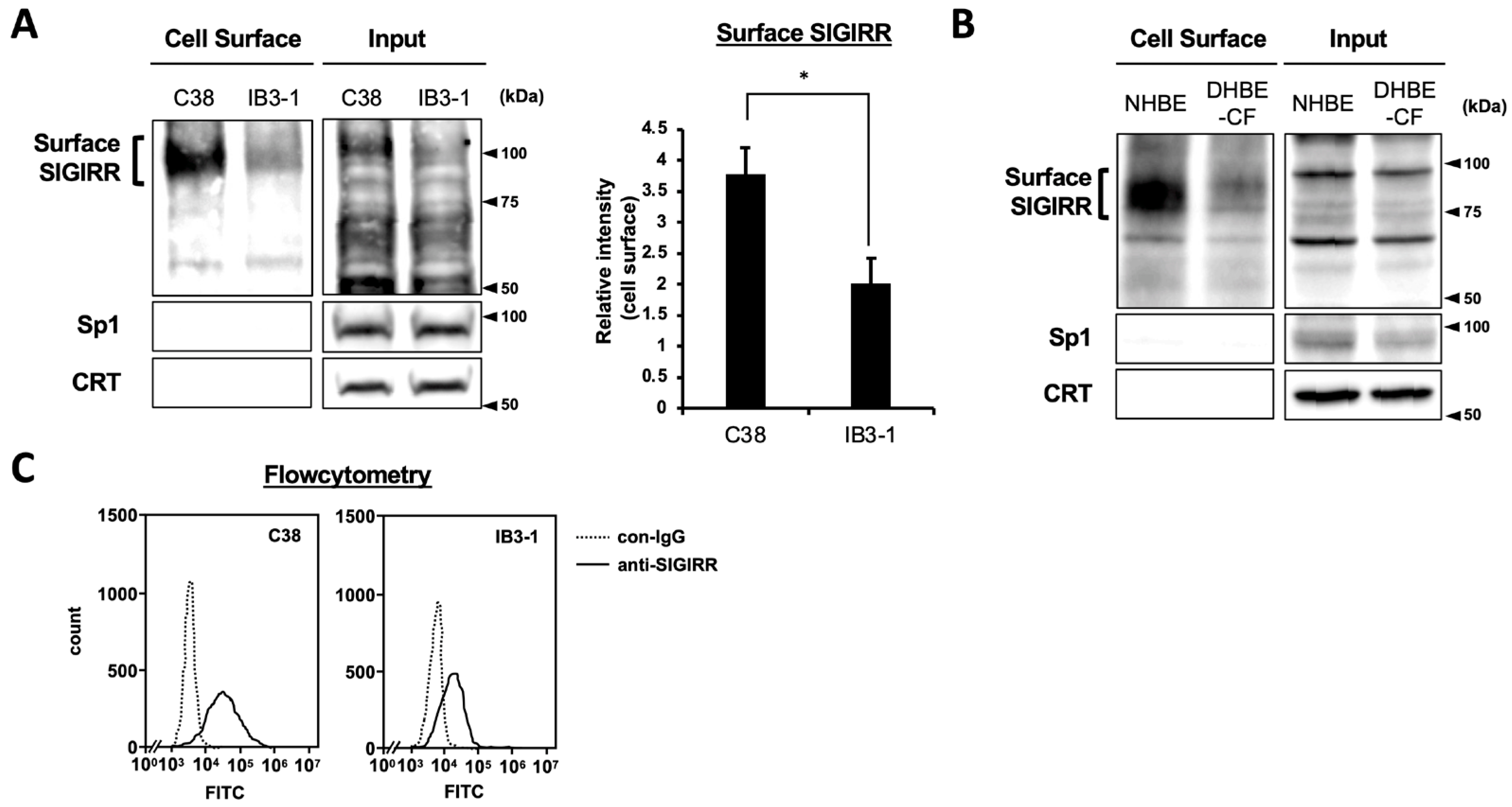
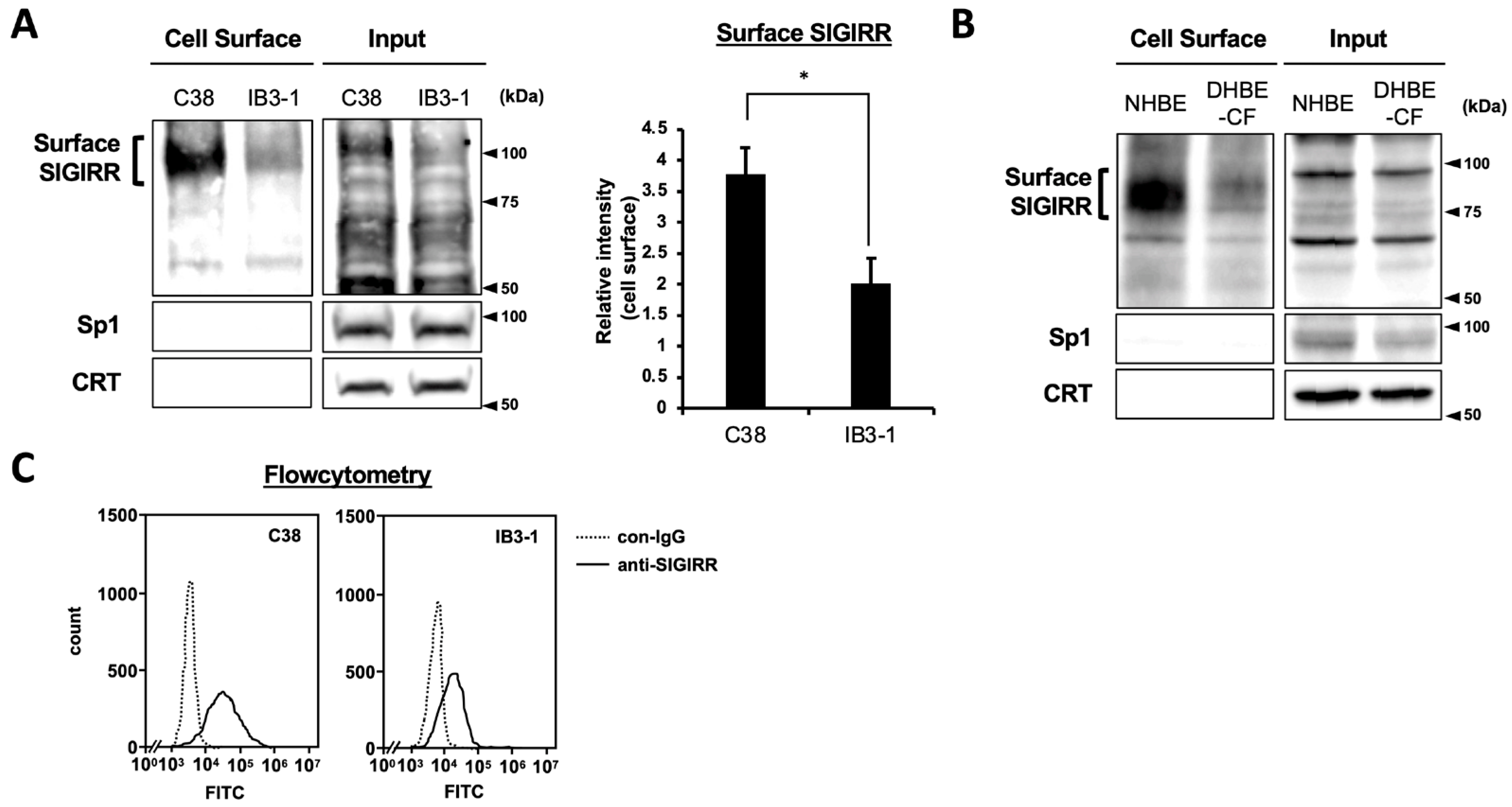
2.1. The Expression of Cell Surface SIGIRR Is Downregulated in CF Cells
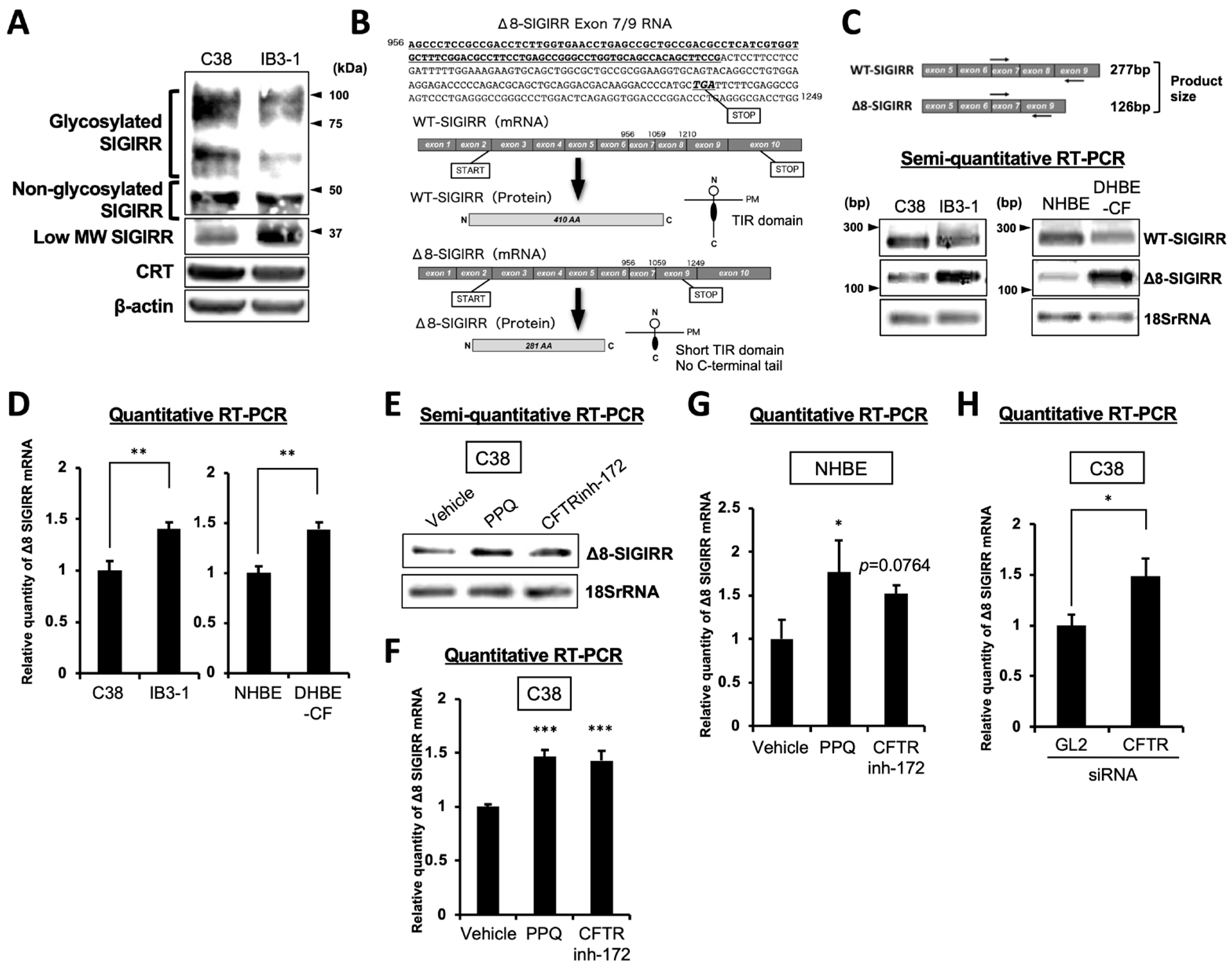
2.2. Enhanced Expression of Alternative Isoform of SIGIRR in CF Cells
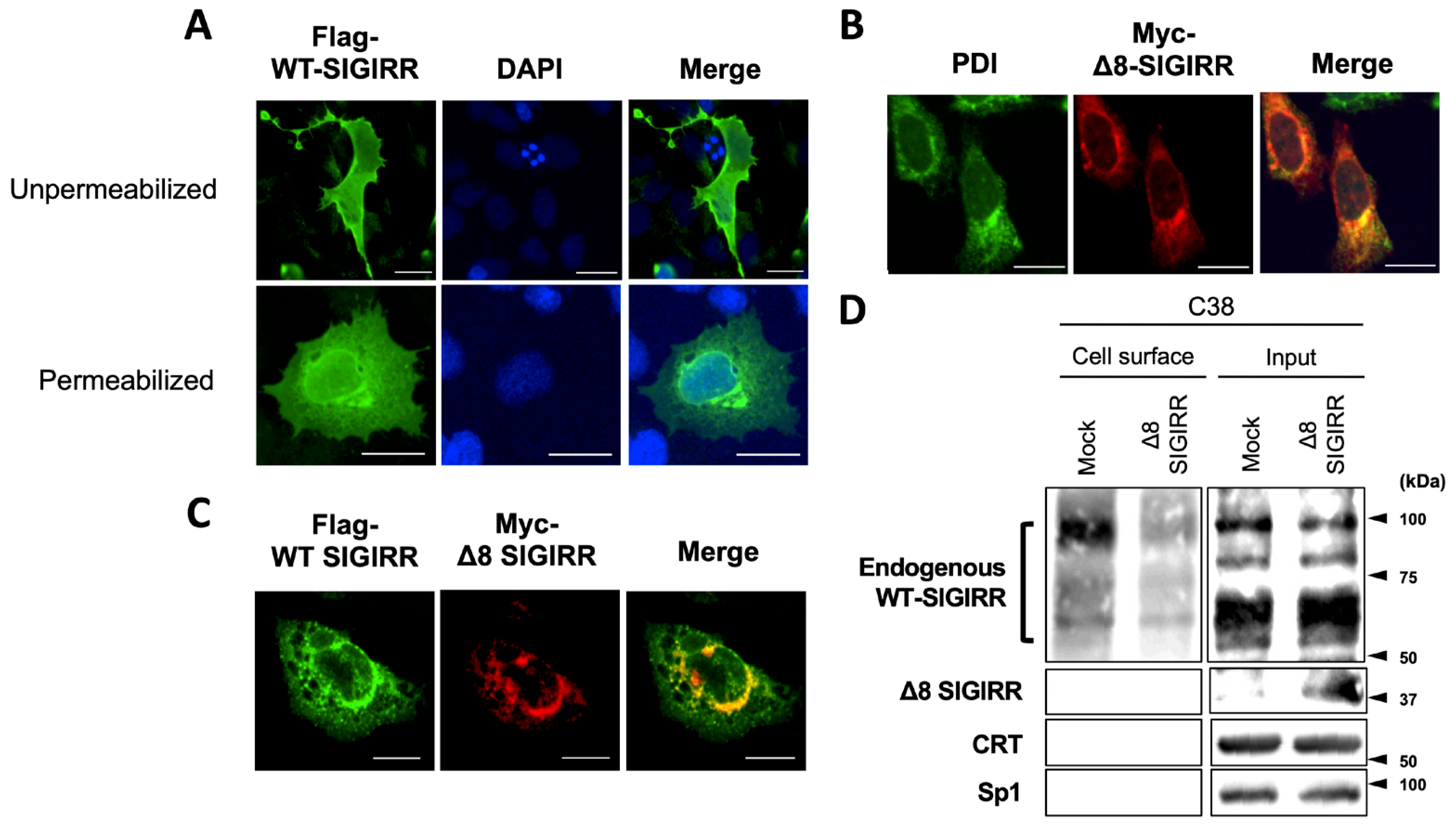
2.3. Δ8-SIGIRR Impedes Cell Surface Expression of WT-SIGIRR
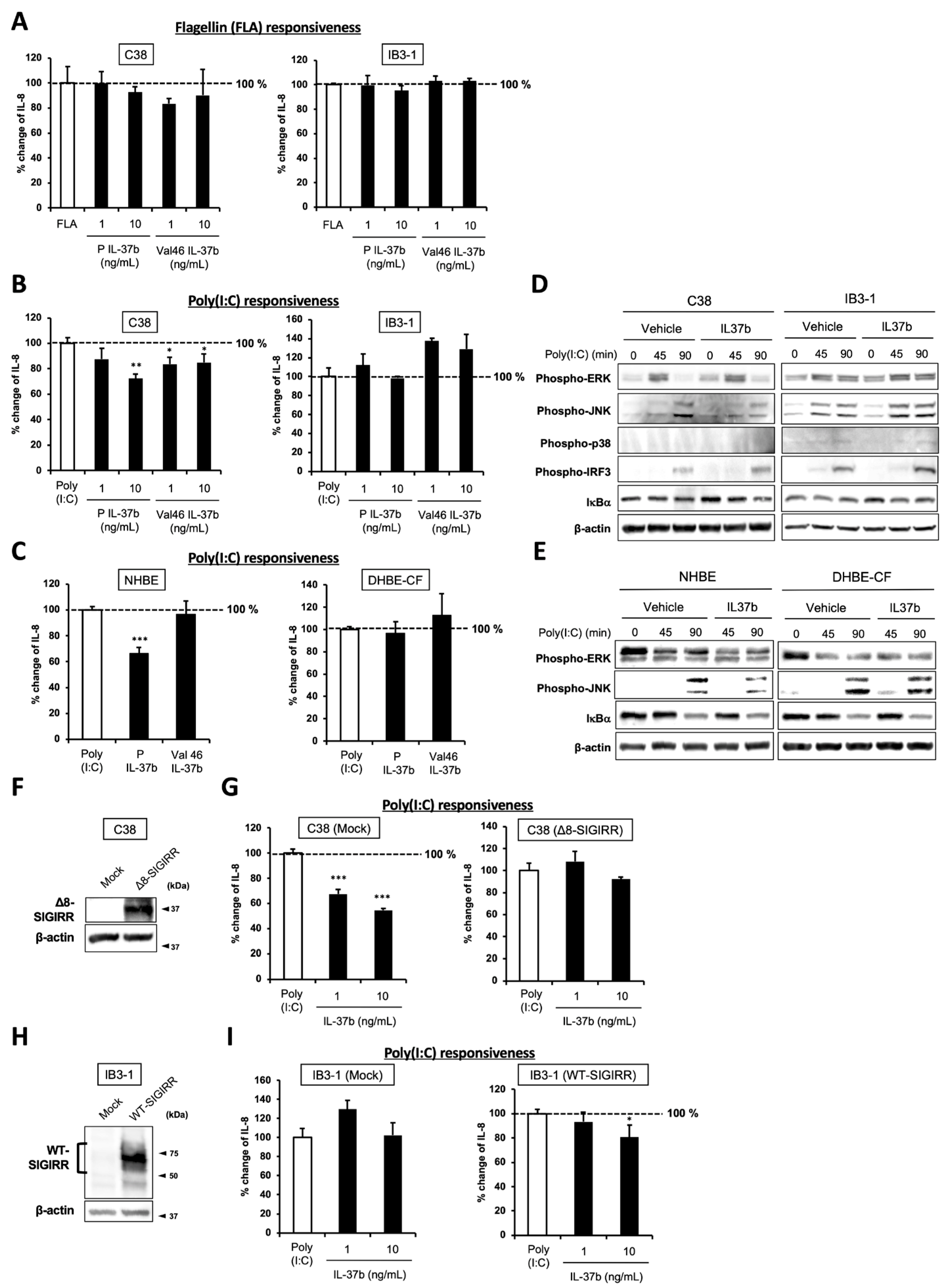
2.4. IL-37b, a Ligand for SIGIRR, Fails to Suppress Poly(I:C)-Dependent IL-8 Production and JNK Phosphorylation in CF Cells
2.5. Cell Surface-Expressed WT-SIGIRR Contributes to IL-37b-Dependent IL-8 Suppression
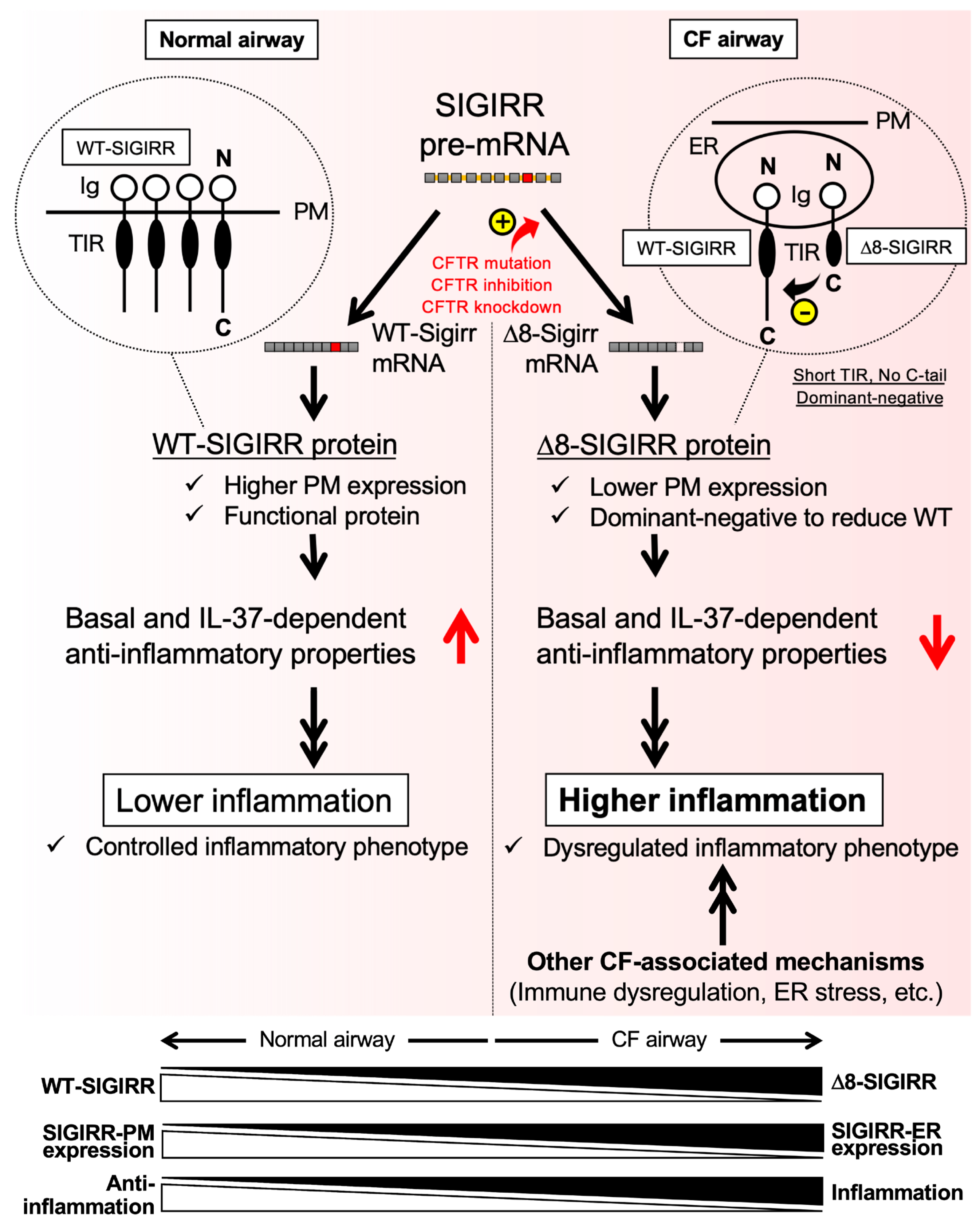
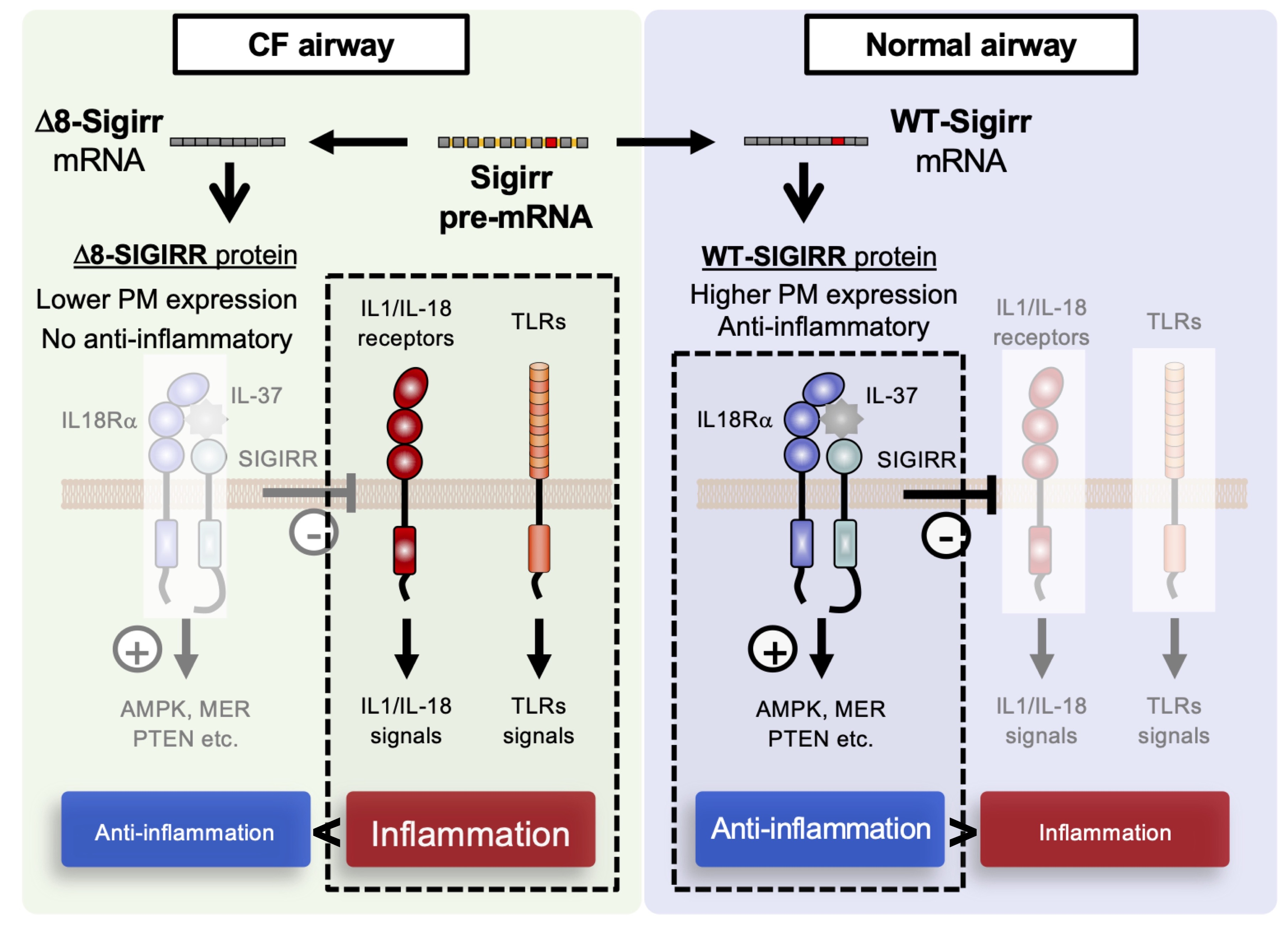
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. RNA Isolation, cDNA Synthesis, and Quantitative and Semi-Quantitative RT-PCR Analysis
4.3. Immunofluorescence (IF) Staining
4.4. Flowcytometry
4.5. Constructs and Transfection
4.6. Immunoblot Analysis
4.7. Biotinylation Assay
4.8. Enzyme-Linked Immunosorbent Assay (ELISA)
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jundi, K.; Greene, C.M. Transcription of interleukin-8: How altered regulation can affect cystic fibrosis lung disease. Biomolecules 2015, 5, 1386–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, M.; Casey, M.; Gabillard-Lefort, C.; Alharbi, A.; Teo, Y.Q.J.; McElvaney, N.G.; Reeves, E.P. Current evidence on the effect of highly effective CFTR modulation on interleukin-8 in cystic fibrosis. Expert Rev. Respir. Med. 2022, 16, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.S.; Alon, N.O.A.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.I.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Do Carmo, T.I.T.; Soares, V.E.M.; Wruck, J.; dos Anjos, F.; de Resende e Silva, D.T.; de Oliveira Maciel, S.F.V.; Bagatini, M.D. Hyperinflammation and airway surface liquid dehydration in cystic fibrosis: Purinergic system as therapeutic target. Inflamm. Res. 2021, 70, 633–649. [Google Scholar] [CrossRef] [PubMed]
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in cystic fibrosis: An update. Pediatr. Pulmonol. 2018, 53, S30–S50. [Google Scholar] [CrossRef] [Green Version]
