
The Capillary Morphogenesis Gene 2 Triggers the Intracellular Hallmarks of Collagen VI-Related Muscular Dystrophy

 ,
, 
 , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
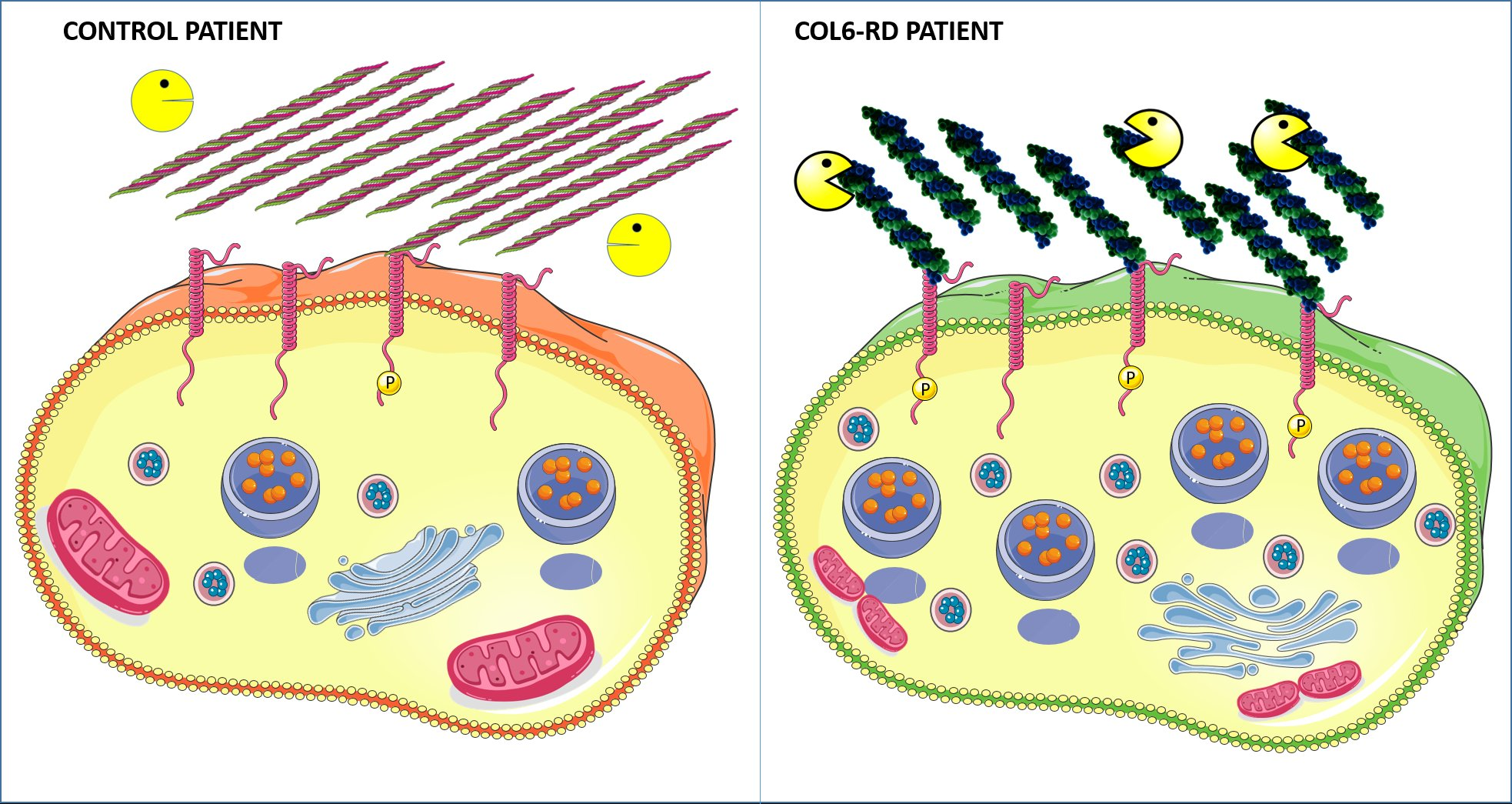
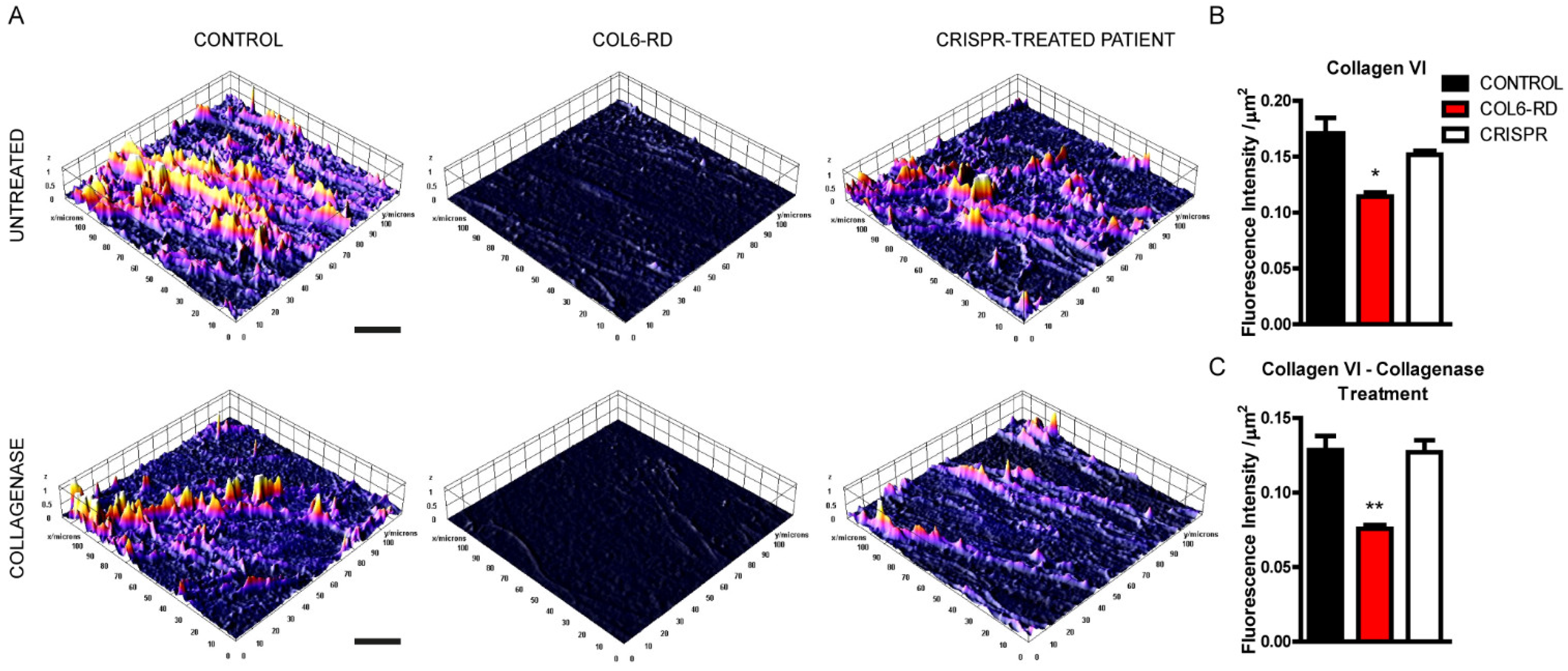
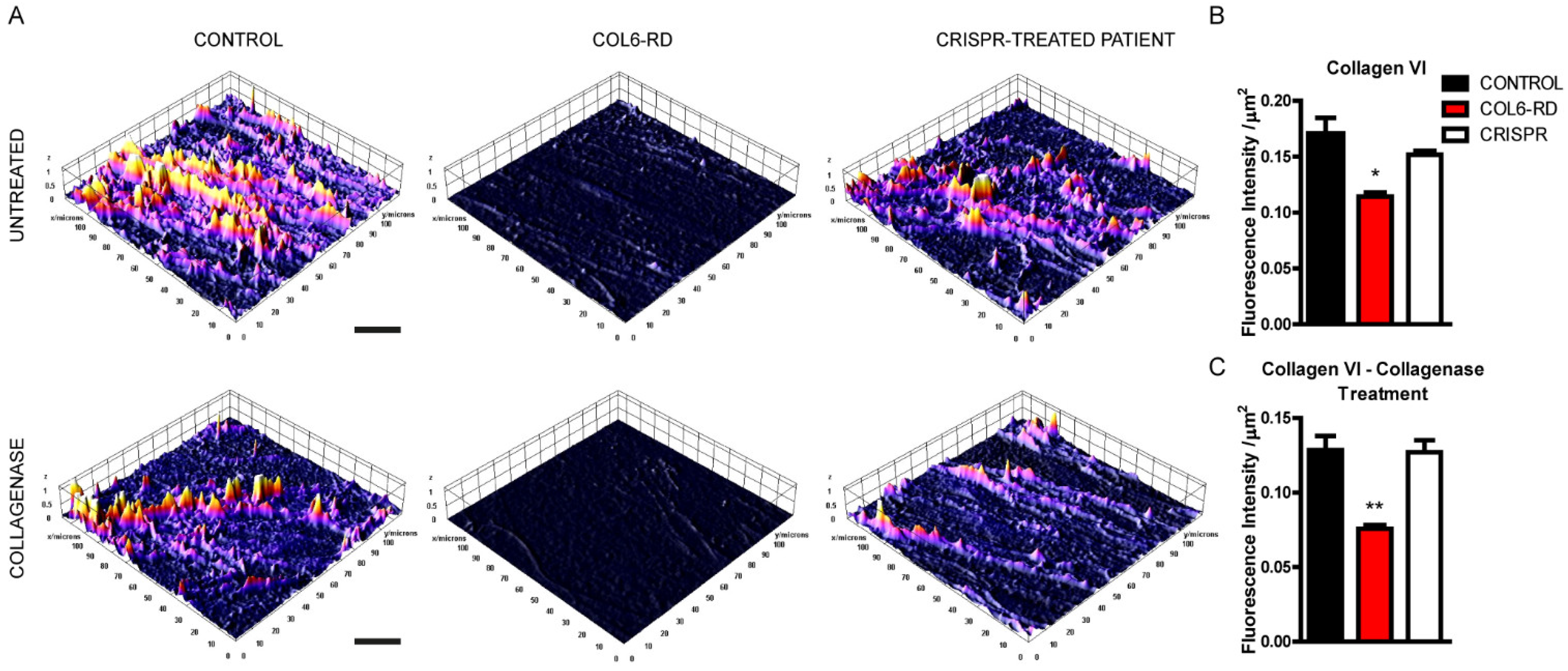
2.1. Patient-Derived Fibroblasts Cannot Assemble the Extracellular Collagen VI Network
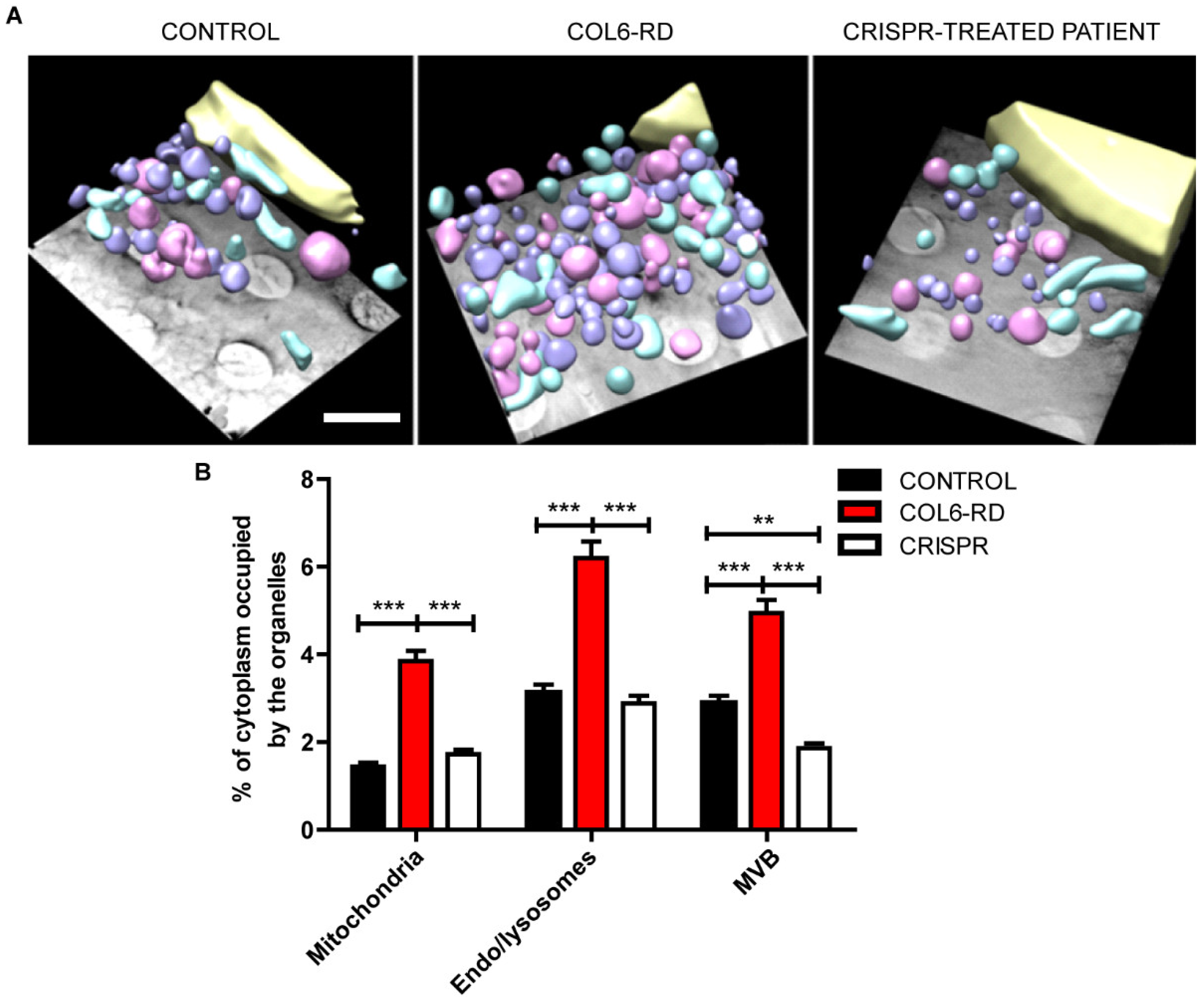
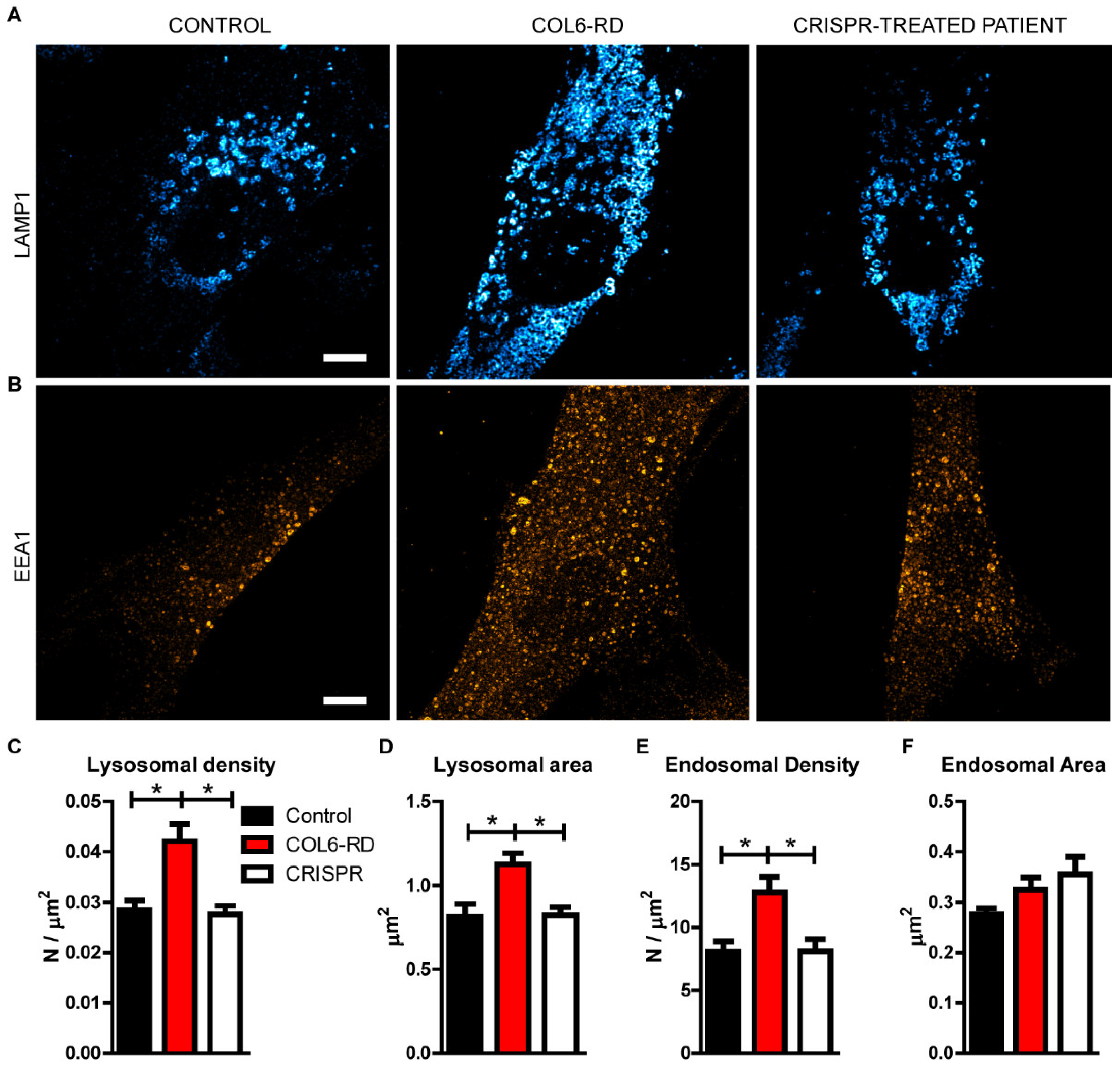
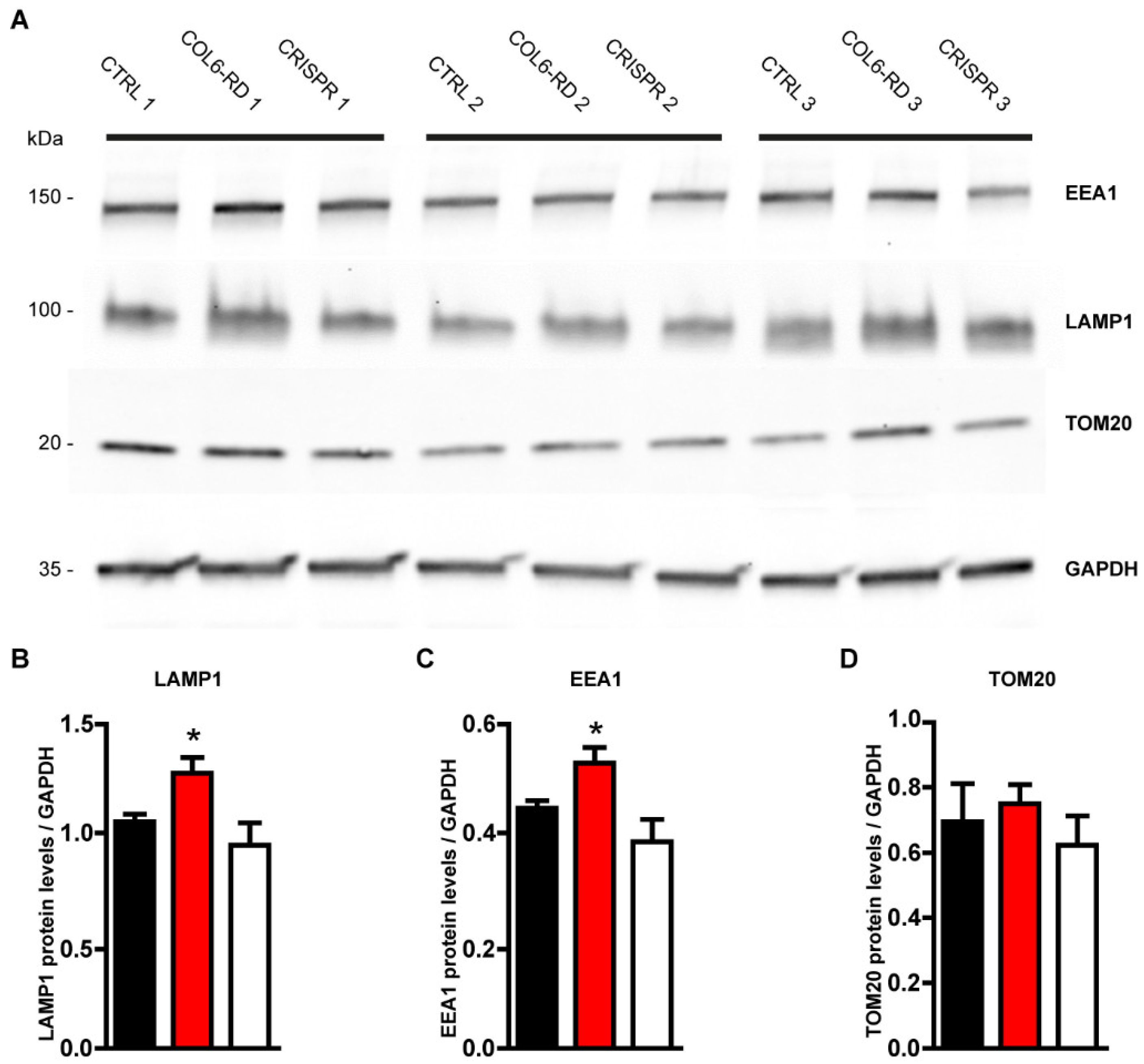
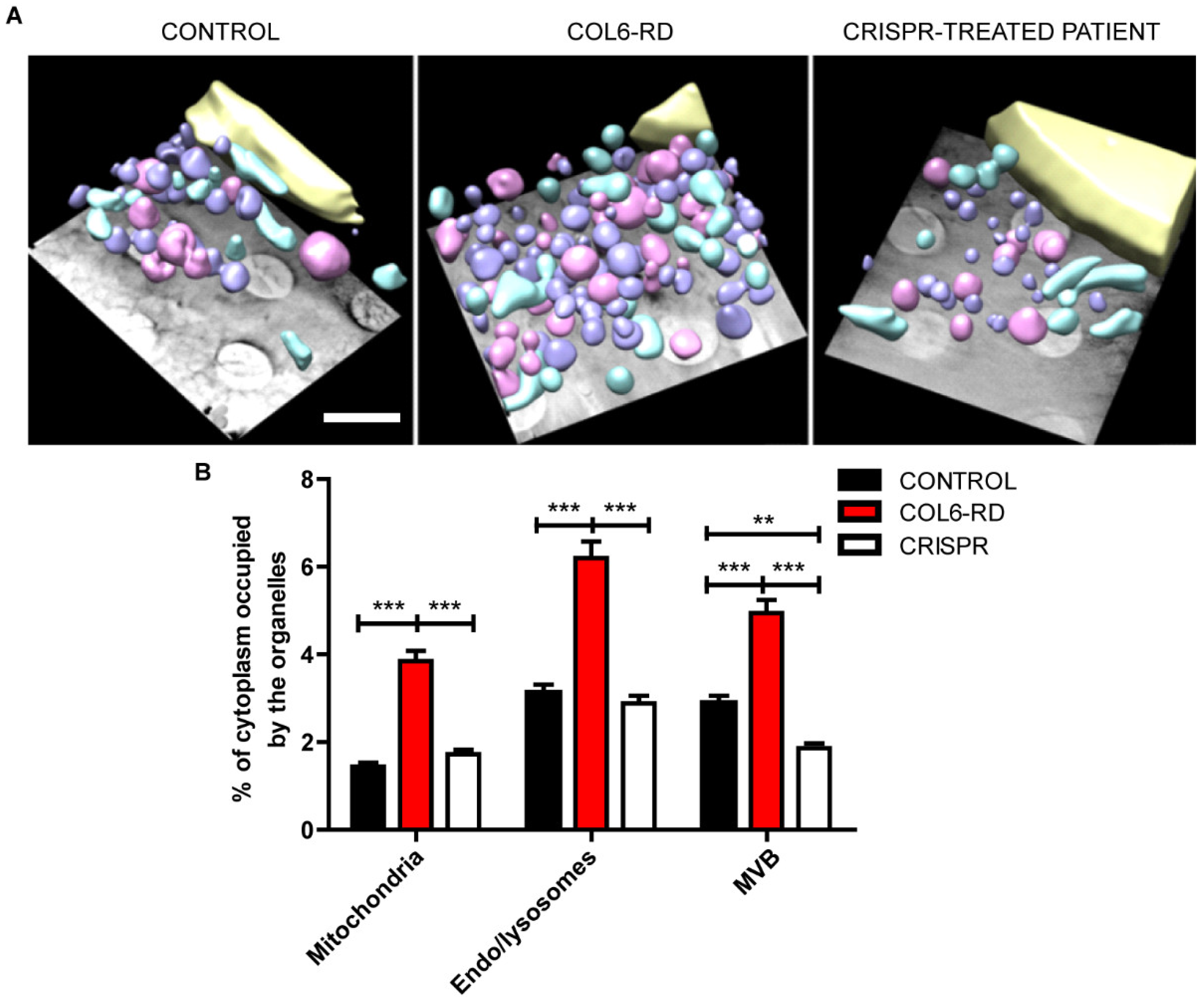
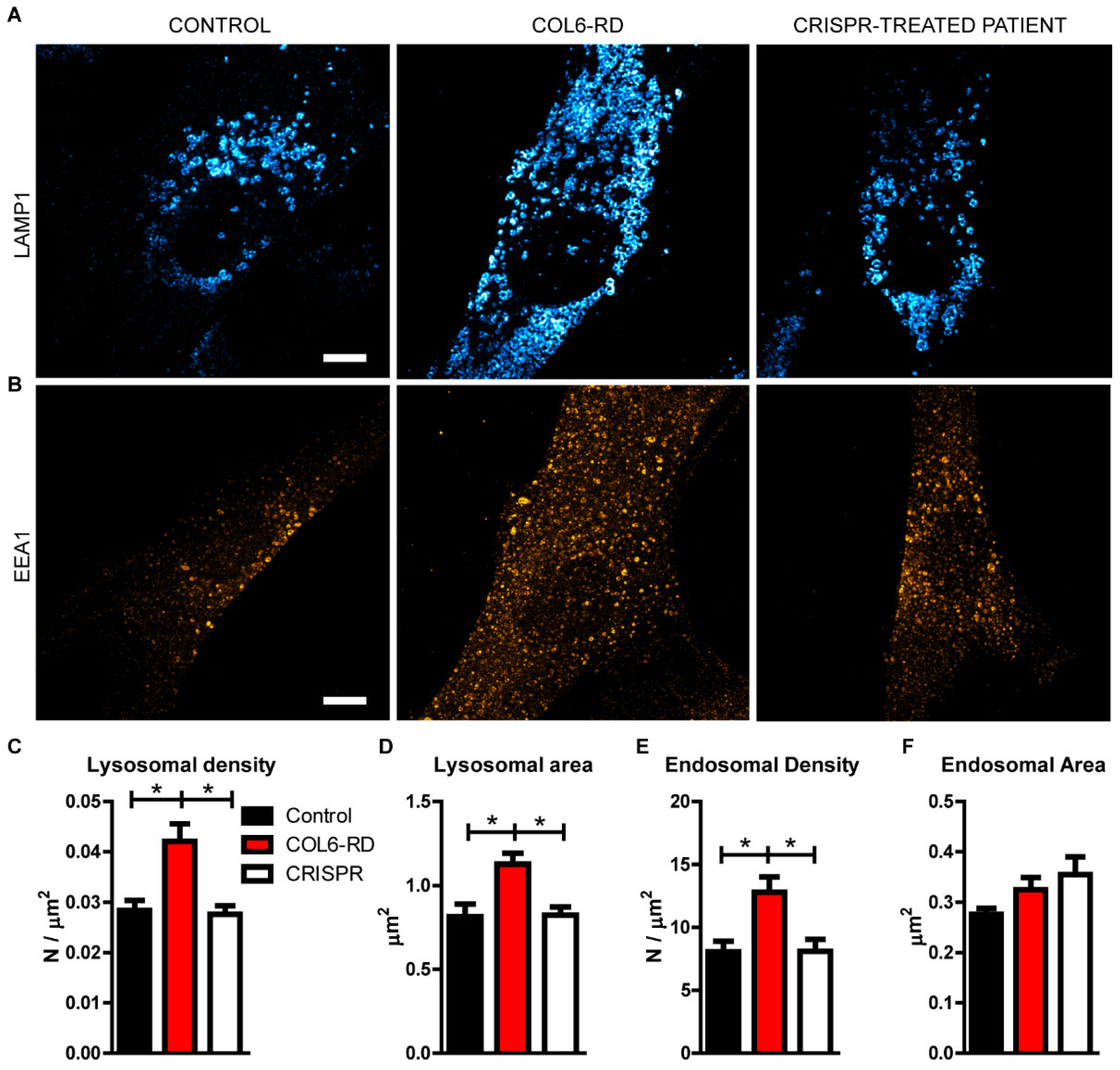
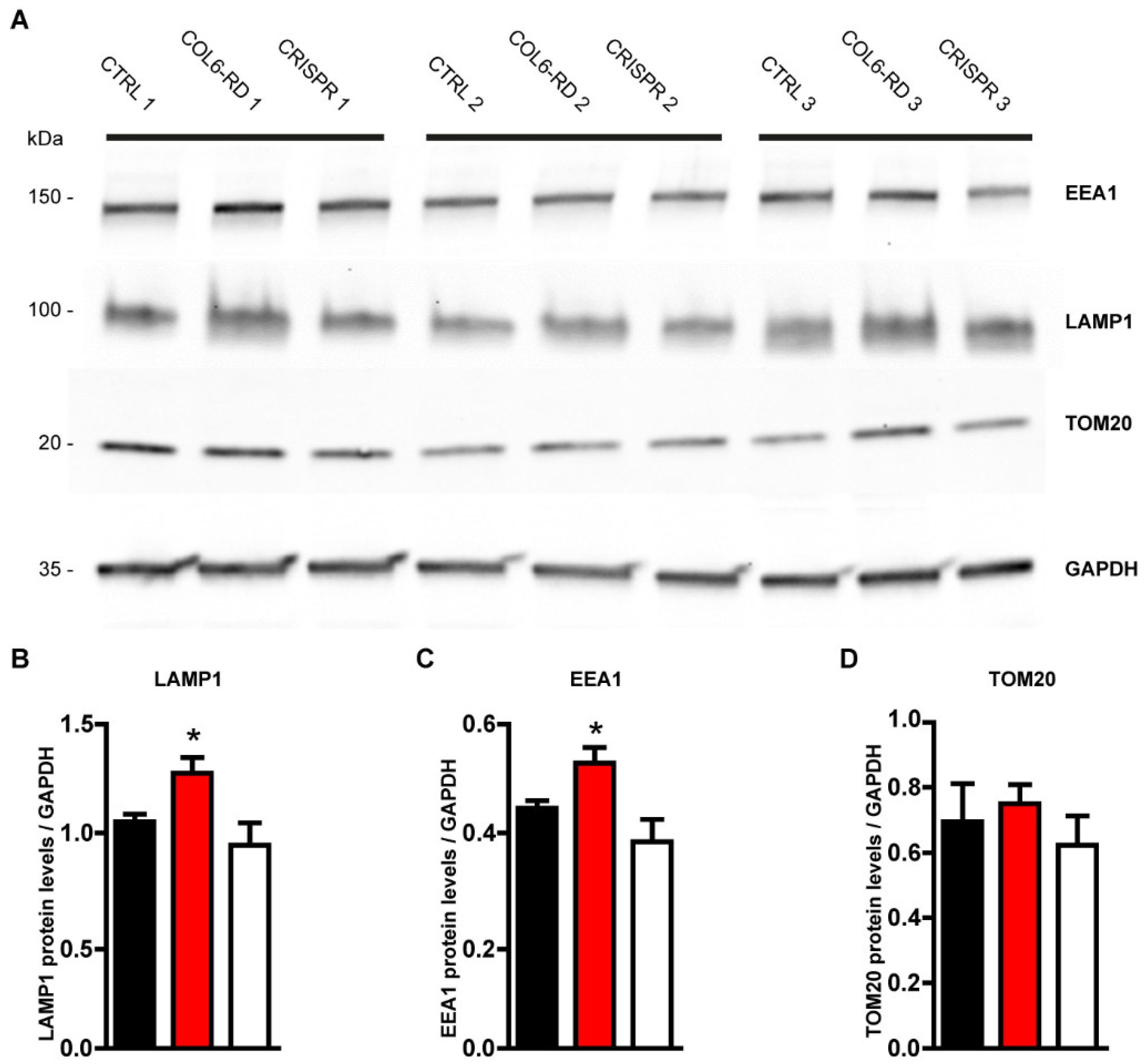
2.2. The Endo-Lysosomal System Homeostasis Is Altered in COL6-RD Patient-Derived Fibroblasts
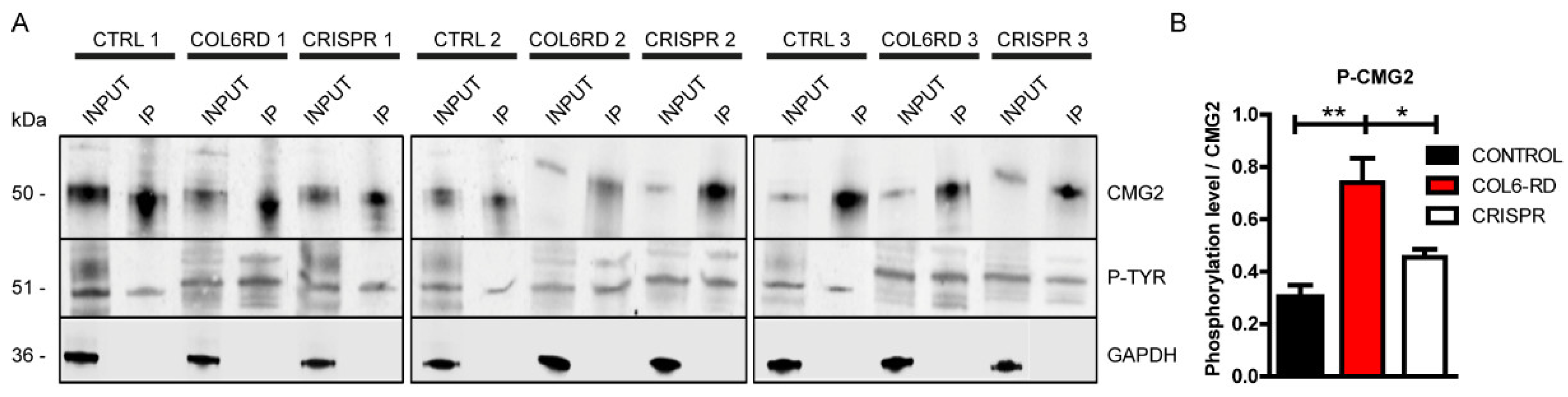
2.3. Col VI Mutation Induces Hyper-Phosphorylation of the CMG2 Receptor in Humans
3. Discussion
4. Materials and Methods
4.1. Human Fibroblasts
4.2. CRISPR-Cas9
4.3. Immunofluorescence and Stimulated Emission Depletion (STED) Microscopy
4.4. Soft X-ray Cryo-Tomography
4.5. Western Blotting and Immunoprecipitation
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bönnemann, C.G. The collagen VI-related myopathies: Muscle meets its matrix. Nat. Rev. Neurol. 2011, 7, 379–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natera-de Benito, D.; Reghan Foley, A.; Domínguez-González, C.; Ortez, C.; Jain, M.; Mebrahtu, A.; Donkervoort, S.; Hu, Y.; Fink, M.; Yun, P.; et al. Association of Initial Maximal Motor Ability With Long-term Functional Outcome in Patients With COL6-Related Dystrophies. Neurology 2021, 96, e1413–e1424. [Google Scholar] [CrossRef] [PubMed]
- Mohassel, P.; Reghan Foley, A.; Bönnemann, C.G. Extracellular matrix-driven congenital muscular dystrophies. Matrix Biol. 2018, 71–72, 188–204. [Google Scholar] [CrossRef]
- Tonelotto, V.; Trapani, V.; Bretaud, S.; Heumuller, S.E.; Wagener, R.; Ruggiero, F.; Bonaldo, P. Spatio-temporal expression and distribution of collagen VI during zebrafish development. Sci. Rep. 2019, 9, 19851. [Google Scholar] [CrossRef] [PubMed]
- Paco, S.; Casserras, T.; Rodríguez, M.A.; Jou, C.; Puigdelloses, M.; Ortez, C.I.; Diaz-Manera, J.; Gallardo, E.; Colomer, J.; Nascimento, A.; et al. Transcriptome analysis of ullrich congenital muscular dystrophy fibroblasts reveals a disease extracellular matrix signature and key molecular regulators. PLoS ONE 2015, 10, e0145107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paco, S.; Kalko, S.G.; Jou, C.; Rodríguez, M.A.; Corbera, J.; Muntoni, F.; Feng, L.; Rivas, E.; Torner, F.; Gualandi, F.; et al. Gene Expression Profiling Identifies Molecular Pathways Associated with Collagen VI Deficiency and Provides Novel Therapeutic Targets. PLoS ONE 2013, 8, e77430. [Google Scholar] [CrossRef]
