
Transcriptomic and Metabolomic Analysis of Quality Changes during Sweet Cherry Fruit Development and Mining of Related Genes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
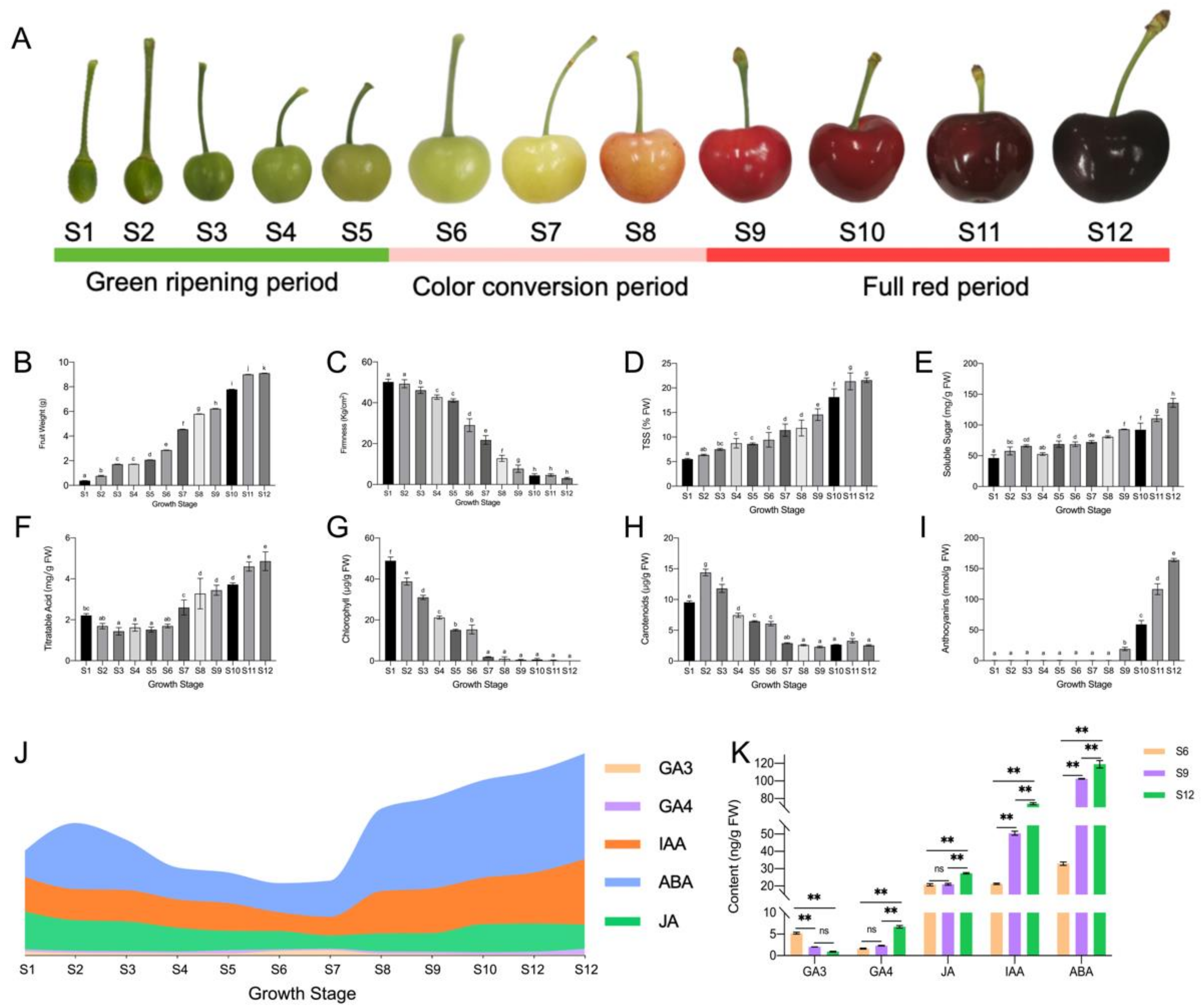
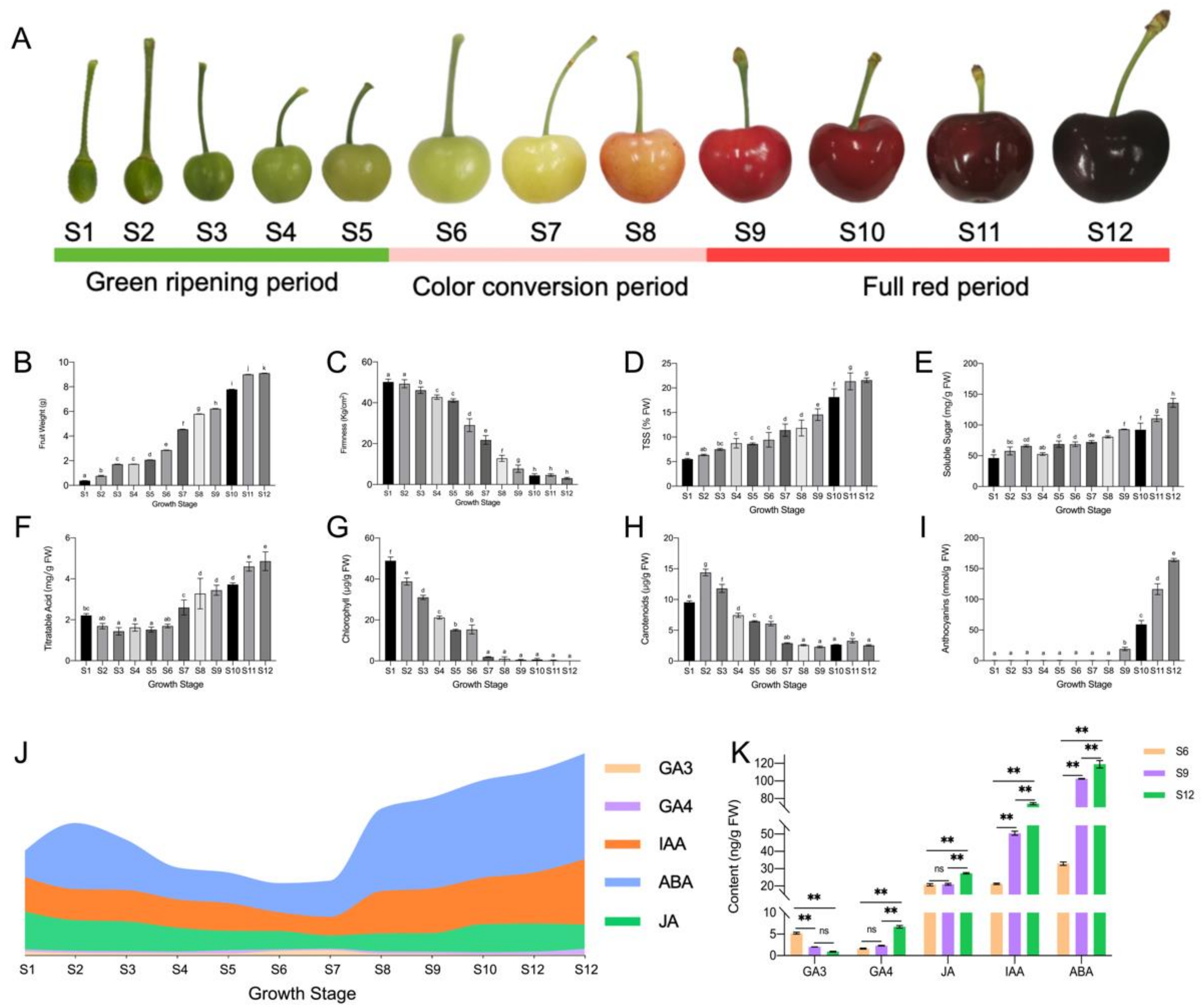
2.1. Changes in the Dynamics of Substance Accumulation during Sweet Cherry Development
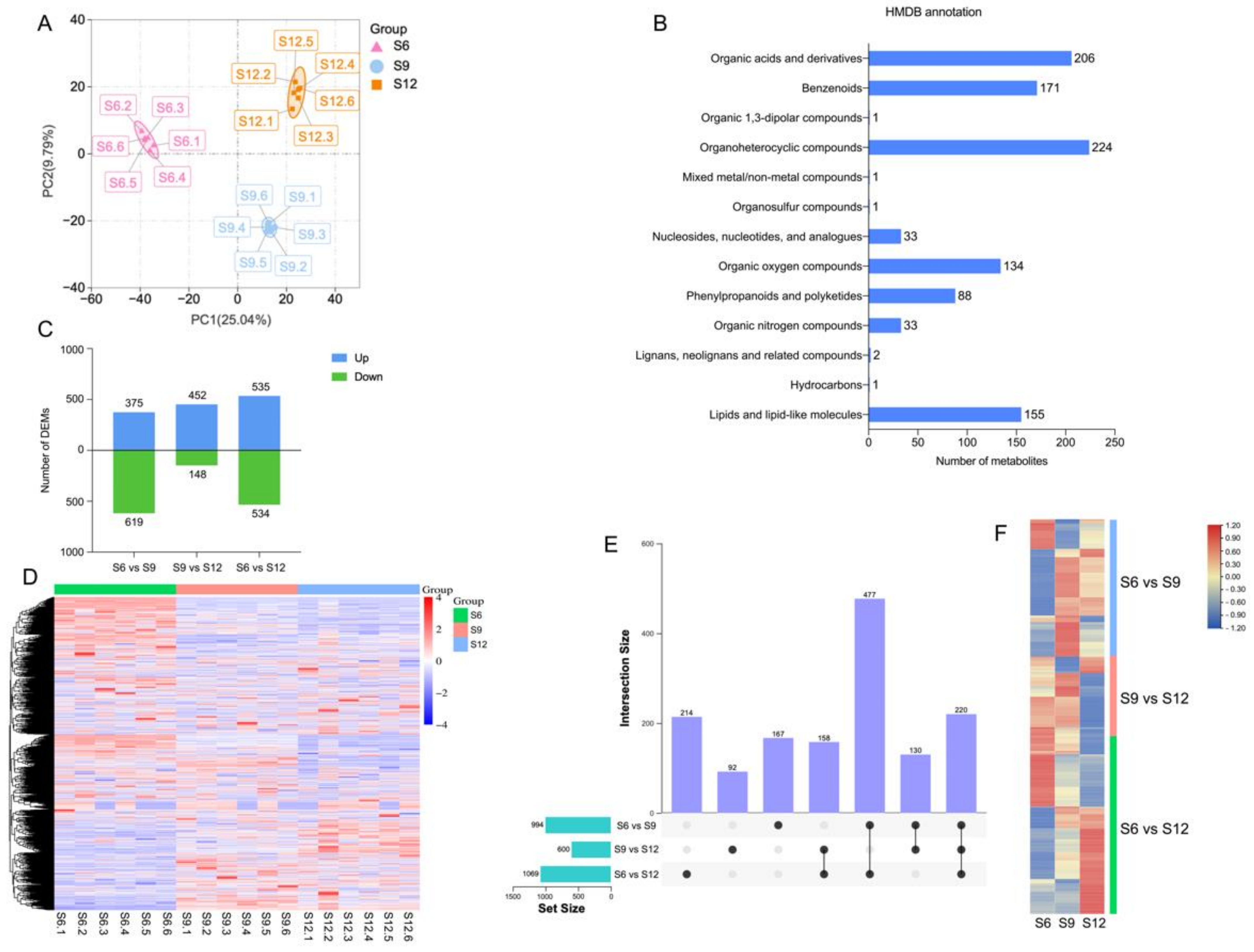
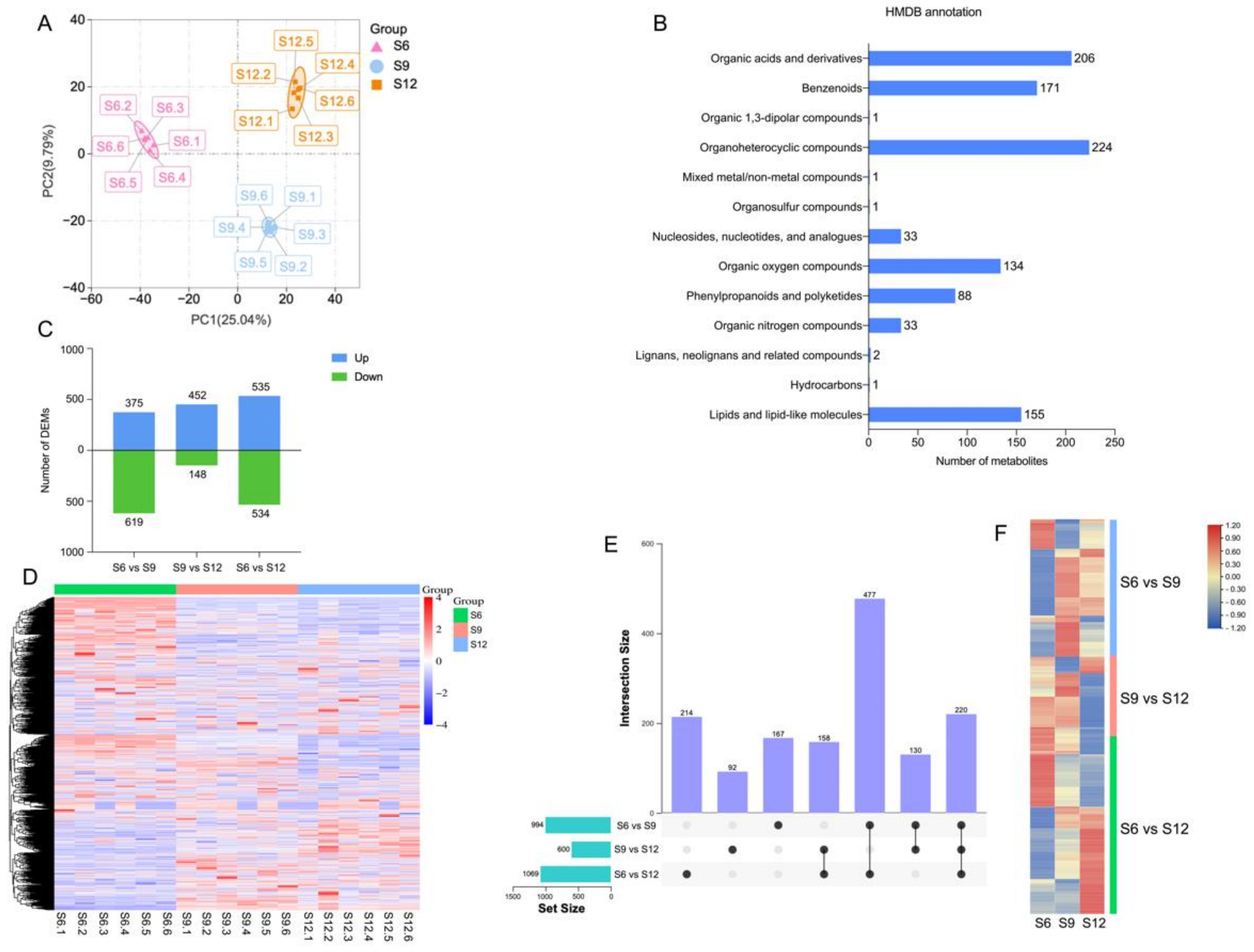
2.2. Metabolome Analysis
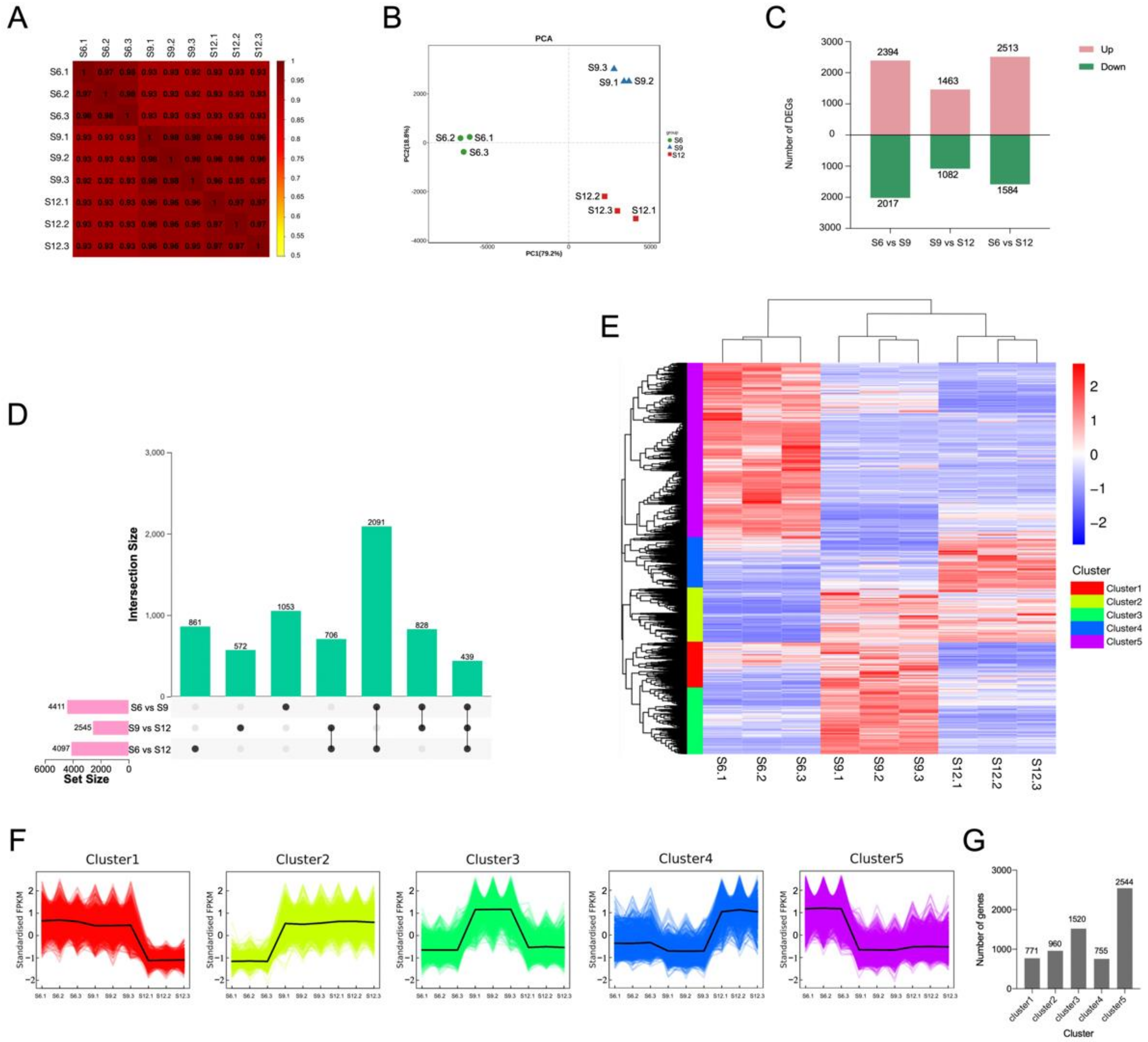
2.3. Transcriptome Identification
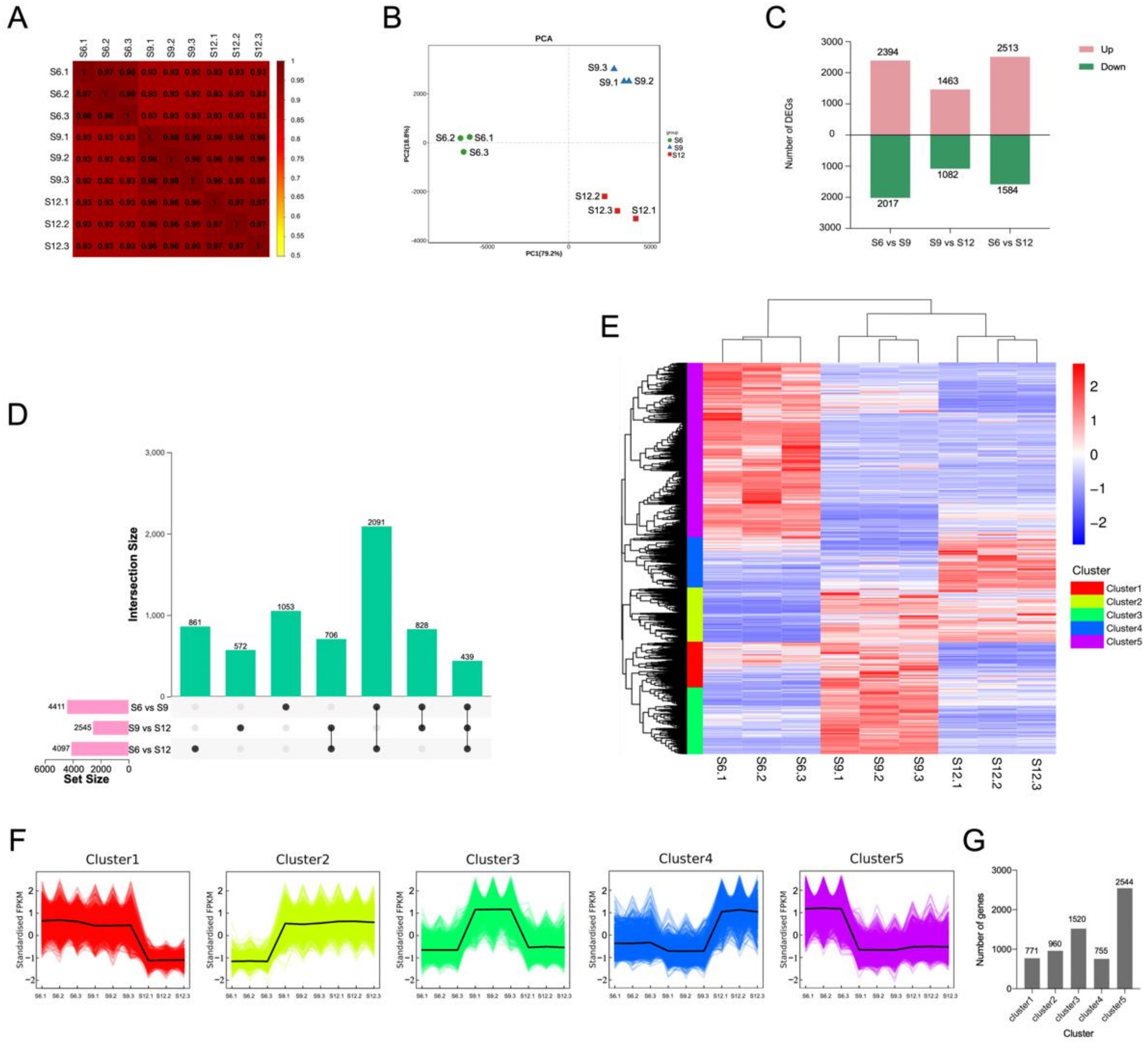
2.3.1. Results of Overall Transcriptome Analysis
2.3.2. Identification of Differentially Expressed Genes
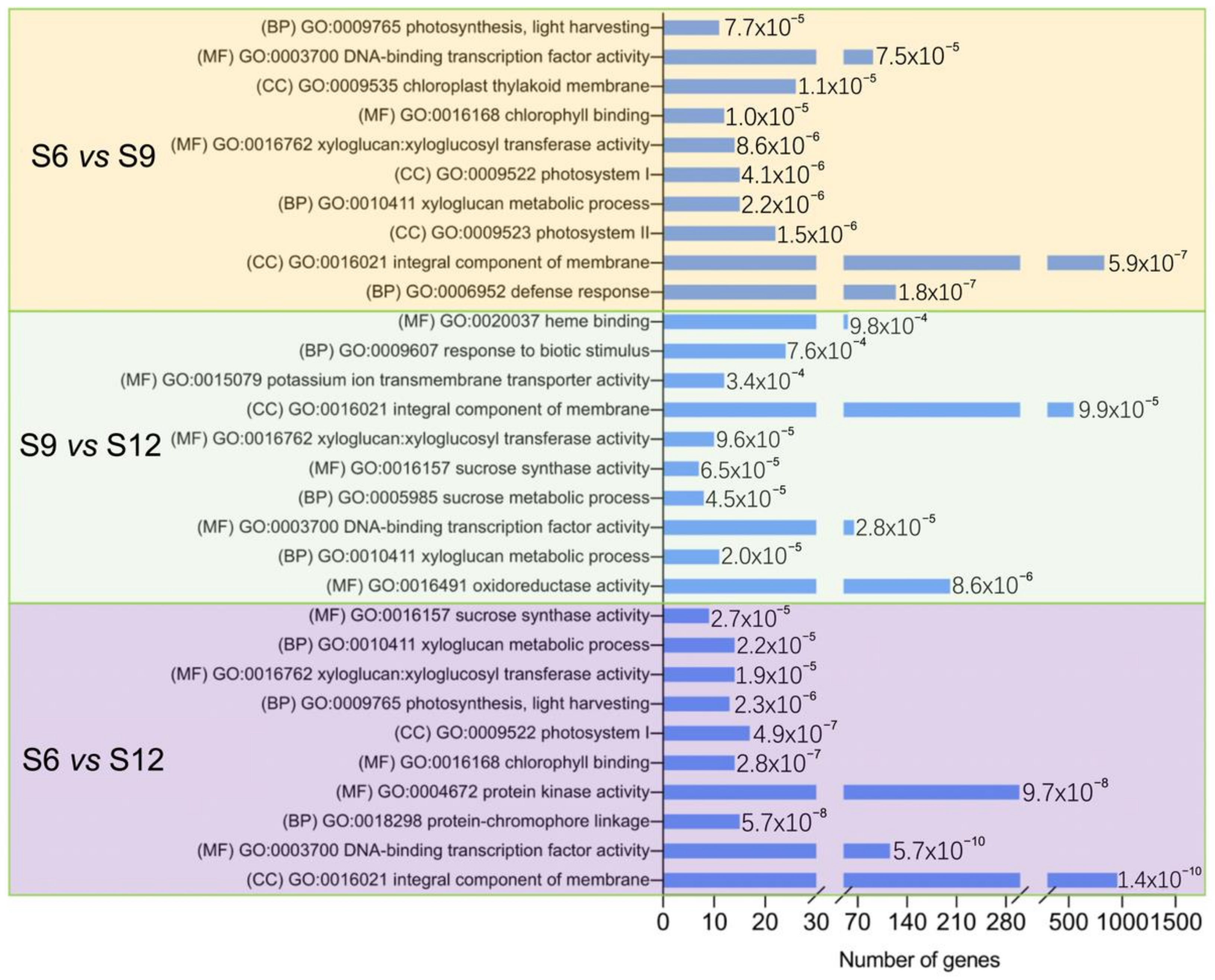
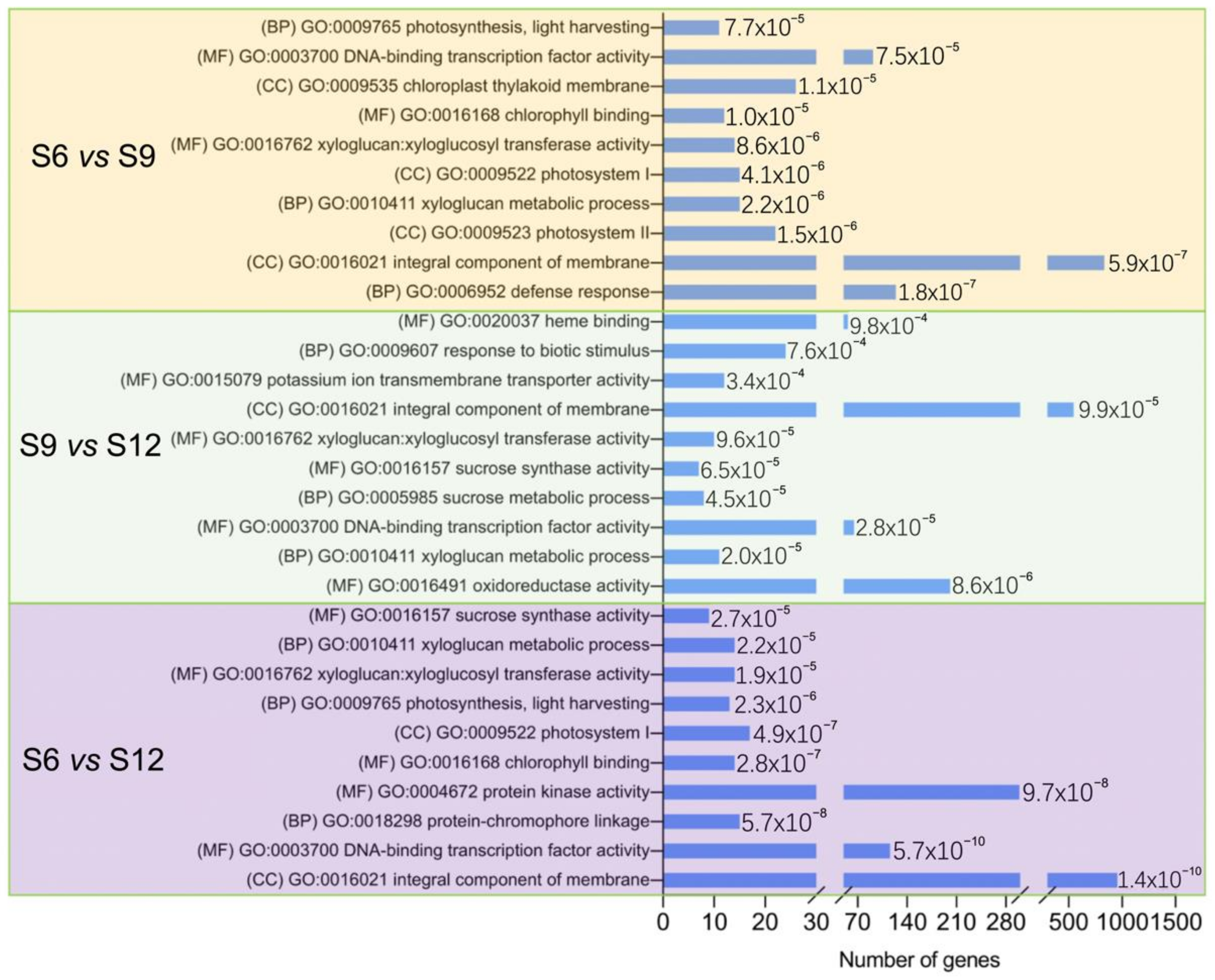
2.3.3. GO Enrichment Analysis
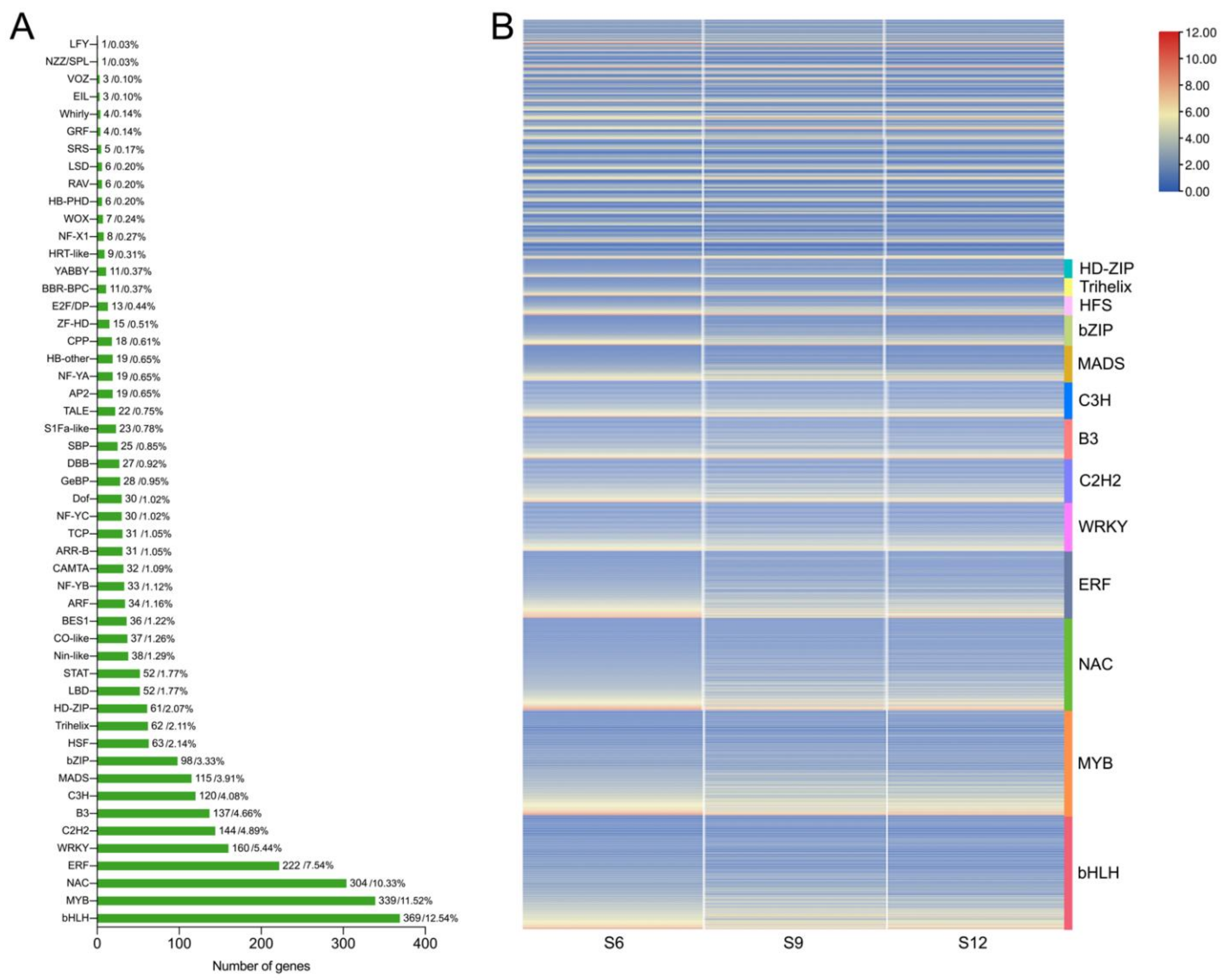
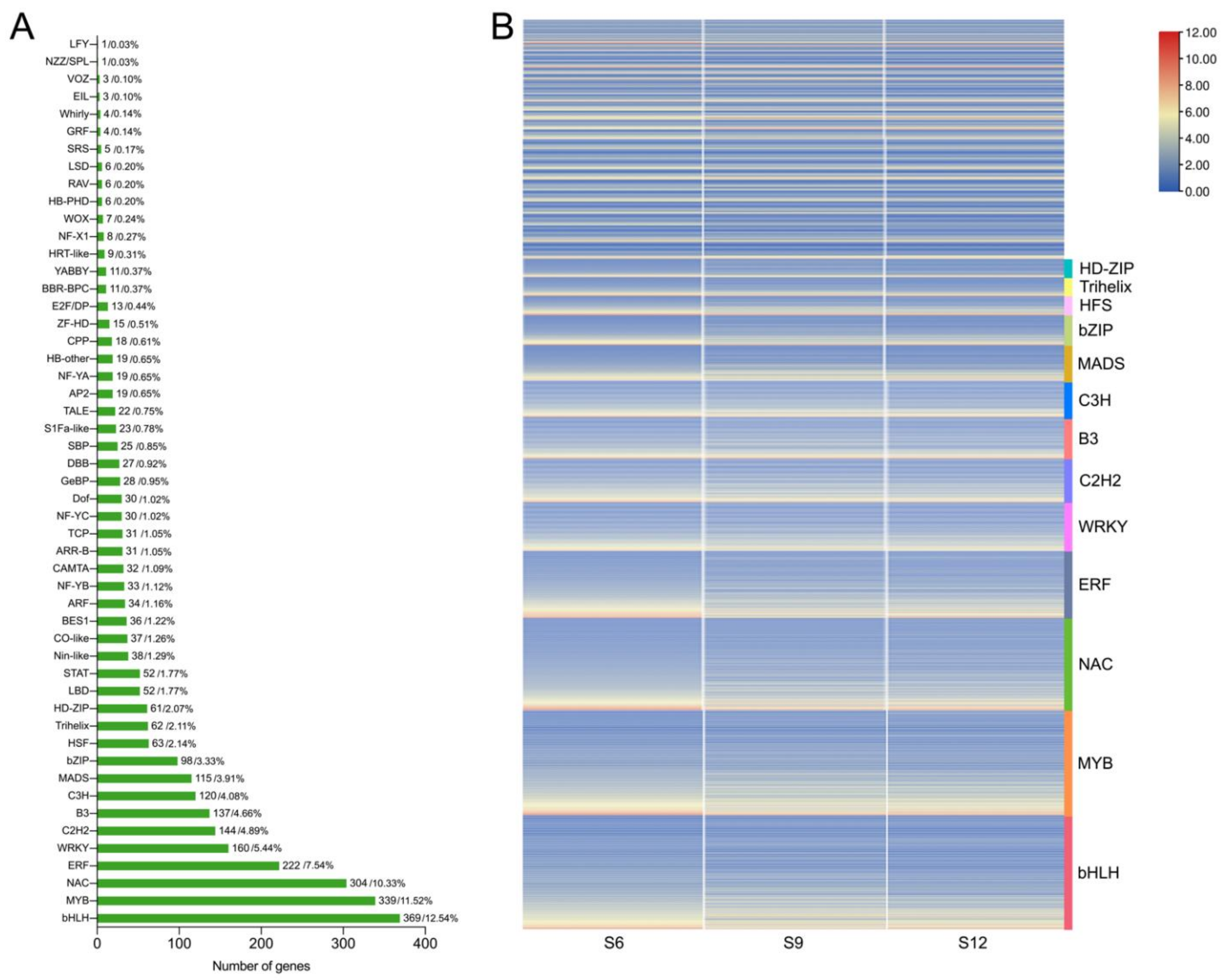
2.4. Transcription Factor Family Analysis
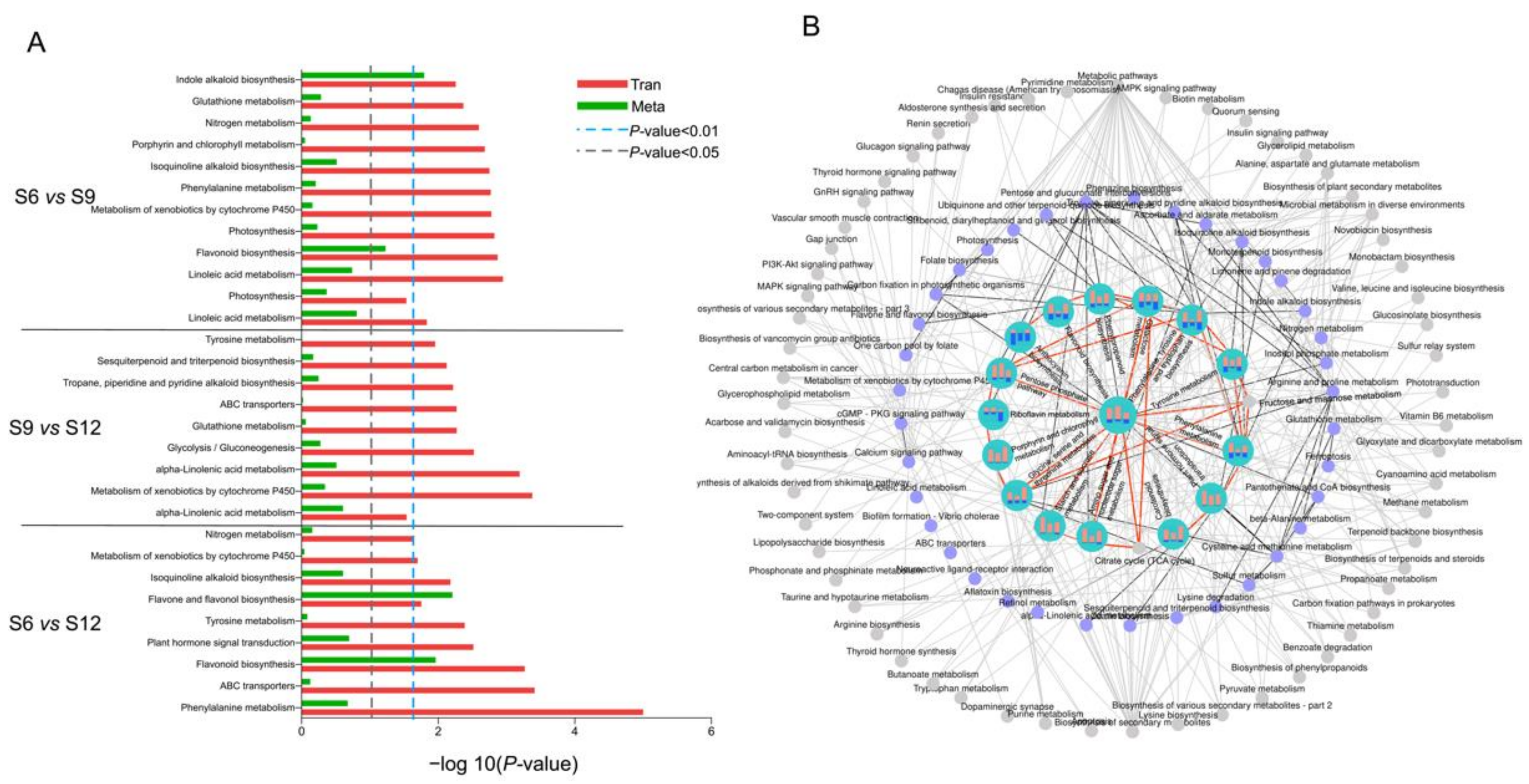
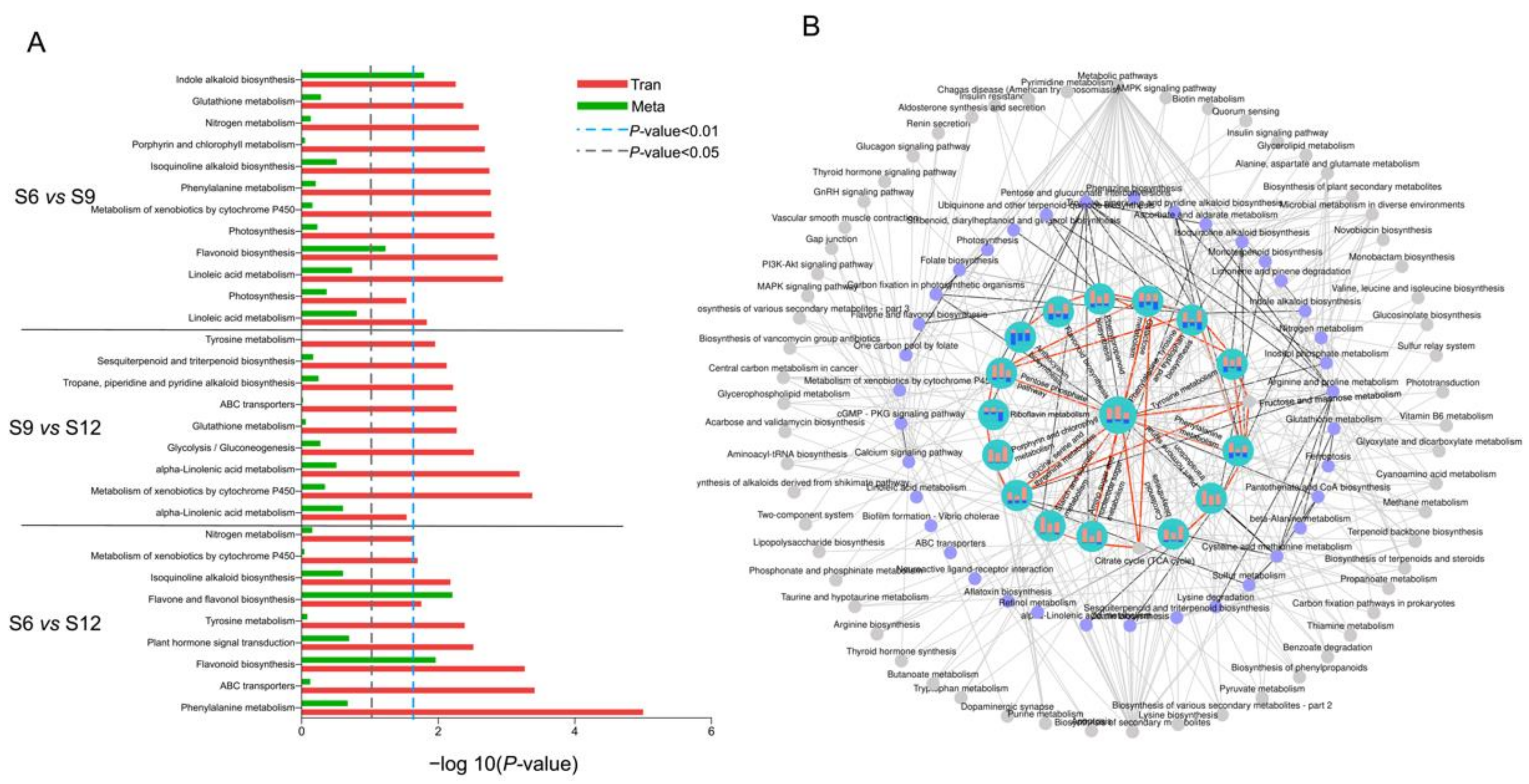
2.5. Transcriptome and Metabolome Pathway Enrichment Analysis
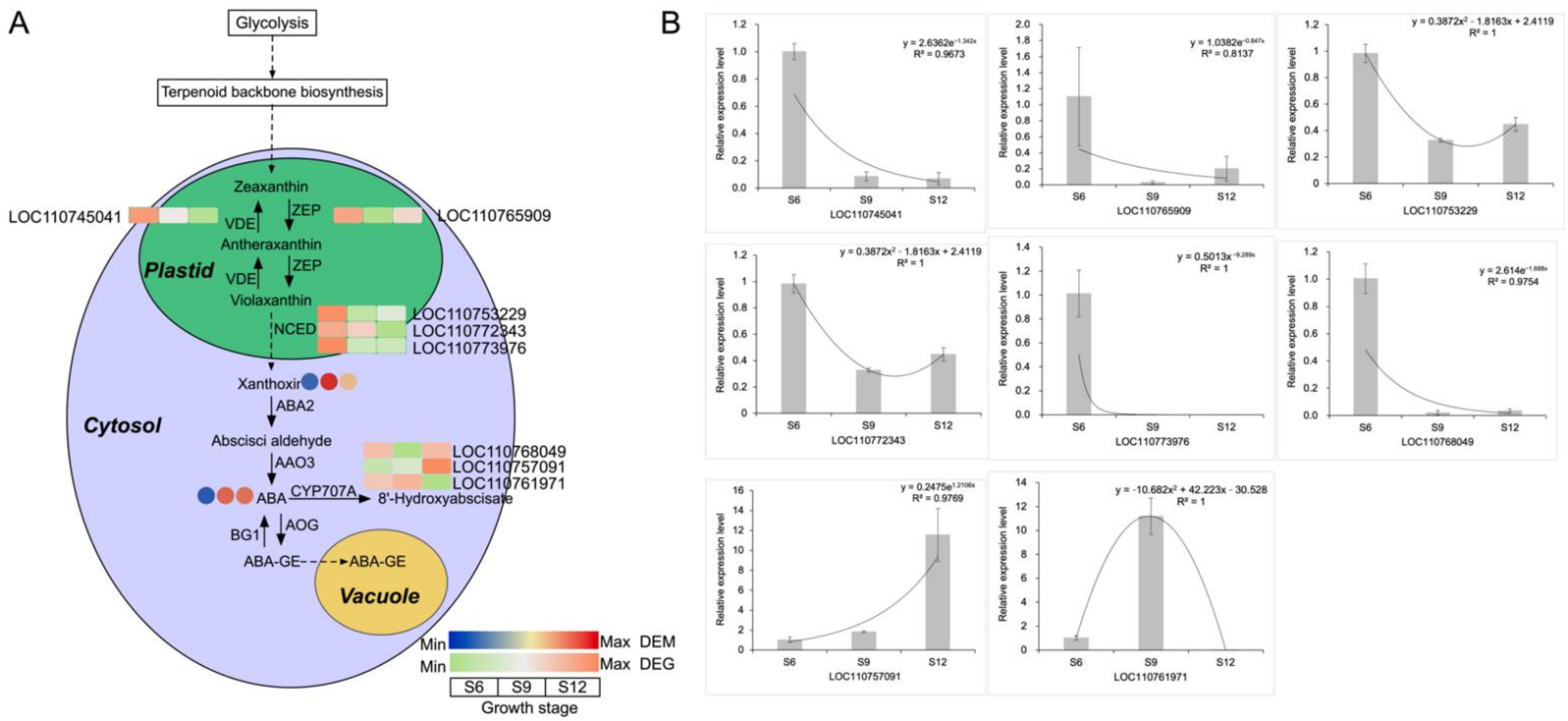
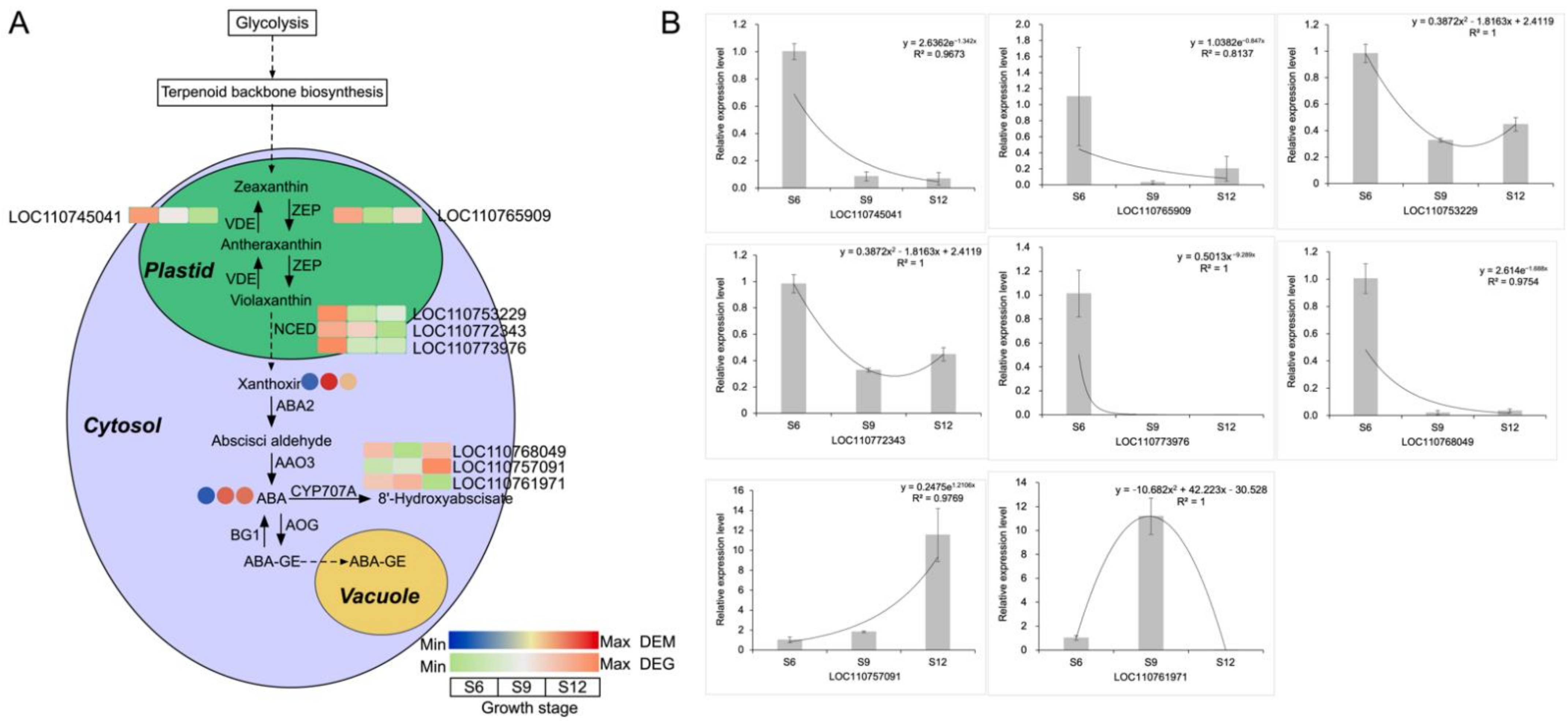
2.5.1. Analysis of the ABA Synthesis Pathway
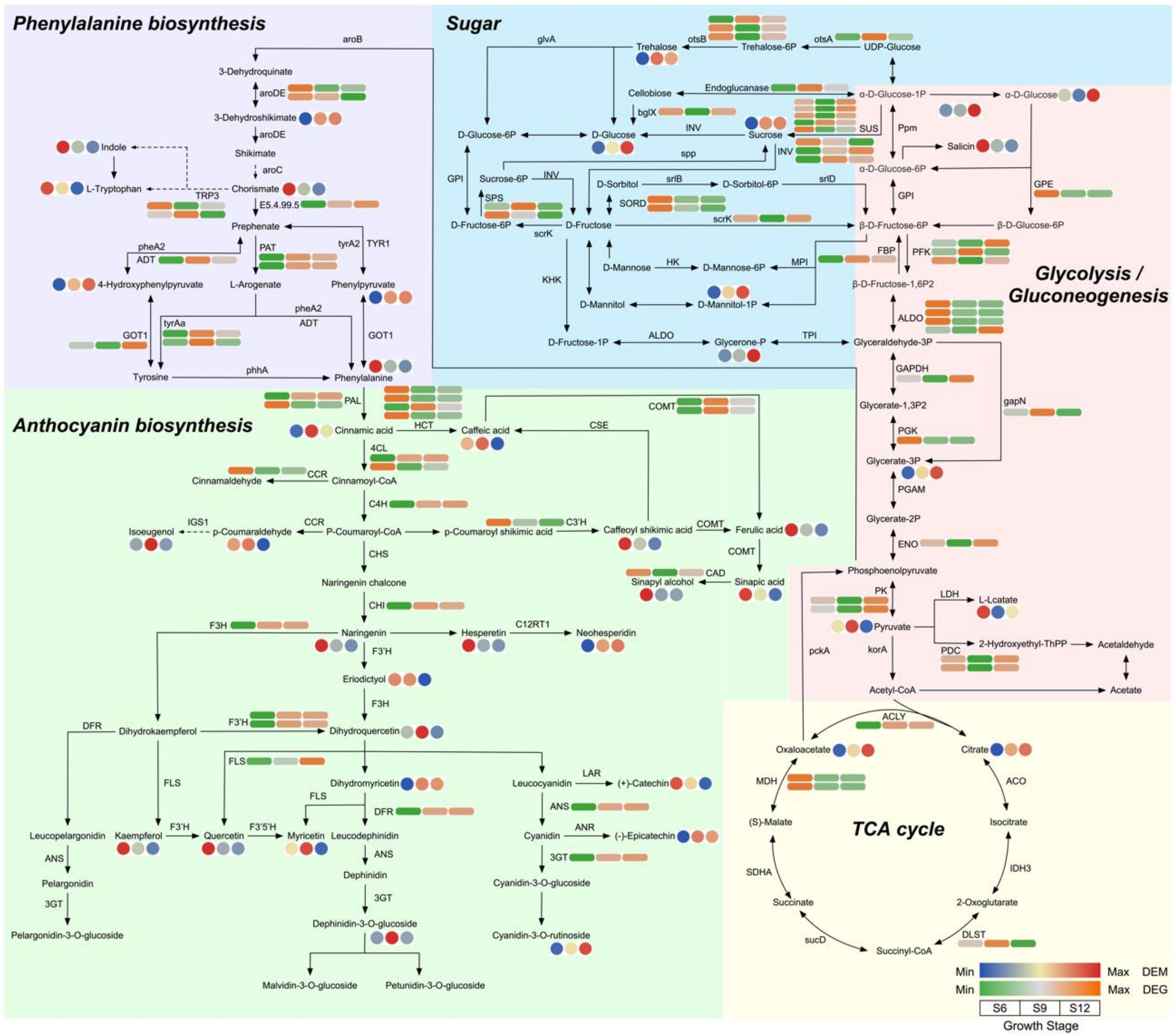
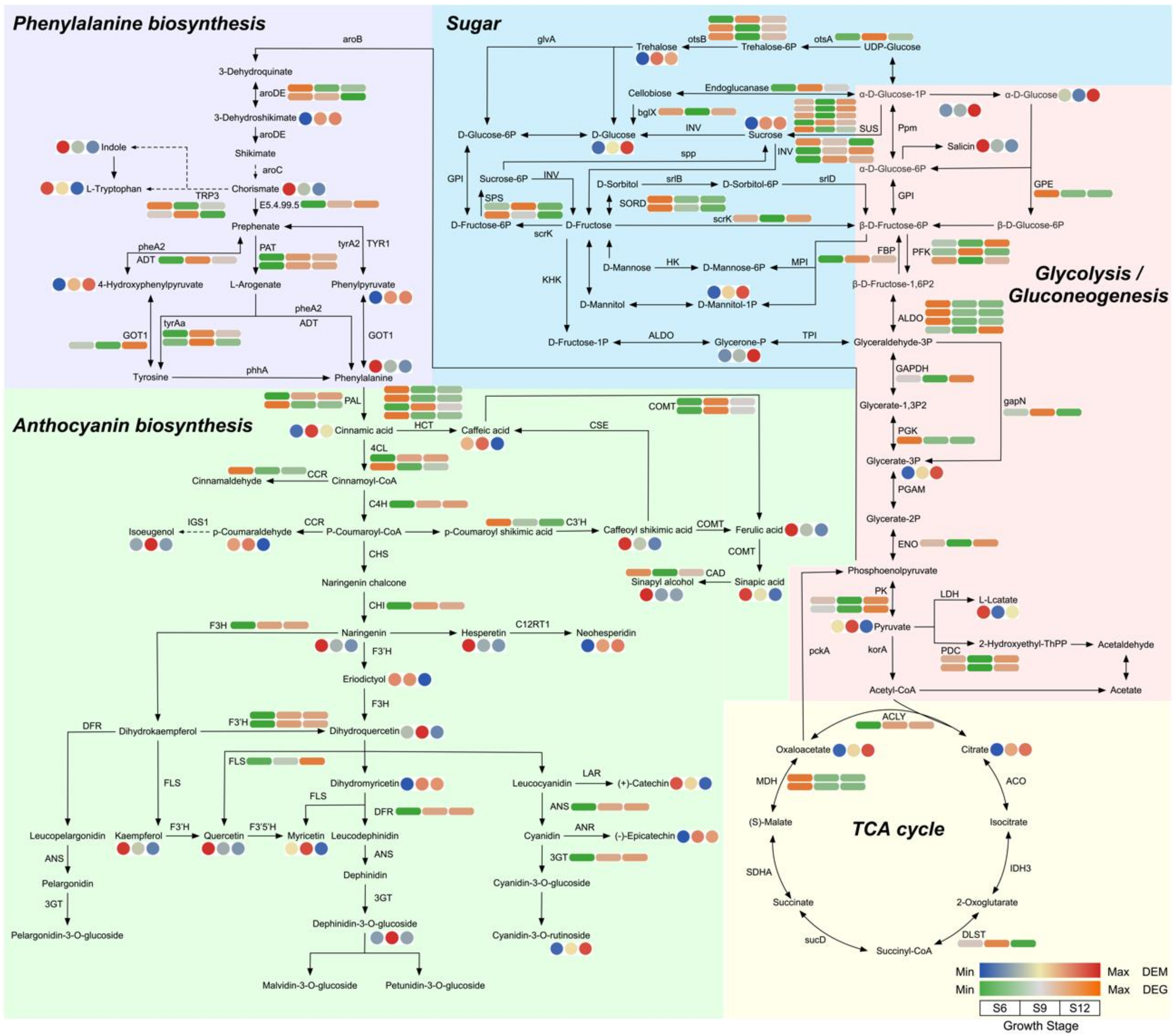
2.5.2. Metabolic Pathway Analysis
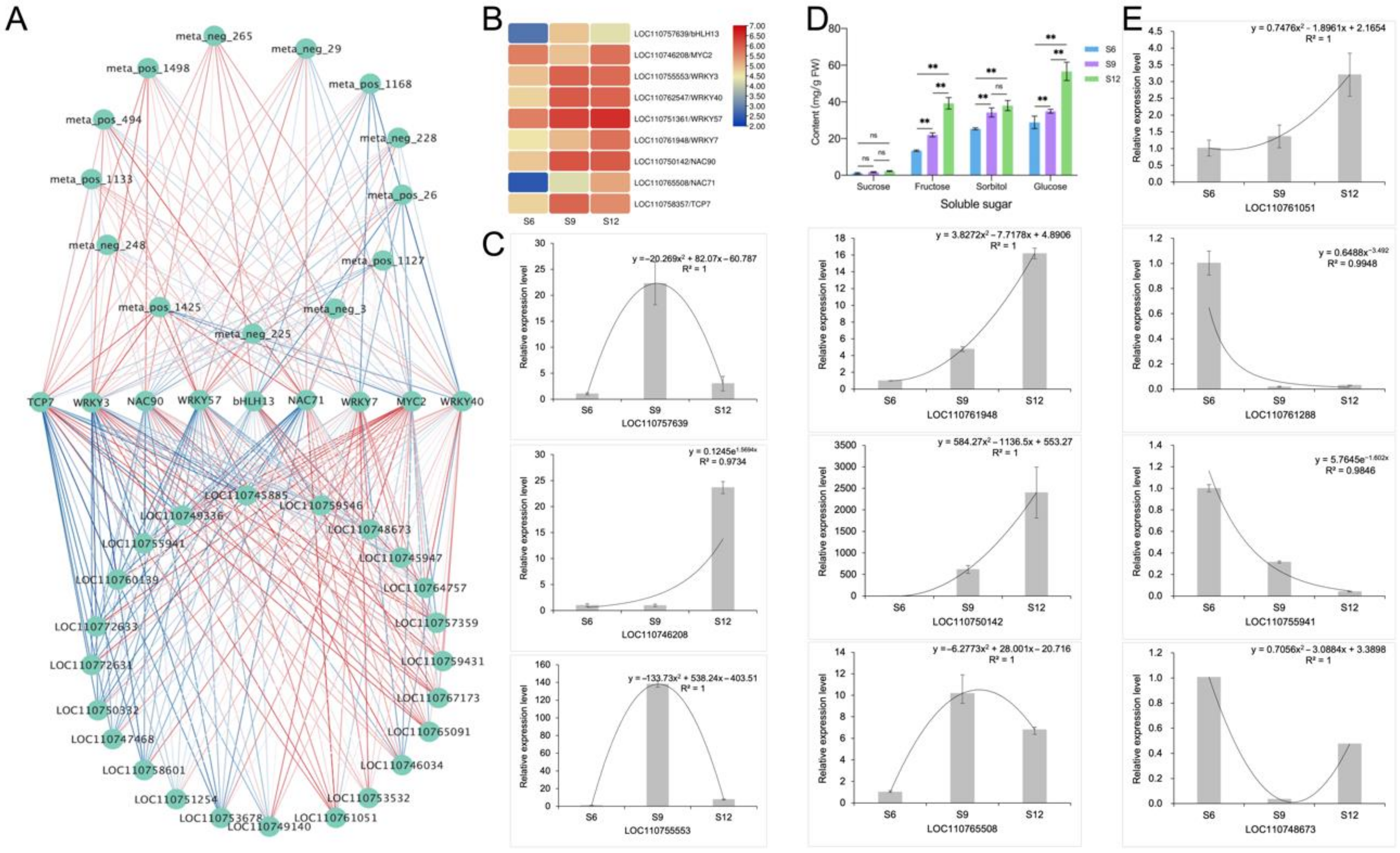
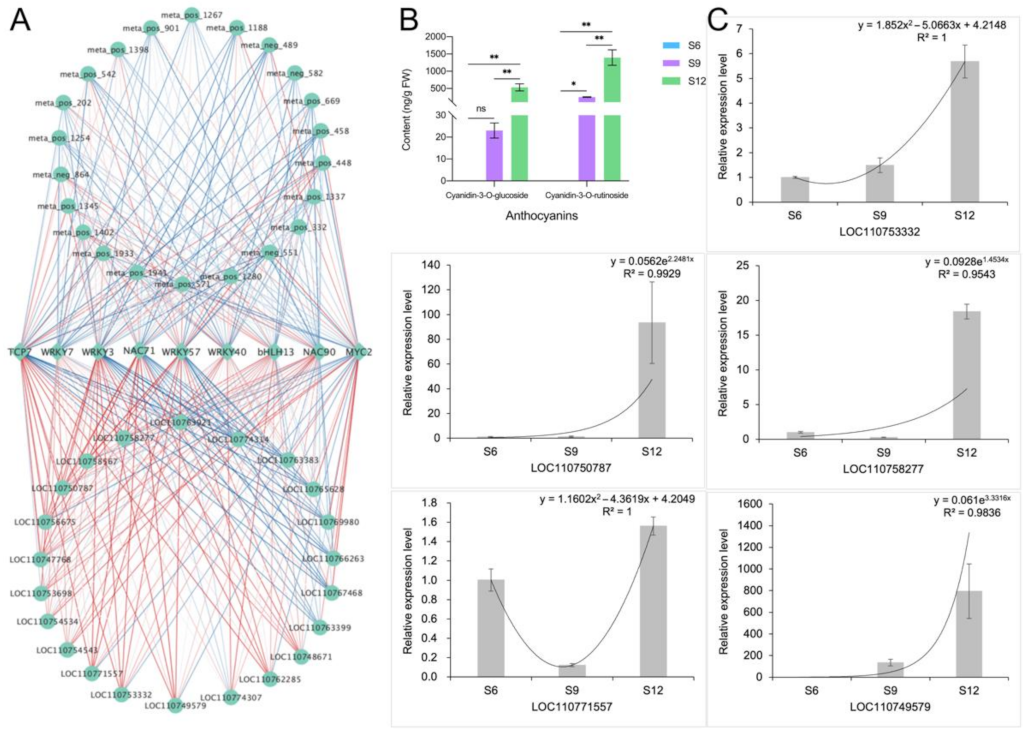
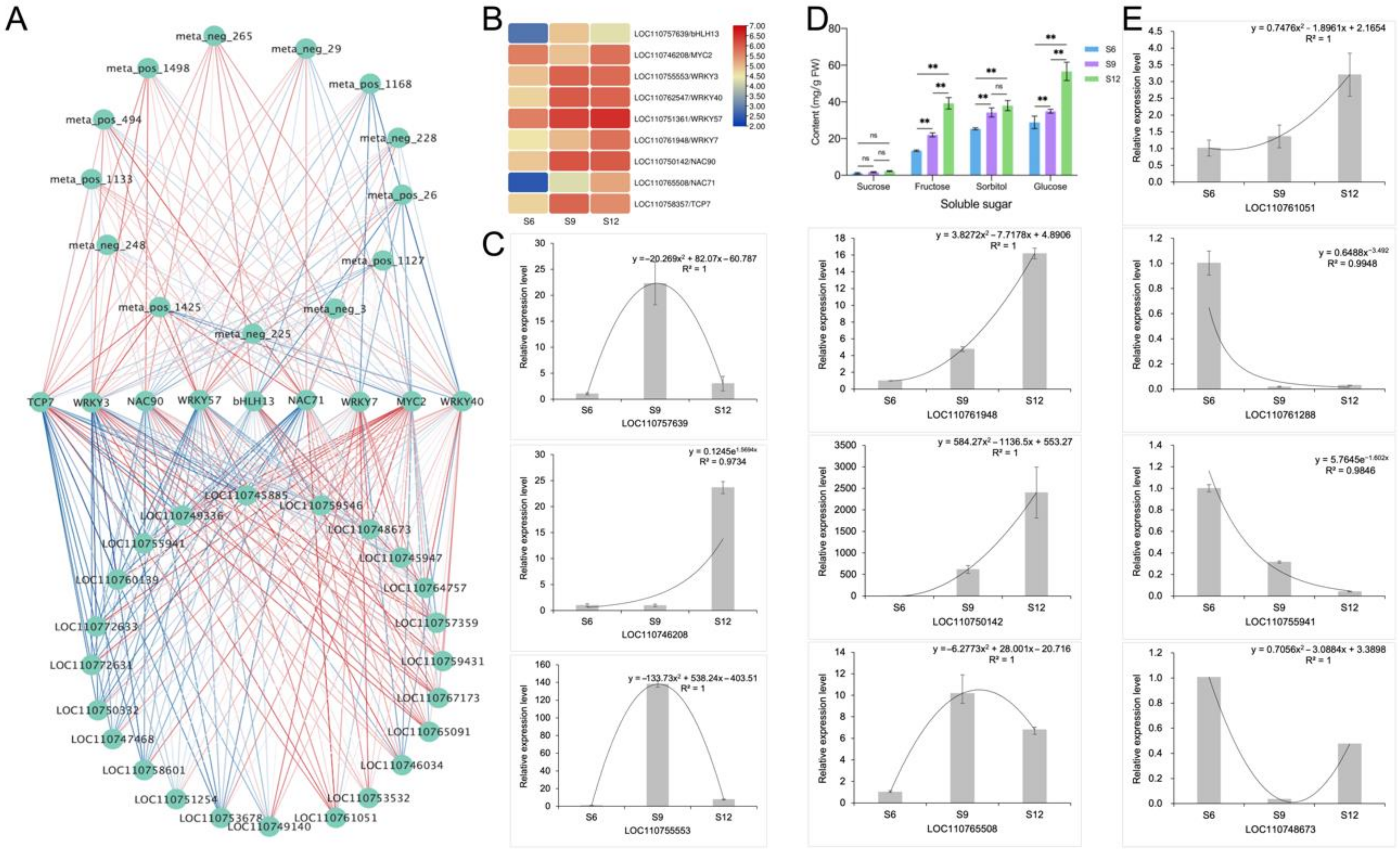
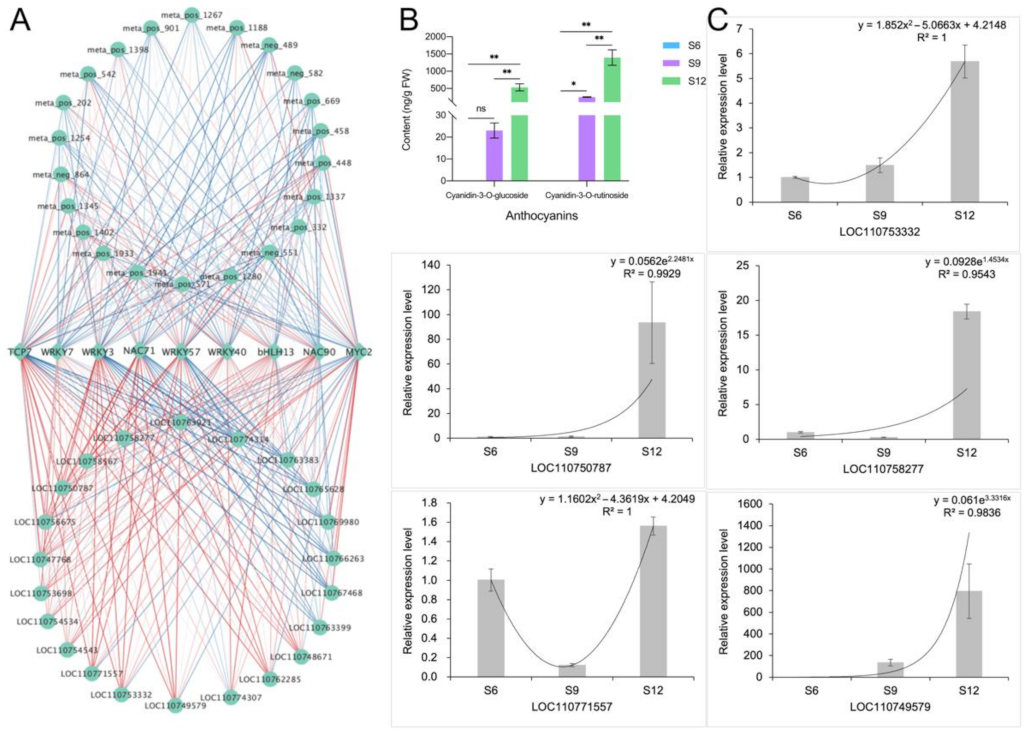
2.6. Correlation Network Analysis and Validation
2.6.1. Identification and Validation of Glucose Anabolic Correlation Networks
2.6.2. Identification and Characterization of Anthocyanin Synthesis Correlation Network
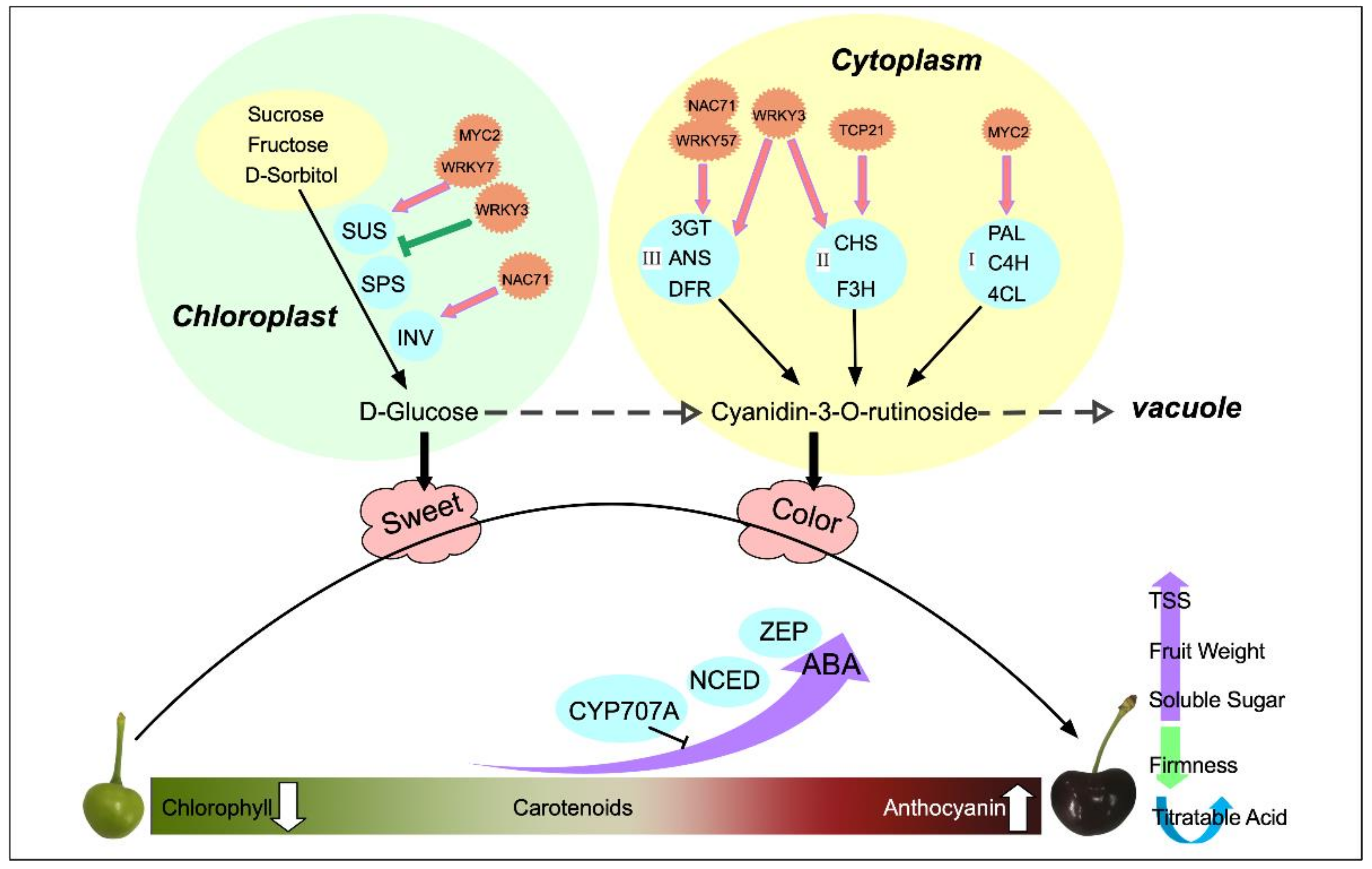
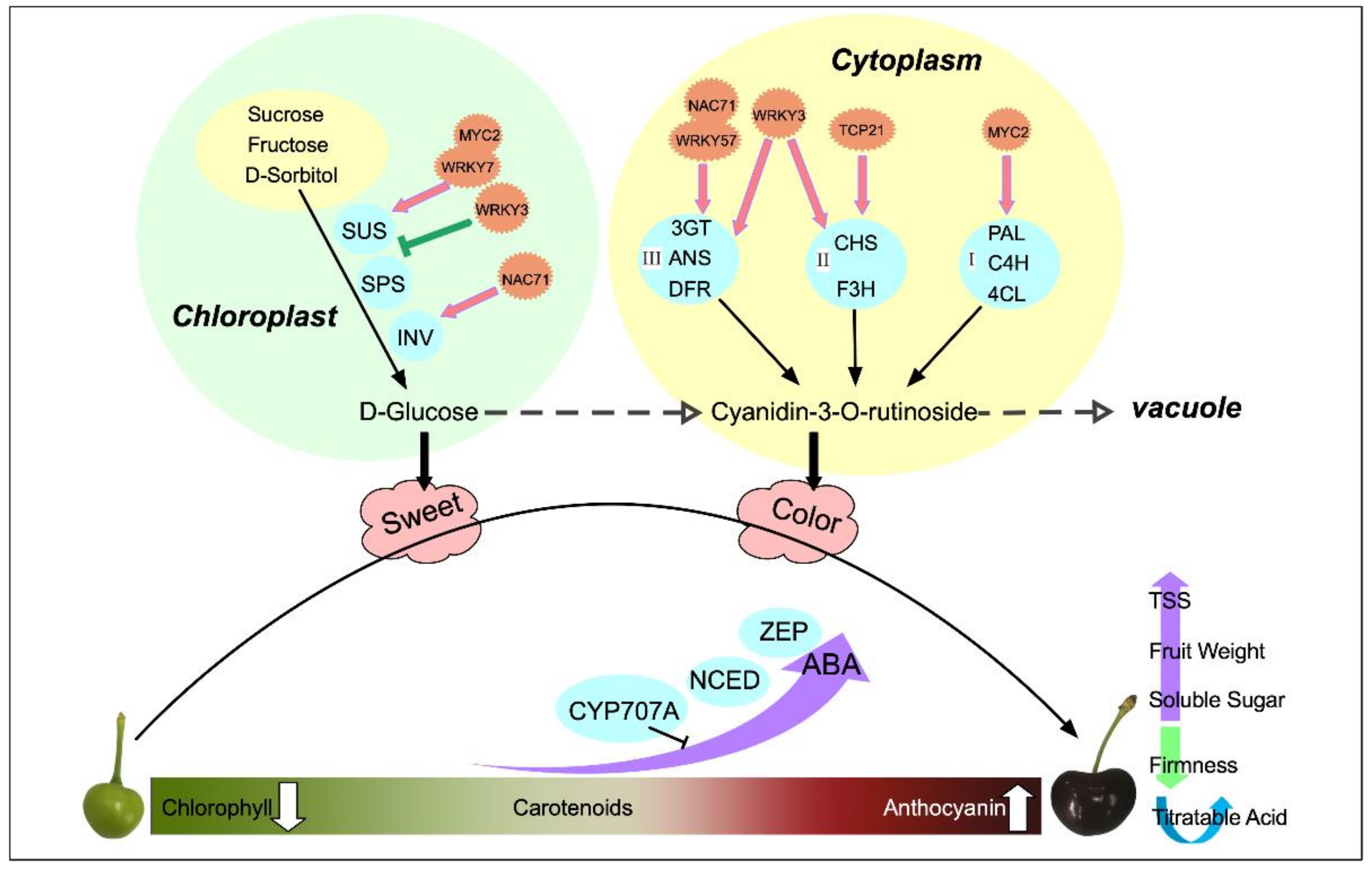
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Physiological Parameters
4.3. Metabolome Analysis
4.3.1. Metabolome Extraction
4.3.2. Metabolite Detection and Analysis
4.4. Transcriptome Sequencing
4.4.1. RNA Extraction and Transcriptome Sequencing Library Preparation