- Bartoszewski, R.; Rab, A.; Jurkuvenaite, A.; Mazur, M.; Wakefield, J.; Collawn, J.F.; Bebok, Z. Activation of the unfolded protein response by DeltaF508 CFTR. Am. J. Respir. Cell Mol. Biol. 2008, 39, 448–457. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Pan, A.; Jia, S.; Ideozu, J.E.; Woods, K.; Murkowski, K.; Hessner, M.J.; Simpson, P.M.; Levy, H. Cystic Fibrosis Plasma Blunts the Immune Response to Bacterial Infection. Am. J. Respir. Cell Mol. Biol. 2019, 61, 301–311. [Google Scholar] [CrossRef]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Akira, S. TLR signaling. Semin. Immunol. 2007, 19, 24–32. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Firoved, A.M.; Ornatowski, W.; Deretic, V. Microarray analysis reveals induction of lipoprotein genes in mucoid Pseudomonas aeruginosa: Implications for inflammation in cystic fibrosis. Infect. Immun. 2004, 72, 5012–5018. [Google Scholar] [CrossRef] [Green Version]
- Muir, A.; Soong, G.; Sokol, S.; Reddy, B.; Gomez, M.I.; Van Heeckeren, A.; Prince, A. Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 30, 777–783. [Google Scholar] [CrossRef] [Green Version]
- Furuta, T.; Shuto, T.; Shimasaki, S.; Ohira, Y.; Suico, M.A.A.; Gruenert, D.C.C.; Kai, H. DNA demethylation-dependent enhancement of toll-like receptor-2 gene expression in cystic fibrosis epithelial cells involves SP1-activated transcription. BMC Mol. Biol. 2008, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Blohmke, C.J.; Victor, R.E.; Hirschfeld, A.F.; Elias, I.M.; Hancock, D.G.; Lane, C.R.; Davidson, A.G.F.; Wilcox, P.G.; Smith, K.D.; Overhage, J.; et al. Innate Immunity Mediated by TLR5 as a Novel Antiinflammatory Target for Cystic Fibrosis Lung Disease. J. Immunol. 2008, 180, 7764–7773. [Google Scholar] [CrossRef] [Green Version]
- Fleurot, I.; López-Gálvez, R.; Barbry, P.; Guillon, A.; Si-Tahar, M.; Bähr, A.; Klymiuk, N.; Sirard, J.C.; Caballero, I. TLR5 signalling is hyper-responsive in porcine cystic fibrosis airways epithelium. J. Cyst. Fibros. 2022, 21, e117–e121. [Google Scholar] [CrossRef]