- Jobsis, G.J.; Keizers, H.; Vreijling, J.P.; de Visser, M.; Speer, M.C.; Wolterman, R.A.; Baas, F.; Bolhuis, P.A. Type VI collagen mutations in Bethlem myopathy, an autosomal dominant myopathy with contractures. Nat. Genet. 1996, 14, 113–115. [Google Scholar] [CrossRef]
- Bolduc, V.; Foley, A.R.; Solomon-Degefa, H.; Sarathy, A.; Donkervoort, S.; Hu, Y.; Chen, G.S.; Sizov, K.; Nalls, M.; Zhou, H.; et al. A recurrent COL6A1 pseudoexon insertion causes muscular dystrophy and is effectively targeted by splice-correction therapies. JCI Insight 2019, 4, e124403. [Google Scholar] [CrossRef] [Green Version]
- Vanegas, O.C.; Bertini, E.; Zhang, R.Z.; Petrini, S.; Minosse, C.; Sabatelli, P.; Giusti, B.; Chu, M.L.; Pepe, G. Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc. Natl. Acad. Sci. USA 2001, 98, 7516–7521. [Google Scholar] [CrossRef] [Green Version]
- Irwin, W.A.; Bergamin, N.; Sabatelli, P.; Reggiani, C.; Megighian, A.; Merlini, L.; Braghetta, P.; Columbaro, M.; Volpin, D.; Bressan, G.M.; et al. Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat. Genet. 2003, 35, 367–371. [Google Scholar] [CrossRef]
- Alexopoulos, L.G.; Youn, I.; Bonaldo, P.; Guilak, F. Developmental and osteoarthritic changes in Col6a1-knockout mice: Biomechanics of type VI collagen in the cartilage pericellular matrix. Arthritis Rheum. 2009, 60, 771–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cescon, M.; Gattazzo, F.; Chen, P.; Bonaldo, P. Collagen VI at a glance. J. Cell Sci. 2015, 128, 3525–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Zhang, R.Z.; Sabatelli, P.; Chu, M.L.; Bönnemann, C.G. Muscle interstitial fibroblasts are the main source of collagen VI synthesis in skeletal muscle: Implications for congenital muscular dystrophy types Ullrich and Bethlem. J. Neuropathol. Exp. Neurol. 2008, 67, 144–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bürgi, J.; Kunz, B.; Abrami, L.; Deuquet, J.; Piersigilli, A.; Scholl-Bürgi, S.; Lausch, E.; Unger, S.; Superti-Furga, A.; Bonaldo, P.; et al. CMG2/ANTXR2 regulates extracellular collagen VI which accumulates in hyaline fibromatosis syndrome. Nat. Commun. 2017, 8, 15861. [Google Scholar] [CrossRef] [Green Version]
- Abrami, L.; Kunz, B.; Van Der Goot, F.G. Anthrax toxin triggers the activation of src-like kinases to mediate its own uptake. Proc. Natl. Acad. Sci. USA 2010, 107, 1420–1424. [Google Scholar] [CrossRef] [Green Version]