4.4.2. Data Processing and Analysis
4.4.3. Quantitative Real-Time PCR (qRT-PCR) Analysis
4.5. Combined Transcriptome and Metabolome Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Acero, N.; Gradillas, A.; Beltran, M.; García, A.; Mingarro, D.M. Comparison of phenolic compounds profile and antioxidant properties of different sweet cherry (Prunus avium L.) varieties. Food Chem. 2019, 279, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Valero, D.; Serrano, M. Growth and ripening stage at harvest modulates postharvest quality and bioactive compounds with antioxidant activity. Stewart Postharvest Rev. 2013, 3, 1–8. [Google Scholar] [CrossRef]
- Kader, A.A. Fruit maturity, ripening, and quality relationships. International Symposium Effect of Pre-& Postharvest Factors in Fruit Storage. Acta Hortic. 1999, 485, 203–208. [Google Scholar] [CrossRef]
- Alonso, M.A.S.; Paquin, J.P.; Mangin, J.P.L. Modelling perceived quality in fruit products: Their extrinsic and intrinsic attributes. J. Food Prod Mark. 2002, 8, 29–48. [Google Scholar] [CrossRef]
- Calle, A.; Wünsch, A. Multiple-population QTL mapping of maturity and fruit-quality traits reveals LG4 region as a breeding target in sweet cherry (Prunus avium L.). Hortic. Res. 2020, 7, 127. [Google Scholar] [CrossRef]
- Tijero, V.; Teribia, N.; Muñoz, P.; Munné-Bosch, S. Implication of abscisic acid on ripening and quality in sweet cherries: Differential effects during pre-and post-harvest. Front. Plant Sci. 2016, 7, 602. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Chen, P.; Dai, S.J.; Li, P.; Li, Q.; Ji, K.; Wang, Y.P.; Leng, P. Role of abscisic acid and ethylene in sweet cherry fruit maturation: Molecular aspects. N. Z. J. Crop Hortic. Sci. 2011, 39, 161–174. [Google Scholar] [CrossRef]
- Kuhn, N.; Maldonado, J.; Ponce, C.; Arellano, M.; Time, A.; Multari, S.; Martens, S.; Carrera, E.; Donoso, J.M.; Sagredo, B.; et al. RNAseq reveals different transcriptomic responses to GA3 in early and midseason varieties before ripening initiation in sweet cherry fruits. Sci. Rep. 2021, 11, 13075. [Google Scholar] [CrossRef]