- Di Pietro, C.; Zhang, P.X.; O’Rourke, T.K.; Murray, T.S.; Wang, L.; Britto, C.J.; Koff, J.L.; Krause, D.S.; Egan, M.E.; Bruscia, E.M. Ezrin links CFTR to TLR4 signaling to orchestrate anti-bacterial immune response in macrophages. Sci. Rep. 2017, 7, 10882. [Google Scholar] [CrossRef]
- Kelly, C.; Canning, P.; Buchanan, P.J.; Williams, M.T.; Brown, V.; Gruenert, D.C.; Elborn, J.S.; Ennis, M.; Schock, B.C. Toll-like receptor 4 is not targeted to the lysosome in cystic fibrosis airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L371–L382. [Google Scholar] [CrossRef] [Green Version]
- Greene, C.M.; Branagan, P.; McElvaney, N.G. Toll-like receptors as therapeutic targets in cystic fibrosis. Exp. Opin. Ther. Targets 2008, 12, 1481–1495. [Google Scholar] [CrossRef]
- Kiedrowski, M.R.; Bomberger, J.M. Viral-Bacterial Co-infections in the Cystic Fibrosis Respiratory Tract. Front. Immunol. 2018, 9, 3067. [Google Scholar] [CrossRef] [Green Version]
- Voynow, J.A.; Zheng, S. Airway surface liquid and impaired antiviral defense in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 12–13. [Google Scholar] [CrossRef]
- Guven-Maiorov, E.; Keskin, O.; Gursoy, A.; Nussinov, R. A Structural View of Negative Regulation of the Toll-like Receptor-Mediated Inflammatory Pathway. Biophys. J. 2015, 109, 1214–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hu, Y.; Deng, W.W.; Sun, B. Negative regulation of Toll-like receptor signaling pathway. Microbes Infect. 2009, 11, 321–327. [Google Scholar] [CrossRef]
- Kondo, T.; Kawai, T.; Akira, S. Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 2012, 33, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Molgora, M.; Barajon, I.; Mantovani, A.; Garlanda, C. Regulatory role of IL-1R8 in immunity and disease. Front. Immunol. 2016, 7, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wald, D.; Qin, J.; Zhao, Z.; Qian, Y.; Naramura, M.; Tian, L.; Towne, J.; Sims, J.E.; Stark, G.R.; Li, X. SIGIRR, a negative regulator of Toll-like receptor-Interleukin 1 receptor signaling. Nat. Immunol. 