1. Introduction
FLT3 is best described for its pivotal role in the constitutive activation and development of acute myeloid leukemia (AML) in humans [
1]. It is primarily expressed in murine and hematopoietic stem cells and is responsible for the natural development of the immune system. Structurally, FLT3 consists of five immunoglobulin (Ig)-like extracellular domains, a single transmembrane (TM) domain, a juxtamembrane (JM) domain inside the cytoplasm, a cytoplasmic tyrosine kinase domain (TKD) separated by a kinase insert (KI), and a C-terminal intracellular domain (
Figure S1a) [
2,
3]. In the inactive state, FLT3 exists in its unbound, monomeric, and unphosphorylated forms. Upon binding to the indigenous ligand FL, FLT3 undergoes a conformational change. This conformational change occurs by unfolding the receptor and subsequent receptor–receptor homodimerization, bringing the kinase domain in proximity to the intracellular module, allowing the phosphorylation of the tyrosine residues (Y589, Y591, and Y599) in the JM domain. This leads to a cascade of phosphorylation and activation of secondary mediators, including STAT5, PI3K/Akt/mTOR, and Ras/Raf/MAPK oncogenic signal transduction (
Figure S1b) [
1,
4]. The premature activation of transcription factors triggers cell proliferation and impedes cell differentiation and apoptosis in leukemia cells. As simplified in
Figure S1c, the autoinhibited kinase domain (KD) consists of an N and C bi-lobal structure with an activation loop and a JM domain (PDB ID 6JQR) [
5]. The interaction between the KD and JM domains prevented ATP binding. The N-lobe has an α-helix (αC-helix) and five antiparallel β-sheets, namely, β1–β5. The C-lobe, on the other hand, has seven α-helices and three β-sheets, namely, αD–αI and β6 to β8. The activation loop comprised two twisted β-sheets (β10 and β11). An additional β-sheet is present in the JM domain, termed as βJ2. The N and C lobes are connected by a polypeptide stretch, called the hinge loop, which allows the rotational movement of the two lobes relative to each other.
Active and inactive FLT3 kinase domains can be distinguished by their characteristic ‘DFG-in’ and ‘DFG-out’ configurations. The phenylalanine residue of the DFG motif flipped 180° from its active configuration to an inactive conformation facing the active site. This creates an additional hydrophobic pocket for type II inhibitors with an elongated geometry to interact with the residues in the αC-helix [
6]. Several clinical and preclinical studies have found that mutations and the overexpression of FLT3 are associated with a poor prognosis of AML. One of the most common mutations, D835Y, has been detected in the kinase activation loop. Nevertheless, mutations in residues I836, D839, Y842, and the gatekeeper residue F691 adjacent to the active site pocket were also found in patients with a lower frequency [
7,
8]. Mutations in TKD consecutively activate tyrosine kinase, which phosphorylates the intracellular domain at various sites and recruits many cytoplasmic adapter proteins for protein–protein interactions with the FLT3 receptor.
As FLT3 involvement becomes more prevalent in oncogenic conditions, many small molecules that target FLT3 tyrosine kinase have been discovered. Midostaurin, sorafenib, and lestaurtinib were used as multikinase C inhibitors to improve clinical outcomes in patients with AML [
9]. However, their antileukemic activities were limited when used as monotherapy, and adverse cytotoxicity was observed. Therefore, researchers are working to develop next-generation inhibitors that selectively target the FLT3 receptor [
10]. Many of them are currently being evaluated in clinical trials and have higher potencies than multikinase inhibitors, such as gilteritinib, quizartinib, and crenolanib. Crenolanib and gilteritinib fall into the category of type I inhibitors and target both active and inactive kinase states, whereas quizartinib is a type II inhibitor specific to inactive-state conformations (
Figure S1d) [
11]. On the other hand, type I inhibitors have an identical set of chemical interactions in the ATP pocket, forming one to three H-bond interactions similar to that of the adenine moiety of ATP molecules. In addition, they occupy the proximal A-loop or allosteric site, front pocket, and P loop, providing an additional selectivity property for the type I inhibitors compared to that of the type II inhibitors.
Computational drug design is a popular choice for discovering small molecules targeting kinase receptors. In our previous study [
12], we performed the molecular modeling of pyrimidine4,6-diamine derivatives against inactive FLT3 as type II inhibitors. In this paper, we conduct the modeling study of 35 pteridin-7(8
H)-one compounds as type I inhibitors targeting the active FLT3 conformer. The compounds exhibited a wide range of inhibitory activity (pIC
50 5.26–8.80) against FLT3, which was studied by Sun et al. [
13]. The molecular docking and MD simulation studies of the most active compound C31 were conducted together with other structurally diversified compounds from the dataset to study the critical interactions and binding stability of the complexes. We conducted MM-PB/GBSA, FPL, LIE, and FEP calculations to evaluate protein–ligand binding affinity and build the scoring models. We also applied US methods to estimate the effective binding free energy of C31 in complexes with wild-type and mutant receptors through the unbinding pathway. The last 1 ns average MD structure of C31 was retrieved to develop the CoMFA and CoMSIA models for the SAR study. Several new compounds were designed, which were further investigated for inhibitory activity prediction using the CoMSIA model. The binding affinities of the newly designed compounds were evaluated by MM-PB/GBSA, LIE, and FEP calculations.