- Saracoglu, O.; Ozturk, B.; Yildiz, K.; Kucuker, E. Pre-harvest methyl jasmonate treatments delayed ripening and improved quality of sweet cherry fruits. Sci. Hortic. 2017, 226, 19–23. [Google Scholar] [CrossRef]
- Wang, M.; Chen, L.; Liang, Z.; He, X.; Liu, W.; Jiang, B.; Yan, J.; Sun, P.; Cao, Z.; Peng, Q.; et al. Metabolome and transcriptome analyses reveal chlorophyll and anthocyanin metabolism pathway associated with cucumber fruit skin color. BMC Plant Biol. 2020, 20, 386. [Google Scholar] [CrossRef]
- Zhou, W.; Zhao, S.; Xu, M.; Niu, Y.; Nasier, M.; Fan, G.; Quan, S.; Zhang, S.; Wang, Y.; Liao, K. Identification of Key Genes Controlling Carotenoid Metabolism during Apricot Fruit Development by Integrating Metabolic Phenotypes and Gene Expression Profiles. J. Agric. Food Chem. 2021, 69, 9472–9483. [Google Scholar] [CrossRef]
- Ramos, P.; Parra-Palma, C.; Figueroa, C.R.; Zuñiga, P.E.; Valenzuela-Riffo, F.; Gonzalez, J.; Gaete-Eastman, C.; Morales-Quintana, L. Cell wall-related enzymatic activities and transcriptional profiles in four strawberry (Fragaria × ananassa) cultivars during fruit development and ripening. Sci. Hortic. 2018, 238, 325–332. [Google Scholar] [CrossRef]
- Wang, C.; Wang, Y.; Wang, M.; Han, H.; Luo, Y.; Ding, W.; Xu, W.; Zhong, Y.; Huang, H.; Qu, S. Soluble sugars accumulation and related gene expression during fruit development in Cucurbita maxima Duchesne. Sci. Hortic. 2020, 272, 109520. [Google Scholar] [CrossRef]
- Forlani, S.; Mizzotti, C.; Masiero, S. The NAC side of the fruit: Tuning of fruit development and maturation. BMC Plant Biol. 2021, 21, 238. [Google Scholar] [CrossRef]
- Yang, H.; Tian, C.; Li, X.; Gong, H.; Zhang, A. Transcriptome co-expression network analysis identifies key genes and regulators of sweet cherry anthocyanin biosynthesis. Horticulturae 2021, 7, 123. [Google Scholar] [CrossRef]
- He, Y.; Wang, Z.; Ge, H.; Liu, Y.; Chen, H. Weighted gene co-expression network analysis identifies genes related to anthocyanin biosynthesis and functional verification of hub gene SmWRKY44. Plant Sci. 2021, 309, 110935. [Google Scholar] [CrossRef]