2003, 4, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Wei, T.; Stark, R.W.; Jamitzky, F.; Heckl, W.M.; Anders, H.J.; Lech, M.; Rössle, S.C. Inhibition of Toll-like receptors TLR4 and 7 signaling pathways by SIGIRR: A computational approach. J. Struct. Biol. 2010, 169, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Qian, Y.; Yao, J.; Grace, C.; Li, X. SIGIRR inhibits interleukin-1 receptor- and Toll-like receptor 4-mediated signaling through different mechanisms. J. Biol. Chem. 2005, 280, 25233–25241. [Google Scholar] [CrossRef]
- Bulek, K.; Swaidani, S.; Qin, J.; Lu, Y.; Gulen, M.F.; Herjan, T.; Min, B.; Kastelein, R.A.; Aronica, M.; Kosz-Vnenchak, M.; et al. The Essential Role of Single Ig IL-1 Receptor-Related Molecule/Toll IL-1R8 in Regulation of Th2 Immune Response. J. Immunol. 2009, 182, 2601–2609. [Google Scholar] [CrossRef] [Green Version]
- Nold-Petry, C.A.; Lo, C.Y.; Rudloff, I.; Elgass, K.D.; Li, S.; Gantier, M.P.; Lotz-Havla, A.S.; Gersting, S.W.; Cho, S.X.; Lao, J.C.; et al. IL-37 requires the receptors IL-18Rα and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat. Immunol. 2015, 16, 354–365. [Google Scholar] [CrossRef]
- Polentarutti, N.; Rol, G.P.; Muzio, M.; Bosisio, D.; Camnasio, M.; Riva, F.; Zoja, C.; Benigni, A.; Tomasoni, S.; Vecchi, A.; et al. Unique pattern of expression and inhibition of IL‐1 signaling by the IL‐1 receptor family member TIR8/SIGIRR. Eur. Cytokine Netw. 2003, 14, 211–218. [Google Scholar]
- Zhao, J.; Bulek, K.; Gulen, M.F.; Zepp, J.A.; Karagkounis, G.; Martin, B.N.; Zhou, H.; Yu, M.; Liu, X.; Huang, E.; et al. Human Colon Tumors Express a Dominant-Negative Form of SIGIRR That Promotes Inflammation and Colitis-Associated Colon Cancer in Mice. Gastroenterology 2015, 149, 1860–1871.e8. [Google Scholar] [CrossRef] [Green Version]
- Dinarello, C.A.; Nold-Petry, C.; Nold, M.; Fujita, M.; Li, S.; Kim, S.; Bufler, P.; Riordan, J.R.; Rommens, J.M.; Kerem, B.S.; et al. Suppression of innate inflammation and immunity by interleukin-37. Eur. J. Immunol. 2016, 46, 1535–1544. [Google Scholar] [CrossRef]
- Cavalli, G.; Dinarello, C.A. Suppression of inflammation and acquired immunity by IL-37. Immunol. Rev. 2018, 281, 179–190. [Google Scholar] [CrossRef]