2. Results and Discussion
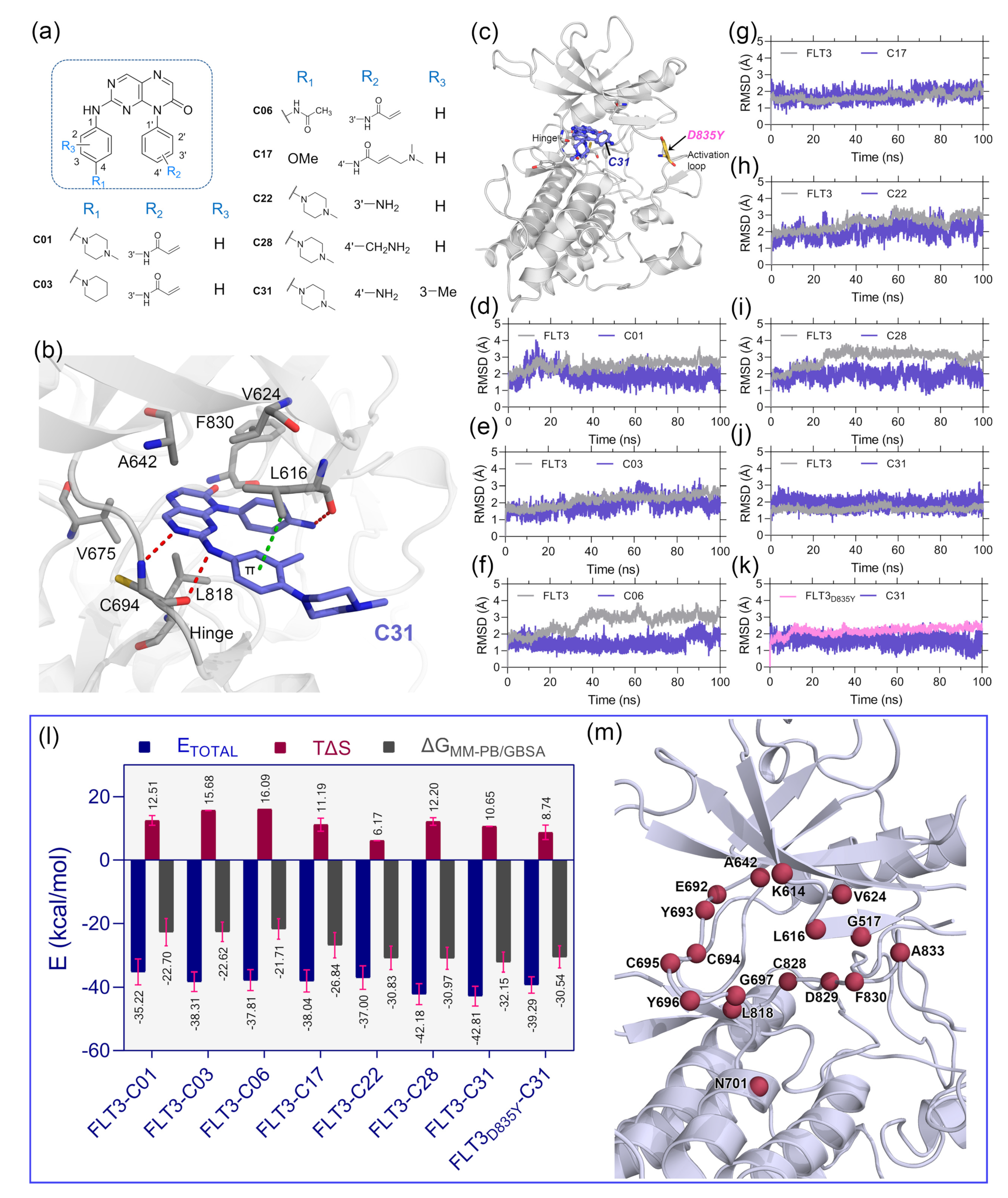
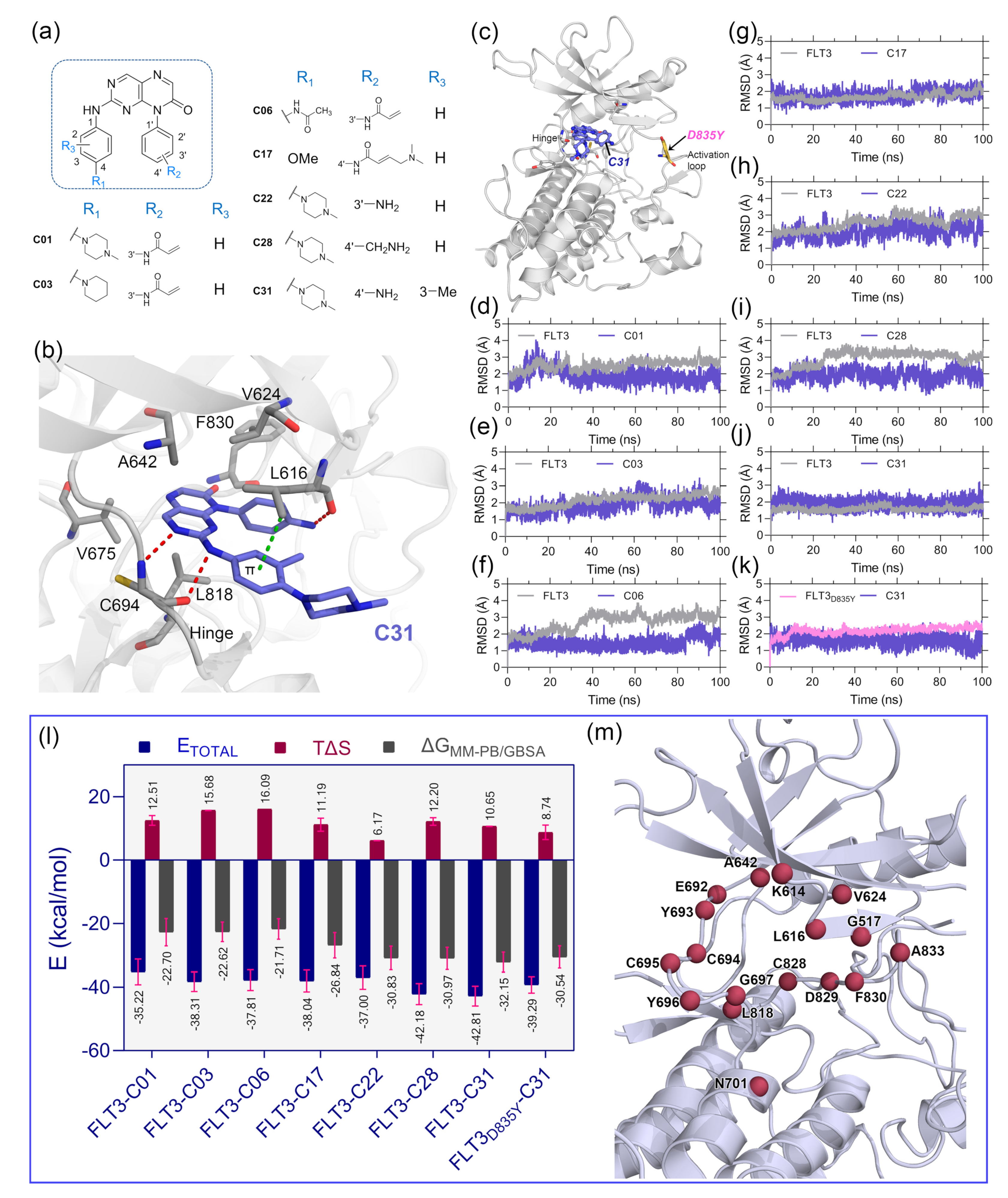
The optimal ligand-binding orientation at the active site was predicted using the molecular docking study. Docking pose verification is a critical step since it is used in molecular simulation, MM-PB/GBSA binding energy evaluation, and lastly, the generation of 3D-QSAR models. The 2D structure of pteridin-7(8
H)-one-based compounds that have been chosen for molecular docking studies is shown in
Figure 1a. None of the compounds in the dataset had FLT3 co-crystal forms; the most active compound C31 was cross-docked into the FLT3 binding pocket, which the gilteritinib molecule had previously occupied. The top-scoring ligand poses in a cross-docking experiment are not always accurate for evaluating the final docking result. Therefore, in addition to the top-scoring solution, we employed RMSD evaluation between the docked pose and gilteritinib crystal pose by the LigRMSD web server [
14]. According to this study [
15], RMSDs between the docked and crystal poses of 2.0–3.0 Å are an acceptable docking solution. The final docked complex should also comply with ECIDALs norms, in which the essential chemical interaction and binding configuration of the analogous ligands were thoroughly inspected across the same protein family from the PDB database. The docking interaction of the top-ranked FLT3-C31 within the binding pocket is shown in
Figure 2b. Compound C31 and gilteritinib are designed to target active FLT3 in a type I inhibitor-like manner. The RMSD between the docked ligand and crystal ligand gilteritinib was found to be 2.2 Å. In addition, compounds C31 and gilteritinib have 2-phenylaminopyrimidine and 2-phenylaminopyrazine moieties in their structure, which share an identical chemical scaffold and form a bidentate H-bond interaction with the residue C694 at the hinge loop. C31 formed two additional H-bond interactions with residue L616 and the keto (-C=O) group of the DFG residue F830. Other notable interactions, such as π–π stacking and π–sigma interactions, were formed between the pteridine ring and residues Y693 and L818, respectively. Residues V624, A642, V675, and C828 formed hydrophobic interactions with the ligand. The comprehensive docking results were summarized in
Table S1, and the interaction diagrams were illustrated in
Figure S3. The RMSDs with giltertinib for compounds C03, C06, C17, C22, and C28 were found to be less than 3.0 Å, indicating reasonable docking accuracy. Overall, the docking analysis suggested a satisfactory docking solution that could be utilized in the binding study of new compounds.