- Huang, T.; Yu, D.; Wang, X. VvWRKY22 transcription factor interacts with VvSnRK1. 1/VvSnRK1. 2 and regulates sugar accumulation in grape. Biochem. Biophys. Res. Commun. 2021, 554, 193–198. [Google Scholar] [CrossRef]
- Umer, M.J.; Bin Safdar, L.; Gebremeskel, H.; Zhao, S.; Yuan, P.; Zhu, H.; Kaseb, M.O.; Anees, M.; Lu, X.; He, N.; et al. Identification of key gene networks controlling organic acid and sugar metabolism during watermelon fruit development by integrating metabolic phenotypes and gene expression profiles. Hortic. Res. 2020, 7, 193. [Google Scholar] [CrossRef]
- Wang, Z.; Cui, Y.; Vainstein, A.; Chen, S.; Ma, H. Regulation of fig (Ficus carica L.) fruit color: Metabolomic and transcriptomic analyses of the flavonoid biosynthetic pathway. Front. Plant Sci. 2017, 8, 1990. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Li, Y.; Lu, Q.; Lu, H.; Li, J. Combined Analysis of the Metabolome and Transcriptome Identified Candidate Genes Involved in Phenolic Acid Biosynthesis in the Leaves of Cyclocarya paliurus. Int. J. Mol. Sci. 2020, 21, 1337. [Google Scholar] [CrossRef] [Green Version]
- Hall, R.D.; D’Auria, J.C.; Ferreira, A.C.S.; Gibon, Y.; Kruszka, D.; Mishra, P.; Van de Zedde, R. High-throughput plant phenotyping: A role for metabolomics? Trends Plant Sci. 2022, 27, 549–563. [Google Scholar] [CrossRef]
- Veerappan, K.; Natarajan, S.; Chung, H.; Park, J. Molecular Insights of Fruit Quality Traits in Peaches, Prunus persica. Plants 2021, 10, 2191. [Google Scholar] [CrossRef]
- Guo, K.B.; Qiao, G.; Qiu, Z.L.; Wen, Z.; Yang, H.; Wen, X.P. The fruit dropping characters of sweet cherry and its interior causes in insufficient chilling zone. Russ J. Plant Physiol. 2020, 67, 94–102. [Google Scholar] [CrossRef]
- Zhang, C.; Whiting, M.D. Improving ‘Bing’ sweet cherry fruit quality with plant growth regulators. Sci. Hortic. 2011, 127, 341–346. [Google Scholar] [CrossRef]