- Bürgi, J.; Abrami, L.; Castanon, I.; Abriata, L.A.; Kunz, B.; Yan, S.E.; Lera, M.; Unger, S.; Superti-Furga, A.; Dal Peraro, M.; et al. Ligand Binding to the Collagen VI Receptor Triggers a Talin-to-RhoA Switch that Regulates Receptor Endocytosis. Dev. Cell 2020, 53, 418–430.e4. [Google Scholar] [CrossRef]
- Jiménez-Mallebrera, C.; Maioli, M.A.; Kim, J.; Brown, S.C.; Feng, L.; Lampe, A.K.; Bushby, K.; Hicks, D.; Flanigan, K.M.; Bönnemann, C.G.; et al. A comparative analysis of collagen VI production in muscle, skin and fibroblasts from 14 Ullrich congenital muscular dystrophy patients with dominant and recessive COL6A mutations. Neuromuscul. Disord. 2006, 16, 571–582. [Google Scholar] [CrossRef]
- Fields, G.B.; Van Wart, H.E.; Birkedal-Hansen, H. Sequence specificity of human skin fibroblast collagenase. Evidence for the role of collagen structure in determining the collagenase cleavage site. J. Biol. Chem. 1987, 262, 6221–6226. [Google Scholar] [CrossRef]
- López-Márquez, A.; Morin, M.; Fernández-Peñalver, S.; Badosa, C.; Hernández-Delgado, A.; Natera-de Benito, D.; Ortez, C.; Nascimiento, A.; Grinberg, D.; Balcells, S.; et al. CRISPR/Cas9-Mediated Allele-Specific Disruption of a Dominant COL6A1 Pathogenic Variant Improves Collagen VI Network in Patient Fibroblasts. Int. J. Mol. Sci. 2022, 23, 4410. [Google Scholar] [CrossRef]
- Cheng, J.S.; Dubal, D.B.; Kim, D.H.; Legleiter, J.; Cheng, I.H.; Yu, G.; Tesseur, I.; Wyss-Coray, T.; Bonaldo, P.; Mucke, L. Collagen VI protects neurons against Aβ toxicity. Nat. Neurosci. 2009, 12, 119–121. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Chiu, R.; Leung, S.M.; Shields, D. Fragmentation of the Golgi apparatus: An early apoptotic event independent of the cytoskeleton. Traffic 2007, 8, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Castagnaro, S.; Chrisam, M.; Cescon, M.; Braghetta, P.; Grumati, P.; Bonaldo, P. Extracellular collagen VI has prosurvival and autophagy instructive properties in mouse fibroblasts. Front. Physiol. 2018, 9, 1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garriga, D.; Chichón, F.J.; Calisto, B.M.; Ferrero, D.S.; Gastaminza, P.; Pereiro, E.; Pérez-Berná, A.J. Imaging of Virus-Infected Cells with Soft X-ray Tomography. Viruses 2021, 13, 2109. [Google Scholar] [CrossRef]
- Groen, J.; Sorrentino, A.; Aballe, L.; Oliete, R.; Valcárcel, R.; Okolo, C.; Kounatidis, I.; Harkiolaki, M.; Pérez-Berná, A.J.; Pereiro, E. A 3D Cartographic Description of the Cell by Cryo Soft X-ray Tomography. J. Vis. Exp. 2021, 2021, 62190. [Google Scholar] [CrossRef] [PubMed]