- Li, J.; Kartha, S.; Iasvovskaia, S.; Tan, A.; Bhat, R.K.; Manaligod, J.M.; Page, K.; Brasier, A.R.; Hershenson, M.B.; Reg, M.B.H. Regulation of human airway epithelial cell IL-8 expression by MAP kinases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, 690–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saatian, B.; Zhao, Y.; He, D.; Georas, S.N.; Watkins, T.; Spannhake, E.W.; Natarajan, V. Transcriptional regulation of lysophosphatidic acid-induced interleukin-8 expression and secretion by p38 MAPK and JNK in human bronchial epithelial cells. Biochem. J. 2006, 393, 657–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veit, G.; Roldan, A.; Hancock, M.A.; da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef] [PubMed]
- Capurro, V.; Tomati, V.; Sondo, E.; Renda, M.; Borrelli, A.; Pastorino, C.; Guidone, D.; Venturini, A.; Giraudo, A.; Bertozzi, S.M.; et al. Partial rescue of f508del-cftr stability and trafficking defects by double corrector treatment. Int. J. Mol. Sci. 2021, 22, 5262. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Pezzulo, A.A.; Tang, X.X.; Hoegger, M.J.; Alaiwa, M.H.A.; Ramachandran, S.; Moninger, T.O.; Karp, P.H.; Wohlford-Lenane, C.L.; Haagsman, H.P.; Van Eijk, M.; et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 2012, 487, 109–113. [Google Scholar] [CrossRef]
- Hoegger, M.J.; Fischer, A.J.; McMenimen, J.D.; Ostedgaard, L.S.; Tucker, A.J.; Awadalla, M.A.; Moninger, T.O.; Michalski, A.S.; Hoffman, E.A.; Zabner, J.; et al. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 2014, 345, 818–822. [Google Scholar] [CrossRef] [Green Version]
- Ghigo, A.; Prono, G.; Riccardi, E.; De Rose, V. Dysfunctional inflammation in cystic fibrosis airways: From mechanisms to novel therapeutic approaches. Int. J. Mol. Sci. 2021, 22, 1952. [Google Scholar] [CrossRef]
- Kelly, C.; Williams, M.T.; Elborn, J.S.; Ennis, M.; Schock, B.C. Expression of the inflammatory regulator A20 correlates with lung function in patients with cystic fibrosis. J. Cyst. Fibros. 2013, 12, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Chillappagari, S.; Venkatesan, S.; Garapati, V.; Mahavadi, P.; Munder, A.; Seubert, A.; Sarode, G.; Guenther, A.; Schmeck, B.T.; Tümmler, B.; et al. Impaired TLR4 and HIF expression in cystic fibrosis bronchial epithelial cells downregulates hemeoxygenase-1 and alters iron homeostasis in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, 791–799. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Volmer, A.S.; Wilkinson, K.J.; Deng, Y.; Jones, L.C.; Yu, D.; Bustamante-Marin, X.M.; Burns, K.A.; Grubb, B.R.; O’Neal, W.K.; et al. Role of spdef in the regulation of muc5b expression in the airways of naive and mucoobstructed mice. Am. J. Respir. Cell Mol. Biol. 2018, 59, 383–396. [Google Scholar] [CrossRef]
- Ribeiro, C.M.P.; Boucher, R.C. Role of endoplasmic reticulum stress in cystic fibrosis-related airway inflammatory responses. Proc. Am. Thorac. Soc. 2010, 7, 387–394. [Google Scholar] [CrossRef]
- Rimessi, A.; Bezzerri, V.; Patergnani, S.; Marchi, S.; Cabrini, G.; Pinton, P. Mitochondrial Ca2+-dependent NLRP3 activation exacerbates the Pseudomonas aeruginosa-driven inflammatory response in cystic fibrosis. Nat. Commun. 2015, 6, 6201. [Google Scholar] [CrossRef]
- Bardin, P.; Foussignière, T.; Rousselet, N.; Rebeyrol, C.; Porter, J.C.; Corvol, H.; Tabary, O. miR-636: A Newly-Identified Actor for the Regulation of Pulmonary Inflammation in Cystic Fibrosis. Front. Immunol. 2019, 10, 2643. [Google Scholar] [CrossRef]
- Machen, T.E. Innate immune response in CF airway epithelia: Hyperinflammatory? Am. J. Physiol. Cell Physiol. 2006, 291, C218–C230. [Google Scholar] [CrossRef]
- Kamei, S.; Fujikawa, H.; Nohara, H.; Ueno-Shuto, K.; Maruta, K.; Nakashima, R.; Kawakami, T.; Matsumoto, C.; Sakaguchi, Y.; Ono, T.; et al. Zinc Deficiency via a Splice Switch in Zinc Importer ZIP2/SLC39A2 Causes Cystic Fibrosis-Associated MUC5AC Hypersecretion in Airway Epithelial Cells. EBioMedicine 2018, 27, 304–316. [Google Scholar] [CrossRef] [Green Version]
- Mall, M.A.; Galietta, L.J.V. Targeting ion channels in cystic fibrosis. J. Cyst. Fibros. 2015, 14, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Shuto, T.; Kamei, S.; Nohara, H.; Fujikawa, H.; Tasaki, Y.; Sugahara, T.; Ono, T.; Matsumoto, C.; Sakaguchi, Y.; Maruta, K.; et al. Pharmacological and genetic reappraisals of protease and oxidative stress pathways in a mouse model of obstructive lung diseases. Sci. Rep. 2016, 6, 39305. [Google Scholar] [CrossRef] [Green Version]