- Teribia, N.; Tijero, V.; Munné-Bosch, S. Linking hormonal profiles with variations in sugar and anthocyanin contents during the natural development and ripening of sweet cherries. New Biotechnol. 2016, 33, 824–833. [Google Scholar] [CrossRef]
- Kondo, S.; Motoyama, M.; Michiyama, H.; Kim, M. Roles of jasmonic acid in the development of sweet cherries as measured from fruit or disc samples. Plant Growth Regul. 2002, 37, 37–44. [Google Scholar] [CrossRef]
- Srivastava, A.; Handa, A.K. Hormonal regulation of tomato fruit development: A molecular perspective. J. Plant Growth Regul. 2005, 24, 67–82. [Google Scholar] [CrossRef] [Green Version]
- McAtee, P.; Karim, S.; Schaffer, R.J.; David, K. A dynamic interplay between phytohormones is required for fruit development, maturation, and ripening. Front. Plant Sci. 2013, 4, 79. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Ge, C.; Ling, Y.; Mo, F.; Yang, M.; Jiang, L.; Chen, Q.; Lin, Y.; Sun, B.; Zhang, Y.; et al. ABA and sucrose co-regulate strawberry fruit ripening and show inhibition of glycolysis. Mol. Genet. Genom. 2020, 295, 421–438. [Google Scholar] [CrossRef]
- Luo, H.; Dai, S.; Ren, J.; Zhang, C.; Ding, Y.; Li, Z.; Sun, Y.; Ji, K.; Wang, Y.; Li, Q.; et al. The role of ABA in the maturation and postharvest life of a nonclimacteric sweet cherry fruit. J. Plant Growth Regul. 2014, 33, 373–383. [Google Scholar] [CrossRef]
- Zhang, M.; Yuan, B.; Leng, P. The role of ABA in triggering ethylene biosynthesis and ripening of tomato fruit. J. Exp. Bot. 2009, 60, 1579–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, L.B.; Zhang, D.; Li, Y.M.; Shen, Y.W.; Zhao, C.P.; Ma, J.J.; An, N.; Han, M.Y. Transcription profiles reveal sugar and hormone signaling pathways mediating flower induction in apple (Malus domestica Borkh.). Plant Cell Physiol. 