Since protein–ligand interaction is a highly thermodynamic process, a single docking experiment has several limitations. In the cross-docking experiment, the binding site was treated as a rigid body, and neither side-chain nor backbone movements were taken into account. Additionally, in the empirical scoring functions, water-mediated hydrogen bonding, de-solvation, and estimation of ligand binding energy by water swapping remain challenging. In many cases, the docked pose is not stable under physiological conditions. Thus, MD simulation studies were employed to validate the docking solutions and the overall stability of the protein–ligand complexes. We manually mutated the residue D835Y in the FLT3 (
Figure 1c) structure to make the FLT3
D835Y–C31 complex. Therefore, eight total protein–ligand systems, i.e., FLT3–C31, FLT3
D835Y–C31, FLT3–C01, FLT3–C03, FLT3–C06, FLT3–C17, FLT3–C17, and FLT3–C28, were subjected to 100 ns production simulations. The oscillation of the backbone αC of proteins and the heavy atoms of the ligands are plotted with respect to the simulation time in
Figure 1d–k. The RMSD plots stated that the systems were converged within the first 20 ns of the simulations. The RMSDs of the protein and ligand were in the range of 1.0–3.5 Å. The FLT3 complexes with compounds C31, C01, and C17 were stable after initial convergence, although the RMSDs of FLT3–C06 and FLT3–C28 suggested multiple state conversions during the MD runs.
The MM-PB/GBSA binding energy is a frequently used method to calculate the end-state binding free energy between protein–ligand complexes. We collected the last 2 ns trajectory or 200 snapshots from each system to compute the MM-PB/GBSA binding free energy. The entropy term (TΔS) was calculated from the last 80 snapshots of the 2 ns trajectory and then subtracted from the ΔTOTAL term to obtain the final MM-PB/GBSA binding energy. The comprehensive assessments of the MM-PB/GBSA binding energy terms are shown in
Table S2. The final
values were estimated to be −32.15 kcal/mol and −30.54 kcal/mol for the C31-bound wild-type and mutant FLT3 complexes, respectively.
The binding energies for the compounds C01, C03, C06, C17, C22, and C28 with wild-type FLT3 were found to be −22.70 kcal/mol, −22.62 kcal/mol, −21.71 kcal/mol, −26.84 kcal/mol, −30.83 kcal/mol, and −30.97 kcal/mol, respectively. Subsequently, we computed the residue-specific binding energy contribution within the 4.0 Å distance from the ligand atoms. The residues K614, L616, G617, V624, A642, E692, Y693, C694, L818, and F830 were found to be the major binding energy contributors in MM-PB/GBSA terms. The residue-specific binding energy decomposition analysis is summarized in
Table S3, and the graphical illustration is shown in
Figure 1m.