- McNamee, E.N.; Masterson, J.C.; Jedlicka, P.; McManus, M.; Grenz, A.; Collins, C.B.; Nold, M.F.; Nold-Petry, C.; Bufler, P.; Dinarello, C.A.; et al. Interleukin 37 expression protects mice from colitis. Proc. Natl. Acad. Sci. USA 2011, 108, 16711–16716. [Google Scholar] [CrossRef] [Green Version]
- Coll-Miró, M.; Francos-Quijorna, I.; Santos-Nogueira, E.; Torres-Espin, A.; Bufler, P.; Dinarello, C.A.; López-Vales, R. Beneficial effects of IL-37 after spinal cord injury in mice. Proc. Natl. Acad. Sci. USA 2016, 113, 1411–1416. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Jiang, B.; Deng, J.; Du, J.; Xiong, W.; Guan, Y.; Wen, Z.; Huang, K.; Huang, Z. IL-37 Alleviates Rheumatoid Arthritis by Suppressing IL-17 and IL-17–Triggering Cytokine Production and Limiting Th17 Cell Proliferation. J. Immunol. 2015, 194, 5110–5119. [Google Scholar] [CrossRef]
- Hybiske, K.; Fu, Z.; Schwarzer, C.; Tseng, J.; Do, J.; Huang, N.; Machen, T.E. Effects of cystic fibrosis transmembrane conductance regulator and DeltaF508CFTR on inflammatory response, ER stress, and Ca2+ of airway epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1250–L1260. [Google Scholar] [CrossRef] [Green Version]
- Ueno-Shuto, K.; Kato, K.; Tasaki, Y.; Sato, M.; Sato, K.; Uchida, Y.; Sakai, H.; Ono, T.; Suico, M.A.A.; Mitsutake, K.; et al. Lipopolysaccharide decreases single immunoglobulin interleukin-1 receptor-related molecule (SIGIRR) expression by suppressing specificity protein 1 (Sp1) via the toll-like receptor 4 (TLR4)-p38 pathway in monocytes and neutrophils. J. Biol. Chem. 2014, 289, 18097–18109. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Tao, X. Current Understanding of IL-37 in Human Health and Disease. Front. Immunol. 2021, 12, 696605. [Google Scholar] [CrossRef]
- Kamei, S.; Maruta, K.; Fujikawa, H.; Nohara, H.; Ueno-Shuto, K.; Tasaki, Y.; Nakashima, R.; Kawakami, T.; Eto, Y.; Suico, M.A.M.A.; et al. Integrative expression analysis identifies a novel interplay between CFTR and linc-SUMF1-2 that involves CF-associated gene dysregulation. Biochem. Biophys. Res. Commun. 2019, 509, 521–528. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Orientation | Sequence |