- Groen, J.; Conesa, J.J.; Valcárcel, R.; Pereiro, E. The cellular landscape by cryo soft X-ray tomography. Biophys. Rev. 2019, 11, 611–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engvall, E.; Hessle, H.; Klier, G. Molecular assembly, secretion, and matrix deposition of type VI collagen. J. Cell Biol. 1986, 102, 703–710. [Google Scholar] [CrossRef] [Green Version]
- Butterfield, R.J.; Reghan Foley, A.; Dastgir, J.; Asman, S.; Dunn, D.M.; Zou, Y.; Hu, Y.; Flanigan, K.M.; Swoboda, K.J.; Winder, T.L.; et al. Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen VI myopathies. Hum. Mutat. 2013, 34, 1558–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villar-Quiles, R.N.; Donkervoort, S.; de Becdelièvre, A.; Gartioux, C.; Jobic, V.; Foley, A.R.; McCarty, R.M.; Hu, Y.; Menassa, R.; Michel, L.; et al. Clinical and Molecular Spectrum Associated with COL6A3 c.7447A>G p.(Lys2483Glu) Variant: Elucidating its Role in Collagen VI-related Myopathies. J. Neuromuscul. Dis. 2021, 8, 633–645. [Google Scholar] [CrossRef]
- Bella, J.; Eaton, M.; Brodsky, B.; Berman, H.M. Crystal and molecular structure of a collagen-like peptide at 1.9 A resolution. Science 1994, 266, 75–81. [Google Scholar] [CrossRef]
- Long, C.G.; Braswell, E.; Zhu, D.; Apigo, J.; Baum, J.; Brodsky, B. Characterization of collagen-like peptides containing interruptions in the repeating Gly-X-Y sequence. Biochemistry 1993, 32, 11688–11695. [Google Scholar] [CrossRef]
- Everts, V.; Korper, W.; Niehof, A.; Jansen, I.; Beertsen, W. Typer VI collagen is phagocytosed by fibroblasts and digested in the lysosomal apparatus: Involvement of collagenase, serine proteinases and lysosomal enzymes. Matrix Biol. 1995, 14, 665–675. [Google Scholar] [CrossRef]
- Shoulders, M.D.; Raines, R.T. Collagen structure and stability. Annu. Rev. Biochem. 2009, 78, 929–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, O.; Difeo, A.; Ramirez, M.C.; Tukel, T.; Narla, G.; Bonafe, L.; Kayserili, H.; Yuksel-Apak, M.; Paller, A.S.; Norton, K.; et al. Mutations in Capillary Morphogenesis Gene-2 Result in the Allelic Disorders Juvenile Hyaline Fibromatosis and Infantile Systemic Hyalinosis. Am. J. Hum. Genet. 2003, 73, 957–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piper, R.C.; Katzmann, D.J. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 2007, 23, 519–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, P.I.; Cashikar, A. Multivesicular body morphogenesis. Annu. Rev. Cell Dev. Biol. 2012, 28, 337–362. [Google Scholar] [CrossRef]