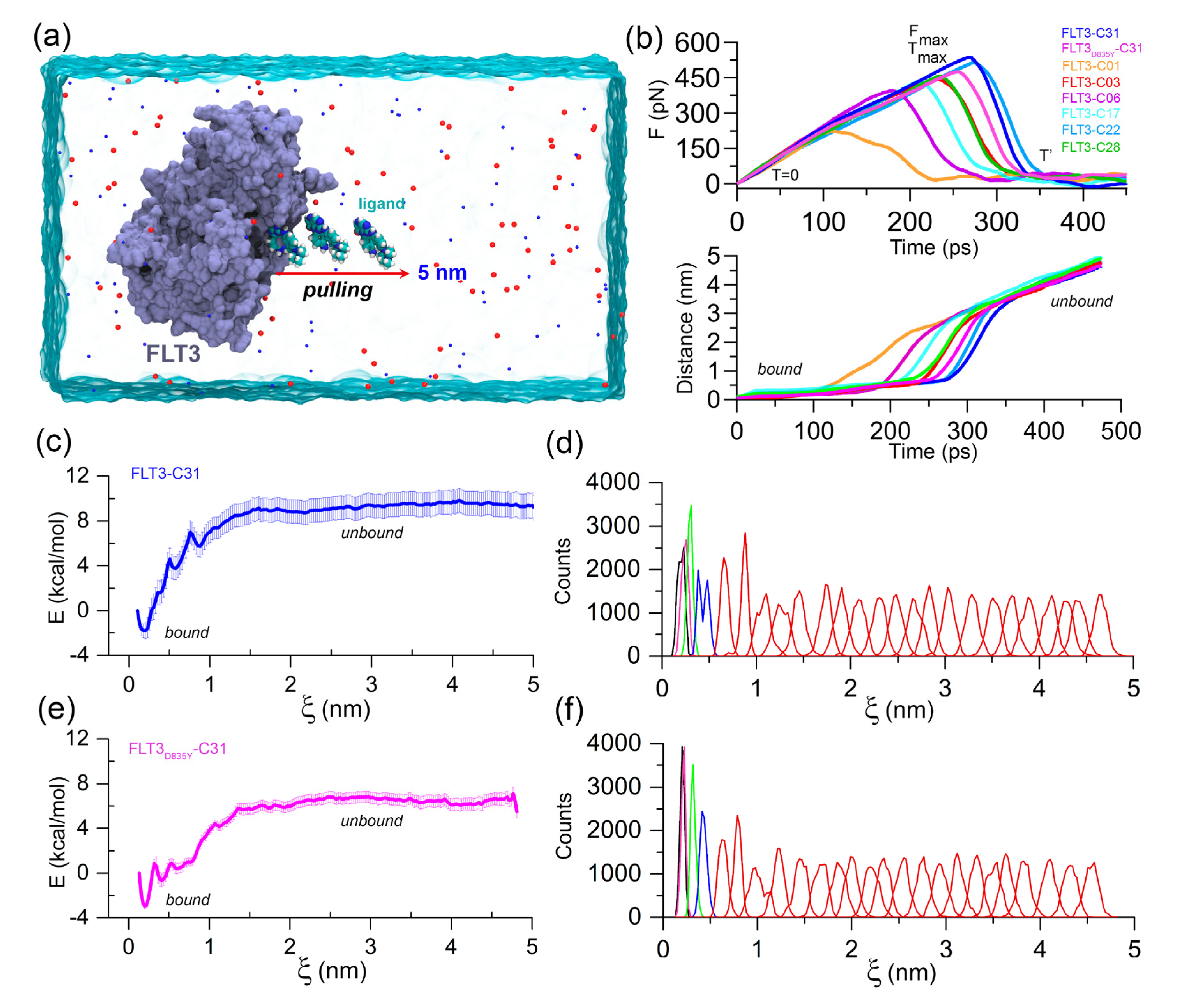
FPL simulation along with the unbinding pathway was conducted, which is based on the SMD principle. It is also a relatively straightforward approach for estimating the binding affinity between protein–ligand complexes. In this method, the ligands were forced to dissociate from the center of mass (COM) distance of the DFG residues through the caver-predicted unbinding tunnels (
Figure S4) at a distance of about 5 nm toward the
X-axis (
Figure 2a). Initially (T = 0 ps), the pulling force was minimal, and the ligand was bound to the active site cavity, referred to as the
bound state. Over the simulation, the pulling force was gradually increased until the ligands began to dissociate from the binding pocket. At that time (T =
), the pulling force reached its peak, the ligand was separated from the cavity and mobilized into the solvent, termed rupture force (
). The external force abruptly decreased and maintained a consistent plateau, referred to as the
unbound state. Theoretically, the ligand with higher inhibitory activity poses a higher relative binding affinity. Thus,
could be applied to rank the inhibitor compounds. The external pulling forces and separation distances over time are shown in
Figure 2b. The average F
max values for compounds C01, C03, C06, C17, C22, C28, and C31 were estimated to be 221.40, pN, 441.13 pN, 391.61 pN, 428.75 pN, 475.17 pN, 441.13 pN and 537.07 pN, respectively. In contrast, a lower
value (
= 453.51 pN) was obtained for the FLT3
D835Y–C31 complex compared to the C22 and C31 systems.
Next, we calculated the LIE approximation over the two quasi-equilibrium states (
bound and
unbound) by computing the van der Waals and electrostatic interactions. For the compounds C01, C03, C06, C17, C22, C28, and C31, the absolute binding energies using the LIE approximation were determined to be −28.76 kcal/mol, −27.18 kcal/mol, −28.71 kcal/mol, −30.65 kcal/mol, −30.97 kcal/mol, −30.35 kcal/mol, and −28.92 kcal/mol, respectively. In comparison to the wild-type FLT3, compound C31 had rather lower absolute binding free energy (
= −28.17 kcal/mol) to the mutant receptor. In-depth
and LIE calculations are summarized in
Table S4. Following that, the US simulation was applied to the C31-bound FLT3 and FLT3
D835Y complexes to evaluate the effective binding free energy profile along with their dissociation pathway. A total of 25 evenly distributed overlapping windows were extracted from the FPL trajectories and used for biased sampling simulations. The binding free energy and sufficient sampling could be traced by analyzing the PMF curve and the umbrella histogram with respect to the reaction coordinates (ξ), as shown in
Figure 2c–f. In PMF, the free energy began from zero and then dropped to a minimum value. Subsequently, the energies were gradually increased to attain a stable value, where non-covalent interactions between protein and ligands were completely broken. The binding free energies from the US simulation (
) of the most active compound C31 for wild-type and mutant FLT3 were calculated to be −10.73 ± 1.27 kcal/mol and −9.49 ± 0.57 kcal/mol, respectively. The convergence of the calculation could be validated by the histogram profiles of overlapping neighboring windows.