|---|---|---|
| Quantitative RT-PCR | ||
| Human WT-SIGIRR | Forward | 5′-AGACCCATCTTCATCACCTTCG-3′ |
| Reverse | 5′-GCCAGCTGCACTTCTTTCC-3′ | |
| Human Δ8-SIGIRR | Forward | 5′-CCTCTTGGTGAACCTGAGCC-3′ |
| Reverse | 5′-ATCGGAGGAAGGAGTCGG-3′ | |
| Human 18SrRNA | Forward | 5′-GTAACCCGTTGAACCCCATT-3′ |
| Reverse | 5′-CCATCCAATCGGTAGTAGCG-3′ | |
| Semi-quantitative RT-PCR | ||
| Human SIGIRR (Exon 7) | Forward | 5′-TCTTGGTGAACCTGAGCCG-3′ |
| Human SIGIRR (Exon 9) | Reverse | 5′-GCCAGCTGCACTTCTTTCC-3′ |
| Human 18SrRNA | Forward | 5′-CGGCTACCACATCCAAGGAA-3′ |
| Reverse | 5′-GCTGGAATTACCGCGGCT-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ueno-Shuto, K.; Kamei, S.; Hayashi, M.; Fukuyama, A.; Uchida, Y.; Tokutomi, N.; Suico, M.A.; Kai, H.; Shuto, T. A Splice Switch in SIGIRR Causes a Defect of IL-37-Dependent Anti-Inflammatory Activity in Cystic Fibrosis Airway Epithelial Cells. Int. J. Mol. Sci. 2022, 23, 7748. https://doi.org/10.3390/ijms23147748
Ueno-Shuto K, Kamei S, Hayashi M, Fukuyama A, Uchida Y, Tokutomi N, Suico MA, Kai H, Shuto T. A Splice Switch in SIGIRR Causes a Defect of IL-37-Dependent Anti-Inflammatory Activity in Cystic Fibrosis Airway Epithelial Cells. International Journal of Molecular Sciences. 2022; 23(14):7748. https://doi.org/10.3390/ijms23147748
Chicago/Turabian StyleUeno-Shuto, Keiko, Shunsuke Kamei, Megumi Hayashi, Ayami Fukuyama, Yuji Uchida, Naofumi Tokutomi, Mary Ann Suico, Hirofumi Kai, and Tsuyoshi Shuto. 2022. "A Splice Switch in SIGIRR Causes a Defect of IL-37-Dependent Anti-Inflammatory Activity in Cystic Fibrosis Airway Epithelial Cells" International Journal of Molecular Sciences 23, no. 14: 7748. https://doi.org/10.3390/ijms23147748
APA StyleUeno-Shuto, K., Kamei, S., Hayashi, M., Fukuyama, A., Uchida, Y., Tokutomi, N., Suico, M. A., Kai, H., & Shuto, T. (2022). A Splice Switch in SIGIRR Causes a Defect of IL-37-Dependent Anti-Inflammatory Activity in Cystic Fibrosis Airway Epithelial Cells. International Journal of Molecular Sciences, 23(14), 7748. https://doi.org/10.3390/ijms23147748