- Jung, Y.S.; Jun, S.; Kim, M.J.; Lee, S.H.; Suh, H.N.; Lien, E.M.; Jung, H.Y.; Lee, S.; Zhang, J.; Yang, J.I.; et al. TMEM9 promotes intestinal tumorigenesis through vacuolar-ATP-ase activated Wnt/β-catenin signaling. Nat. Cell Biol. 2018, 20, 1421–1433. [Google Scholar] [CrossRef]
- Wagenaar-Miller, R.A.; Engelholm, L.H.; Gavard, J.; Yamada, S.S.; Gutkind, J.S.; Behrendt, N.; Bugge, T.H.; Holmbeck, K. Complementary Roles of Intracellular and Pericellular Collagen Degradation Pathways In Vivo. Mol. Cell. Biol. 2007, 27, 6309–6322. [Google Scholar] [CrossRef] [Green Version]
- Scita, G.; Di Fiore, P.P. The endocytic matrix. Nature 2010, 463, 464–473. [Google Scholar] [CrossRef]
- Rainero, E. Extracellular matrix endocytosis in controlling matrix turnover and beyond: Emerging roles in cancer. Biochem. Soc. Trans. 2016, 44, 1347–1354. [Google Scholar] [CrossRef]
- Imanikia, S.; Taylor, R.C. Previews Molty-Level Regulation: Lysosomes Participate in Developmental ECM Remodeling in C. elegans. Dev. Cell 2020, 52, 1–2. [Google Scholar] [CrossRef]
- Dankovich, T.M.; Kaushik, R.; Olsthoorn, L.H.M.; Cassinelli Petersen, G.; Giro, P.E.; Kluever, V.; Agui-Gonzalez, P.; Grewe, K.; Bao, G.; Beuermann, S.; et al. Extracellular matrix remodeling through endocytosis and resurfacing of Tenascin-R. Nat. Commun. 2021, 12, 7129. [Google Scholar] [CrossRef]
- Groen, J.; Palanca, A.; Aires, A.; Conesa, J.J.; Maestro, D.; Rehbein, S.; Harkiolaki, M.; Villar, A.V.; Cortajarena, A.L.; Pereiro, E. Correlative 3D cryo X-ray imaging reveals intracellular location and effect of designed antifibrotic protein–nanomaterial hybrids. Chem. Sci. 2021, 12, 15090–15103. [Google Scholar] [CrossRef] [PubMed]
- Merlini, L.; Angelin, A.; Tiepolo, T.; Braghetta, P.; Sabatelli, P.; Zamparelli, A.; Ferlini, A.; Maraldi, N.M.; Bonaldo, P.; Bernardi, P. Cyclosporin A corrects mitochondrial dysfunction and muscle apoptosis in patients with collagen VI myopathies. Proc. Natl. Acad. Sci. USA 2008, 105, 5225–5229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiepolo, T.; Angelin, A.; Palma, E.; Sabatelli, P.; Merlini, L.; Nicolosi, L.; Finetti, F.; Braghetta, P.; Vaugniaux, G.; Dumont, J.M.; et al. The cyclophilin inhibitor Debio 025 normalizes mitochondrial function, muscle apoptosis and ultrastructural defects in Col6a1-/- myopathic mice. Br. J. Pharmacol. 2009, 157, 1045–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zulian, A.; Rizzo, E.; Schiavone, M.; Palma, E.; Tagliavini, F.; Blaauw, B.; Merlini, L.; Maraldi, N.M.; Sabatelli, P.; Braghetta, P.; et al. NIM811, a cyclophilin inhibitor without immunosuppressive activity, is beneficial in collagen VI congenital muscular dystrophy models. Hum. Mol. Genet. 2014, 23, 5353–5363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubb, K.; Holzer, T.; Nolte, J.L.; Kruger, M.; Wilson, R.; Schotzer-Schrehardt, U.; Brinckmann, J.; Altmuller, J.; Aszodi, A.; Fleischhauer, L.; et al. Mitochondrial respiratory chain function promotes extracellular matrix integrity in cartilage. J. Biol. Chem. 2021, 297, 101224. [Google Scholar] [CrossRef]