The FEP simulation was performed with the last 1 ns average MD structure of the protein–ligand complexes. The vdW and coulombic interactions of the ligands were sequentially turned-on in the solute in a complex and isolated form by alter-λ simulations. The energy convergence plots in the forward and reverse directions are shown in
Figure S5. The first 40% of the trajectory data were discarded to eliminate any convergence error. The remaining data were used to calculate the different binding energy terms in the calculation of the FEP and are summarized in
Table S5. The final absolute binding free energy (
) values from the FEP simulation were determined to be −14.83 kcal/mol, −14.64 kcal/mol, −13.68 kcal/mol, −16.77 kcal/mol, −15.04 kcal/mol, −17.61 kcal/mol, and −17.87 kcal/mol for compounds C01, C03, C06, C17, C22, C28, and C31, respectively. Compared to the wild-type complex, the FLT3
D835Y–C31 complex exhibited lower binding free energy (
= −16.82 kcal/mol) in the FEP calculation.
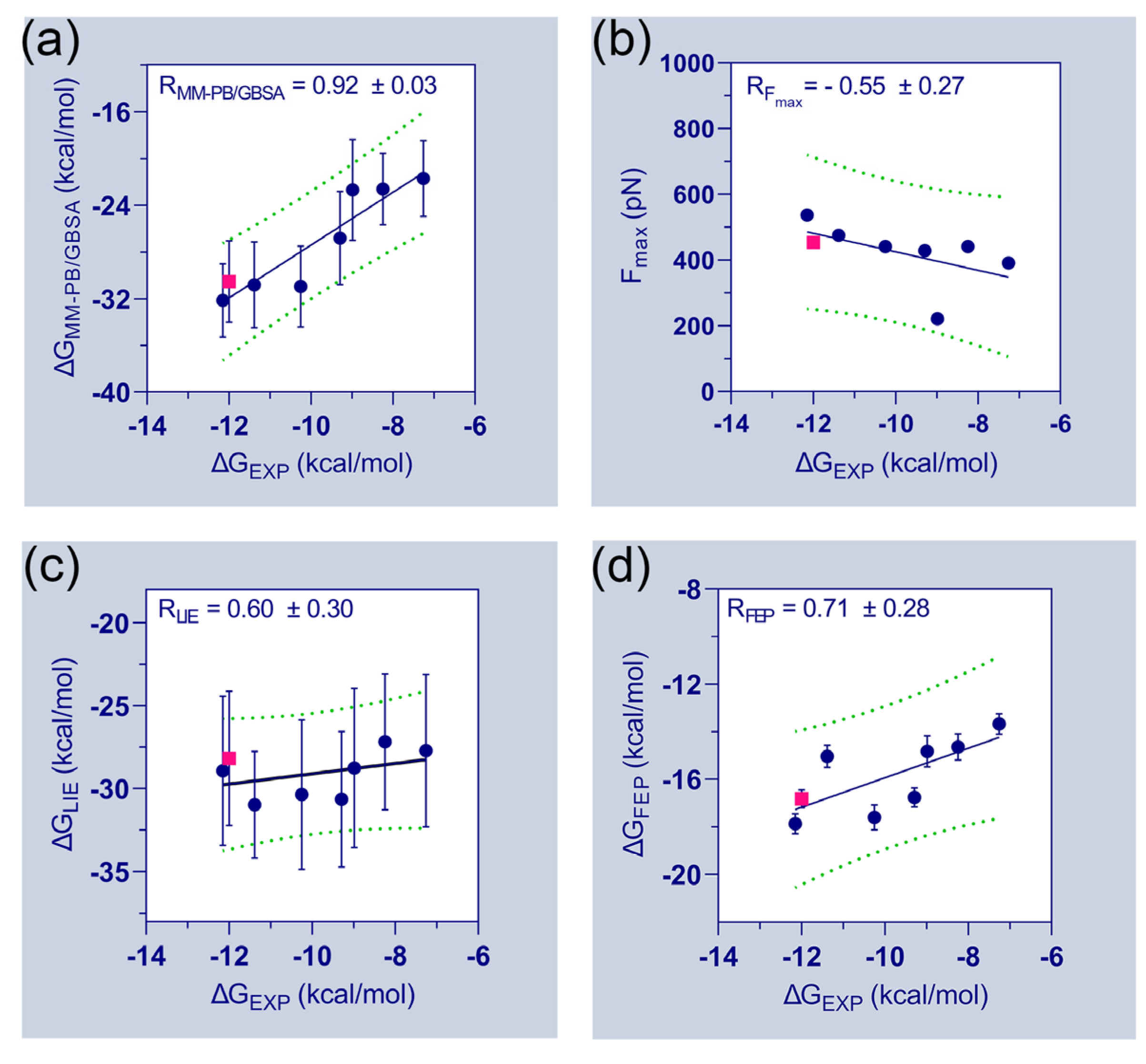
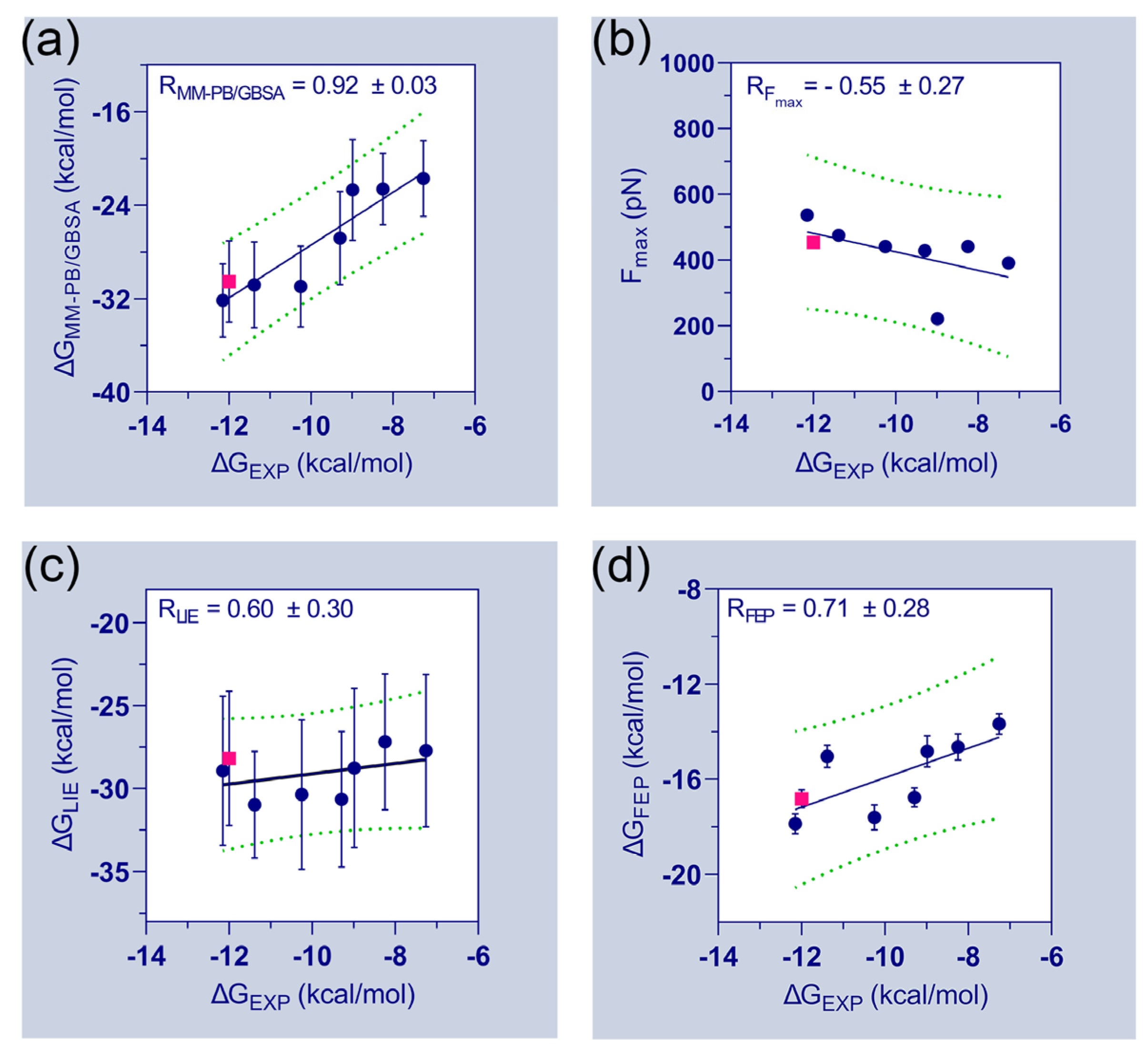
Table 1 further emphasizes the final binding free energies of the protein–ligand complexes, which are derived using MM-PB/GBSA, FPL, LIE, and FEP methods. The experimental binding energies (
) of the compounds were deduced from their inhibitory activity (IC
50) and attempted to correlate with the computed binding free energies. During the correlation analysis, the binding energies of the FLT3
D835–C31 complexes were ignored. The correlation plots between the experimental binding energies and computed binding energies of the seven compounds are shown in
Figure 3. A good correlation coefficient (
= 0.92) was obtained between the
and
. However, the binding energies were overestimated by the MM-PB/GBSA method. In the FPL model, the
values are poorly correlated with the
values (
= −0.55). The correlation coefficient (
) between
and