2015, 56, 2052–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellarin, S.D.; Gambetta, G.A.; Wada, H.; Shackel, K.A.; Matthews, M.A. Fruit ripening in Vitis vinifera: Spatiotemporal relationships among turgor, sugar accumulation, and anthocyanin biosynthesis. J. Exp. Bot. 2011, 62, 4345–4354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, H.; Yun, S.K.; Yoon, I.K.; Nam, E.Y.; Kwon, J.H.; Jun, J.H. Assessment of organic acid and sugar composition in apricot, plumcot, plum, and peach during fruit development. J. Appl. Bot. Food Qual. 2014, 87, 24–29. [Google Scholar] [CrossRef]
- Duan, Y.; Yang, L.; Zhu, H.; Zhou, J.; Sun, H.; Gong, H. Structure and Expression Analysis of Sucrose Phosphate Synthase, Sucrose Synthase and Invertase Gene Families in Solanum lycopersicum. Int. J. Mol. Sci. 2021, 22, 4698. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, L.; Song, C.; Leng, X.; Kayesh, E.; Sun, X.; Fang, J. Mining and comparison of the genes encoding the key enzymes involved in sugar biosynthesis in apple, grape, and sweet orange. Sci. Hortic. 2014, 165, 311–318. [Google Scholar] [CrossRef]
- Zhang, D.; Lee, D.J.; Tippetts, B.J.; Lillywhite, K.D. Date maturity and quality evaluation using color distribution analysis and back projection. J. Food Eng. 2014, 131, 161–169. [Google Scholar] [CrossRef]
- Crecente-Campo, J.; Nunes-Damaceno, M.; Romero-Rodríguez, M.A.; Vázquez-Odériz, M.L. Color, anthocyanin pigment, ascorbic acid and total phenolic compound determination in organic versus conventional strawberries (Fragaria × ananassa Duch, cv Selva). J. Food Compos. Anal. 2012, 28, 23–30. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, N.; Ma, Z.; Che, F.; Mao, J.; Chen, B. The Changes in Color, Soluble Sugars, Organic Acids, Anthocyanins and Aroma Components in “Starkrimson” during the Ripening Period in China. Molecules 2016, 21, 812. [Google Scholar] [CrossRef] [Green Version]
- Revilla, E.; Garcίa-Beneytez, E.; Cabello, F.; Martίn-Ortega, G.; Ryan, J.M. Value of high-performance liquid chromatographic analysis of anthocyanins in the differentiation of red grape cultivars and red wines made from them. J. Chromatogr. A 2001, 915, 53–60. [Google Scholar] [CrossRef]