- Hicks, S.W.; Machamer, C.E. Golgi structure in stress sensing and apoptosis. Biochim. Biophys. Acta 2005, 1744, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Lane, J.D.; Lucocq, J.; Pryde, J.; Barr, F.A.; Woodman, P.G.; Allan, V.J.; Lowe, M. Caspase-mediated cleavage of the stacking protein GRASP65 is required for Golgi fragmentation during apoptosis. J. Cell Biol. 2002, 156, 495–509. [Google Scholar] [CrossRef] [Green Version]
- Lowe, M.; Lane, J.D.; Woodman, P.G.; Allan, V.J. Caspase-mediated cleavage of syntaxin 5 and giantin accompanies inhibition of secretory traffic during apoptosis. J. Cell Sci. 2004, 117, 1139–1150. [Google Scholar] [CrossRef] [Green Version]
- Mancini, M.; Machamer, C.E.; Roy, S.; Nicholson, D.W.; Thornberry, N.A.; Casciola-Rosen, L.A.; Rosen, A. Caspase-2 is localized at the Golgi complex and cleaves golgin-160 during apoptosis. J. Cell Biol. 2000, 149, 603–612. [Google Scholar] [CrossRef] [Green Version]
- Chiu, R.; Novikov, L.; Mukherjee, S.; Shields, D. A caspase cleavage fragment of p115 induces fragmentation of the Golgi apparatus and apoptosis. J. Cell Biol. 2002, 159, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, J.E.; Brown, A.L.; Bayrd, E.D.; Pease, G.L. Ultrastructure of the myeloma cell. Cancer 1966, 19, 1613–1627. [Google Scholar] [CrossRef]
- Kellokumpu, S.; Sormunen, R.; Kellokumpu, I. Abnormal glycosylation and altered Golgi structure in colorectal cancer: Dependence on intra-Golgi pH. FEBS Lett. 2002, 516, 217–224. [Google Scholar] [CrossRef] [Green Version]
- Bui, S.; Mejia, I.; Diaz, B.; Wang, Y. Adaptation of the Golgi Apparatus in Cancer Cell Invasion and Metastasis. Front. Cell Dev. Biol. 2021, 9, 3582. [Google Scholar] [CrossRef] [PubMed]
- Maag, R.S.; Mancini, M.; Rosen, A.; Machamer, C.E. Caspase-resistant Golgin-160 disrupts apoptosis induced by secretory pathway stress and ligation of death receptors. Mol. Biol. Cell 2005, 16, 3019–3027. [Google Scholar] [CrossRef] [PubMed]
- Ujihara, Y.; Kanagawa, M.; Mohri, S.; Takatsu, S.; Kobayashi, K.; Toda, T.; Naruse, K.; Katanosaka, Y. Elimination of fukutin reveals cellular and molecular pathomechanisms in muscular dystrophy-associated heart failure. Nat. Commun. 2019, 10, 5754. [Google Scholar] [CrossRef]