was calculated to be 0.60. Thus, the observations suggested a significant limitation and required special attention when utilizing the FPL and LIE models for ligand ranking. Although, both models were able to distinguish the binding affinity differences between C31 and FLT3 variants. In the FEP model, the correlation coefficient (
) between
and
was estimated to be 0.71, which is statistically reasonable and could be utilized to assess the binding affinities of unknown compounds.
Table S6 and Figure S6 represent the dataset compounds and their molecular alignments on C31. The statistical analysis of CoMFA and CoMSIA is summarized in
Table 2. The acceptable parameters of each statistical term are listed in the ‘Threshold values’ column based on the previously published literature. In CoMFA analysis, q
2 and r
2 were obtained as 0.768 and 0.982, greater than 0.5 and 0.6, respectively, at the ONC of 3. The steric and electrostatic contributions of the CoMFA scheme were found to be 54.2% and 45.8%, respectively. To generate the best statistically significant CoMSIA model, we used five descriptor fields, such as steric (S), electrostatic (E), hydrophobic (H), H-bond donor (D), and H-bond acceptor (A) in the permutation-combination process as shown in
Table S7. The best q
2 and r
2 values of 0.844 and 0.972 were obtained in SH combination at ONC of 4, and, therefore, SH was selected as the final CoMSIA model. The final contributions of the steric and hydrophobic fields were found to be 46.8% and 53.2%, respectively. However, any QSAR models are insufficient without being externally validated by test set compounds that were not used during model development. Thus, the external validation was conducted by estimating the predictive correlation coefficient or
. In CoMFA and CoMSIA, the values of
were determined to be 0.919 and 0.918, respectively, greater than the constrained value of 0.6, signifying that both models were statistically reliable and had good predictability. The predicted activity of the dataset compounds, which includes both training and test set compounds from CoMFA and CoMSIA studies are reported in
Table S8.