- Blackhall, M.L.; Berry, R.; Davies, N.W.; Walls, J.T. Optimized extraction of anthocyanins from Reid Fruits’ Prunus avium ‘Lapins’ cherries. Food Chem. 2018, 256, 280–285. [Google Scholar] [CrossRef]
- Singh, K.; Kumar, A.; Kajal, M.; Singh, B. Characterization and expression analysis of chalcone synthase and chalcone isomerase genes in Phyllanthus emblica (L.). J. Plant Biochem. Biot. 2019, 28, 105–113. [Google Scholar] [CrossRef]
- Kaur, R.; Aslam, L.; Kapoor, N.; Mahajan, R. Identification and comparative expression analysis of chalcone synthase, flavanone 3-hydroxylase and dihydroflavonol 4-reductase genes in wild pomegranate (Punica granatum L.) organs. Braz. J. Bot. 2020, 43, 883–896. [Google Scholar] [CrossRef]
- Shi, M.; Ali, M.M.; He, Y.; Ma, S.; Rizwan, H.M.; Yang, Q.; Li, B.; Lin, Z.; Chen, F. Flavonoids Accumulation in Fruit Peel and Expression Profiling of Related Genes in Purple (Passiflora edulis f. edulis) and Yellow (Passiflora edulis f. flavicarpa) Passion Fruits. Plants 2021, 10, 2240. [Google Scholar] [CrossRef]
- Jin, W.; Wang, H.; Li, M.; Wang, J.; Yang, Y.; Zhang, X.; Yan, G.; Zhang, H.; Liu, J.; Zhang, K. The R2R3 MYB transcription factor PavMYB10.1 involves in anthocyanin biosynthesis and determines fruit skin colour in sweet cherry (Prunus avium L.). Plant Biotechnol. J. 2016, 14, 2120–2133. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.; Huang, Z.; Hua, Q.; Shan, W.; Kuang, J.; Lu, W.; Qin, Y.; Chen, J. The WRKY transcription factor HpWRKY44 regulates CytP450-like1 expression in red pitaya fruit (Hylocereus polyrhizus). Hortic. Res. 2017, 4, 17039. [Google Scholar] [CrossRef] [Green Version]
- Martín-Pizarro, C.; Vallarino, J.G.; Osorio, S.; Meco, V.; Urrutia, M.; Pillet, J.; Casañal, A.; Merchante, C.; Amaya, I.; Willmitzer, L.; et al. The NAC transcription factor FaRIF controls fruit ripening in strawberry. Plant Cell 2021, 33, 1574–1593. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, S.; Wang, X.; Mao, T.; Bao, M.; Zhang, J.; Zhang, J. Genome-wide identification and characterization of the bHLH gene family in an ornamental woody plant Prunus mume. Hortic. Plant J. 2022, in press. [Google Scholar] [CrossRef]