- Mendoza-Ferreira, N.; Karakaya, M.; Cengiz, N.; Beijer, D.; Brigatti, K.W.; Gonzaga-Jauregui, C.; Fuhrmann, N.; Holker, I.; Thelen, M.P.; Zetzsche, S.; et al. De Novo and Inherited Variants in GBF1 are Associated with Axonal Neuropathy Caused by Golgi Fragmentation. Am. J. Hum. Genet. 2020, 107, 763–777. [Google Scholar] [CrossRef]
- Sainio, A.; Järveläinen, H. Extracellular matrix-cell interactions: Focus on therapeutic applications. Cell. Signal. 2020, 66, 109487. [Google Scholar] [CrossRef]
- O’Toole, T.E.; Katagiri, Y.; Faull, R.J.; Peter, K.; Tamura, R.; Quaranta, V.; Loftus, J.C.; Shattil, S.J.; Ginsberg, M.H. Integrin cytoplasmic domains mediate inside-out signal transduction. J. Cell Biol. 1994, 124, 1047–1059. [Google Scholar] [CrossRef]
- Kim, S.H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signalling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Afratis, N.A.; Bouris, P.; Skandalis, S.S.; Multhaupt, H.A.; Couchman, J.R.; Theocharis, A.D.; Karamanos, N.K. IGF-IR cooperates with ERα to inhibit breast cancer cell aggressiveness by regulating the expression and localisation of ECM molecules. Sci. Rep. 2017, 7, 40138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanderson, R.D.; Elkin, M.; Rapraeger, A.C.; Ilan, N.; Vlodavsky, I. Heparanase regulation of cancer, autophagy and inflammation: New mechanisms and targets for therapy. FEBS J. 2017, 284, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, Y.; Pluchon, P.; Robens, J.; Herr, K.; Mercade, M.; Thiery, J.; Yu, H.; Viasnoff, V. Extracellular matrix scaffolding guides lumen elongation by inducing anisotropic intercellular mechanical tension. Nat. Cell Biol. 2016, 18, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, L.; Wan, D.; Zhou, L.; Zheng, S.; Lin, S.; Qiao, Y. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct. Target. Ther. 2021, 6, 153. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C.; Franch, M.; Tabas-Madrid, D.; San-León, D.; Montoliu, L.; Cubas, P.; Pazos, F. Breaking-Cas—interactive design of guide RNAs for CRISPR-Cas experiments for ENSEMBL genomes. Nucleic Acids Res. 2016, 44, W267–W271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castroflorio, E.; den Hoed, J.; Svistunova, D.; Finelli, M.J.; Cebran-Serrano, A.; Corrochano, S.; Bassett, A.R.; Davies, B.; Oliver, P.L. The Ncoa7 locus regulates V-ATPase formation and function, neurodevelopment and behaviour. Cell Mol. Life Sci. 2021, 7, 3503–3524. [Google Scholar] [CrossRef]
- Pereiro, E.; Nicolás, J.; Ferrer, S.; Howells, M.R. A soft X-ray beamline for transmission X-ray microscopy at ALBA. J. Synchrotron Radiat. 2009, 16, 505–512. [Google Scholar] [CrossRef]