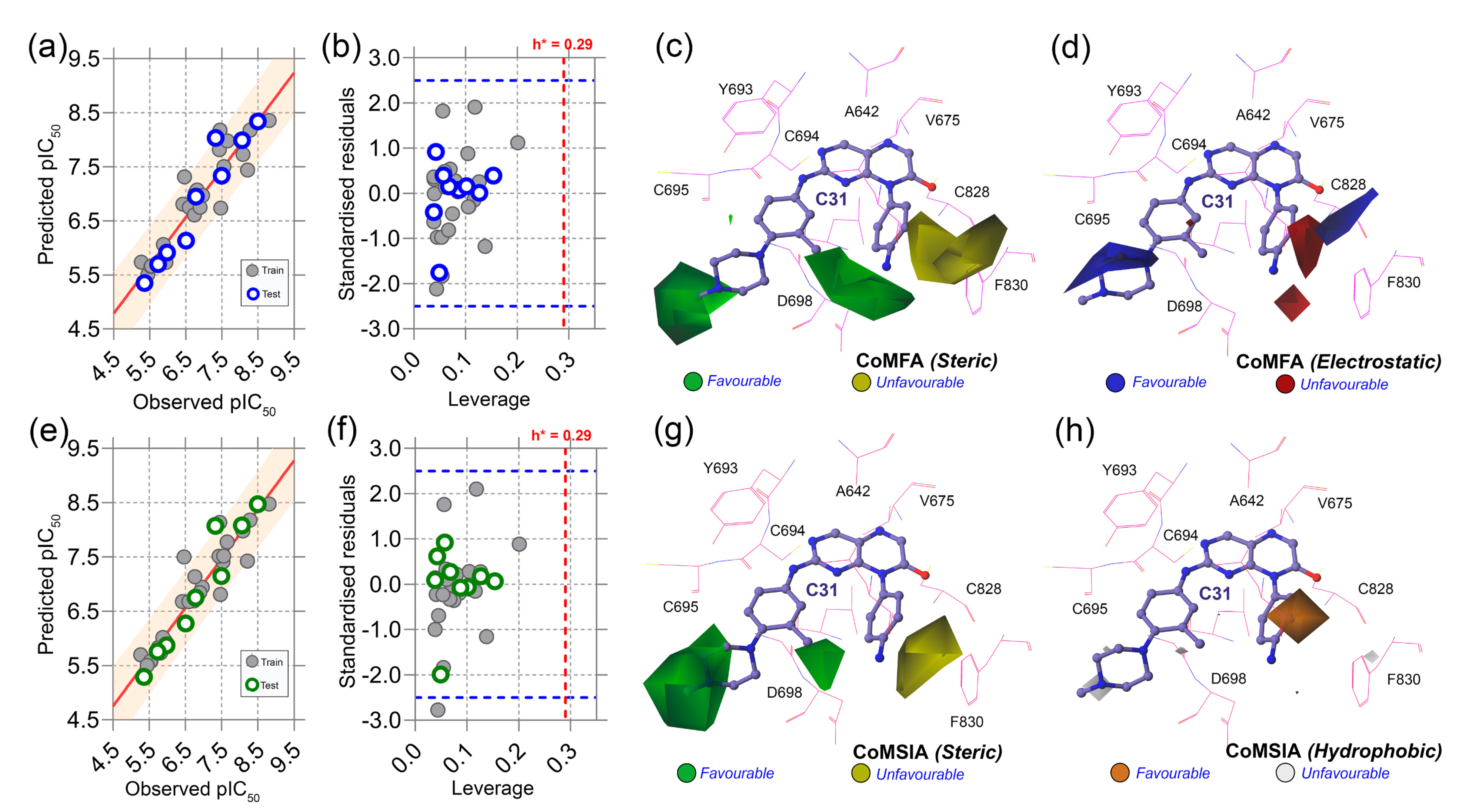
Figure 4a,e represent the PLS regression plots between the observed and predicted activity of compounds from the CoMFA and CoMSIA models. Moreover, we also calculated other statistical parameters, such as
or
,
(n = 1, 2, 3), and
matrices, all of which were calculated to be within the well-accepted parameters.
Next, we conducted the applicability domain analysis to visually detect the outliers [
16]. It is a theoretical chemical space in which the QSAR model could reliably predict the descriptor properties of compounds. The AD analysis of CoMFA and CoMSIA is illustrated in
Figure 4b,f by the distance-based Williams plot. The standardized residual values of the training set and test set compounds were plotted against their leverage values within a square area of σ = ±3 and warning leverage (h*). Compounds with a leverage value greater than h* were considered outliers and significantly affected the regression slope of the QSAR models. In our study, none of the compounds were outside the warning leverage (h* = 0.29), suggesting the robustness of the 3D-QSAR models.
The contour maps analysis from the 3D-QSAR study was conducted to explore the favorable and unfavorable sites for chemical substitution. As shown in
Figure 4c–h, the field effects of the chemical descriptors from CoMFA and CoMSIA were graphically represented by contour polyhedrons around the C31-bound active site. In both CoMFA and CoMSIA, two green contours appeared at the R
1 and R
3 positions near the solvent-exposed area of the active sites, indicating that the presence of bulky steric chemical groups in that region could increase the inhibitor potency. In contrast, a large yellow contour appears near the DFG residues, suggesting a disfavored substitution for bulky steric groups at that position. Compounds C04, C07, C08, C09, and C12 non-steric groups in their R
1 and R
3 positions exhibited lower inhibitory activity (pIC
50 < 0.7) than the other dataset compounds.
On the other hand, compounds C31 and C32 consist of steric groups, such as methyl (-CH
3) or methoxy (-OCH
3) groups, in the
meta-position instead of the
para-position of the piperazine moiety, which allocated them in proximity to the green contours. It might favor the critical inhibitory potency of these two highest active compounds. The blue and red contours (
Figure 4d) suggested a favorable substitution for the electropositive and electronegative chemical groups. In that chemical space, compounds with positively charged nitrogen (N atoms) or amine (-NH
2) groups might enhance the inhibitory activity against FLT3. An orange contour near the aniline moiety at the R
2 position towards the residue F830 suggested that a small hydrophobic substitution could be favorable (
Figure 4h). Taken together, the overall observation was emphasized as a SAR scheme in
Figure 5a.