- Luo, D.L.; Ba, L.J.; Shan, W.; Kuang, J.F.; Lu, W.J.; Chen, J.Y. Involvement of WRKY transcription factors in abscisic-acid-induced cold tolerance of banana fruit. J. Agric. Food Chem. 2017, 65, 3627–3635. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Zhang, Y.; Zhang, A.; You, C.X. Regulation of fleshy fruit ripening: From transcription factors to epigenetic modifications. Hortic. Res. 2022, 9, uhac013. [Google Scholar] [CrossRef]
- Manzoor, M.A.; Manzoor, M.M.; Li, G.; Abdullah, M.; Han, W.; Wenlong, H.; Shakoor, A.; Riaz, W.R.; Rehman, S.; Cai, Y. Genome-wide identification and characterization of bZIP transcription factors and their expression profile under abiotic stresses in Chinese pear (Pyrus bretschneideri). BMC Plant Biol. 2021, 21, 413. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, C.; Chen, H.; Yang, W.; Li, J.; Tang, W.; Gong, R. Transcriptomic and Metabolomic Analysis of Quality Changes during Sweet Cherry Fruit Development and Mining of Related Genes. Int. J. Mol. Sci. 2022, 23, 7402. https://doi.org/10.3390/ijms23137402
Chen C, Chen H, Yang W, Li J, Tang W, Gong R. Transcriptomic and Metabolomic Analysis of Quality Changes during Sweet Cherry Fruit Development and Mining of Related Genes. International Journal of Molecular Sciences. 2022; 23(13):7402. https://doi.org/10.3390/ijms23137402
Chicago/Turabian StyleChen, Chaoqun, Hongxu Chen, Wenlong Yang, Jie Li, Wenjing Tang, and Ronggao Gong. 2022. "Transcriptomic and Metabolomic Analysis of Quality Changes during Sweet Cherry Fruit Development and Mining of Related Genes" International Journal of Molecular Sciences 23, no. 13: 7402. https://doi.org/10.3390/ijms23137402
APA StyleChen, C., Chen, H., Yang, W., Li, J., Tang, W., & Gong, R. (2022). Transcriptomic and Metabolomic Analysis of Quality Changes during Sweet Cherry Fruit Development and Mining of Related Genes. International Journal of Molecular Sciences, 23(13), 7402. https://doi.org/10.3390/ijms23137402