- Sorrentino, A.; Nicolás, J.; Valcárcel, R.; Chichón, F.J.; Rosanes, M.; Avila, J.; Tkachuk, A.; Irwin, J.; Ferrer, S.; Pereiro, E. MISTRAL: A transmission soft X-ray microscopy beamline for cryo nano-tomography of biological samples and magnetic domains imaging. J. Synchrotron Radiat. 2015, 22, 1112–1117. [Google Scholar] [CrossRef]
- Pérez-Berná, A.J.; Rodriguéz, M.J.; Chichón, F.J.; Friesland, M.F.; Sorrentino, A.; Carrascosa, J.L.; Pereiro, E.; Gastaminza, P. Structural Changes in Cells Imaged by Soft X-ray Cryo-Tomography during Hepatitis C Virus Infection. ACS Nano 2016, 10, 6597–6611. [Google Scholar] [CrossRef]
- Kremer, J.R.; Mastronarde, D.N.; McIntosh, J.R. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996, 116, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Agulleiro, J.I.; Fernandez, J.J. Fast tomographic reconstruction on multicore computers. Bioinformatics 2011, 27, 582–583. [Google Scholar] [CrossRef] [PubMed]
- Agulleiro, J.I.; Fernandez, J.J. Tomo3D 2.0—exploitation of advanced vector extensions (AVX) for 3D reconstruction. J. Struct. Biol. 2015, 189, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castroflorio, E.; Pérez Berná, A.J.; López-Márquez, A.; Badosa, C.; Loza-Alvarez, P.; Roldán, M.; Jiménez-Mallebrera, C. The Capillary Morphogenesis Gene 2 Triggers the Intracellular Hallmarks of Collagen VI-Related Muscular Dystrophy. Int. J. Mol. Sci. 2022, 23, 7651. https://doi.org/10.3390/ijms23147651
Castroflorio E, Pérez Berná AJ, López-Márquez A, Badosa C, Loza-Alvarez P, Roldán M, Jiménez-Mallebrera C. The Capillary Morphogenesis Gene 2 Triggers the Intracellular Hallmarks of Collagen VI-Related Muscular Dystrophy. International Journal of Molecular Sciences. 2022; 23(14):7651. https://doi.org/10.3390/ijms23147651
Chicago/Turabian StyleCastroflorio, Enrico, Ana Joaquina Pérez Berná, Arístides López-Márquez, Carmen Badosa, Pablo Loza-Alvarez, Mónica Roldán, and Cecilia Jiménez-Mallebrera. 2022. "The Capillary Morphogenesis Gene 2 Triggers the Intracellular Hallmarks of Collagen VI-Related Muscular Dystrophy" International Journal of Molecular Sciences 23, no. 14: 7651. https://doi.org/10.3390/ijms23147651
APA StyleCastroflorio, E., Pérez Berná, A. J., López-Márquez, A., Badosa, C., Loza-Alvarez, P., Roldán, M., & Jiménez-Mallebrera, C. (2022). The Capillary Morphogenesis Gene 2 Triggers the Intracellular Hallmarks of Collagen VI-Related Muscular Dystrophy. International Journal of Molecular Sciences, 23(14), 7651. https://doi.org/10.3390/ijms23147651