In the context of SAR, we initiated the inhibitor design using substitution growth methods. The contour maps suggested a large steric substitution in the R
1 and R
3 positions, although this is not infinite. The addition of substantially bulky chemical components may result in steric clash and failure of ligand insertion into the binding pocket. Moreover, the designed compounds should satisfy Lipinski’s criterion and have a low complexity in the chemical synthesis route. Similarly, the compounds should be designed with scaffolds similar to the dataset compounds. A rather heterogeneous molecule may not be adequately evaluated by a 3D-QSAR model, causing it to be assigned outside the applicability domain or the chemical space. Earlier modeling studies have reported that D835 mutations alter the conformational changes of the phenylalanine residue (F830) of the conserved DFG motif, affecting the vdW and electrostatic interactions, which influence the binding affinity regardless of the type I or type II inhibitors [
17,
18]. Our multiple binding energy computation schemes estimated a lower binding affinity of the most active compound to the mutant receptor. Therefore, growing molecular probes from the R
2 position towards DFG residues may contribute to additional steric or electrostatic interactions and ultimately improve the binding affinity of the designed compounds. This could be reinforced from the SAR study, as we obtained that the non-steric, hydrophobic, and electronegative groups could be favorable for improving the inhibitory potency of C31. By considering the above factors, we designed up to 50 new compounds (
Table S9), and their activity was predicted by the CoMSIA model. We introduced the steric substitution to the R
3 position as a first step while leaving the other positions unaltered. At the R
2 site, we added hydrophobic and electronegative chemical entities (-C=O, -CF
3), while the R
3 position remained unchanged. Following that, chemical probes were grown in the R
1 position, with varying degrees of substitution in the R
2 and R
3 positions. Beyond the SAR scheme, we also incorporated investigational electronegative groups, such as chlorine and fluorine, as probe moieties. Thirteen designed compounds, namely, D02, D03, D04, D15, D16, D17, D18, D25, D26, D27, D45, D46, and D47, were predicted to have higher pIC
50 values than the most active compound (
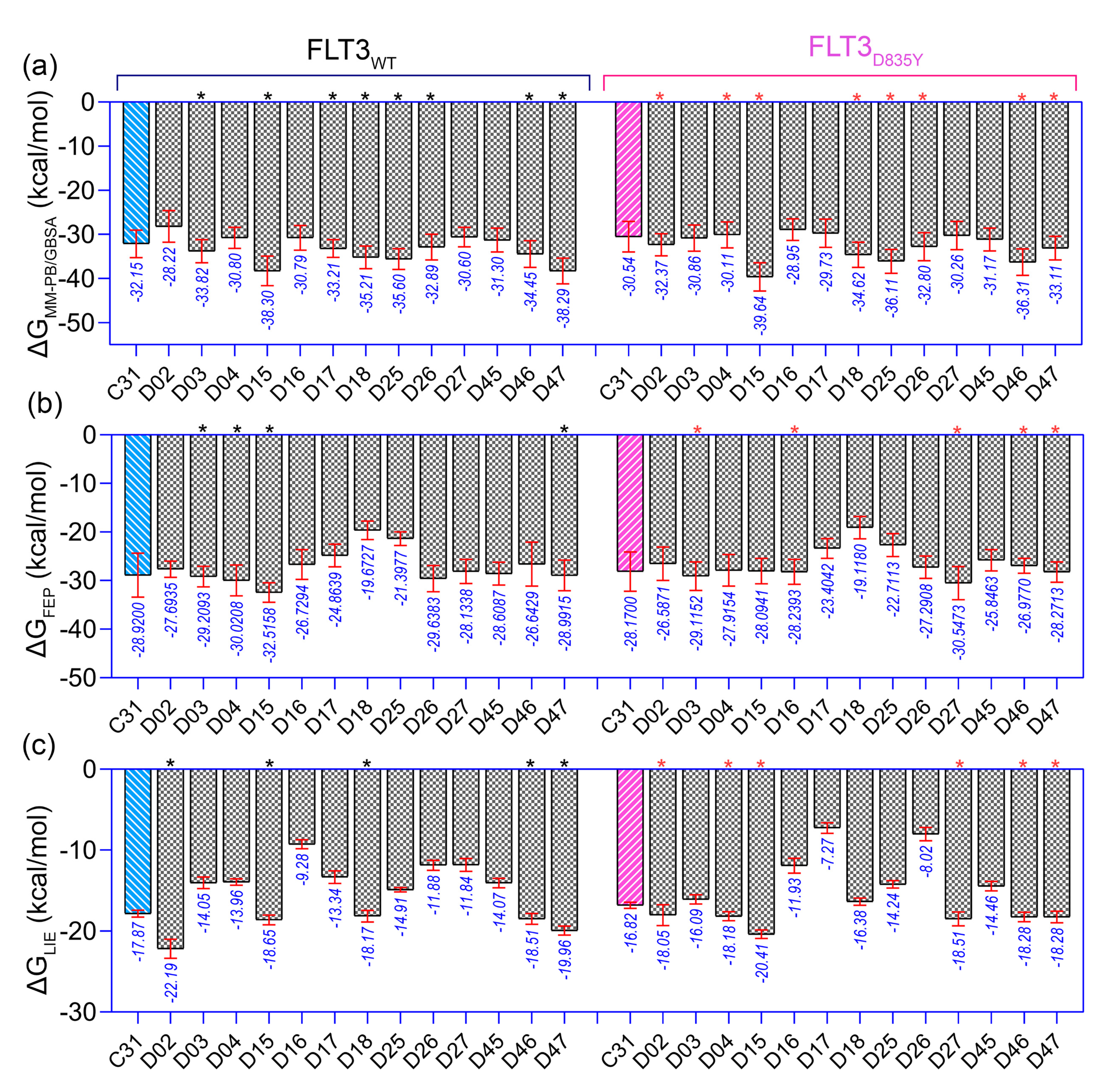
Figure 5b). The binding affinities of these 13 compounds were performed using the MM-PB/GBSA, LIE, and FEP methods targeting wild-type FLT3 and D835Y mutant. RMSD plots of the wild-type and mutant complexes in a complex with the designed compounds are shown in
Figure S7. The last two ns snapshots were extracted from the MD trajectories to calculate the MM-PB/GBSA binding free energy (
Tables S10 and S11). Compounds D03, D15, D17–18, D25–26, and D46–47 had higher binding free energies than C31 in complexes with wild-type receptors. In contrast, D02, D04, D15, D18, D25–26, and D46–47 exhibited higher binding free energies in complexes with the mutant receptor. The last 1 ns average MD complexes of the designed compounds were employed for the FPL and FEP simulation studies. The potential mean force and displacement distance of the ligands over the simulation time are shown in
Figure S8. The calculated LIE terms for the wild-type and mutant complexes are shown in
Tables S12 and S13. Compounds D03, D04, D15–16, D27, and D46–47 had higher binding free energies in complexes with FLT3 receptors than C31. The FEP convergence plots of the designed compounds in complex with FLT3 wild-type and mutant variants are illustrated in
Figures S9 and S10. The first 40% of the data is eliminated during the final FEP energy calculations to avoid the convergence error, as shown in
Table S14. The compounds D02, D04, D15, D18, D27, and D46–47 were shown to have stronger affinity for FLT3 receptors than C31. In
Figure 6, the computed binding free energies from the MM-PB/GBSA, LIE, and FEP models are compared. The designed compounds with higher binding free energies than the most active compounds are designated by asterisks.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}