
Interrelation between α-Cardiac Actin Treadmilling and Myocardin-Related Transcription Factor-A Nuclear Shuttling in Cardiomyocytes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
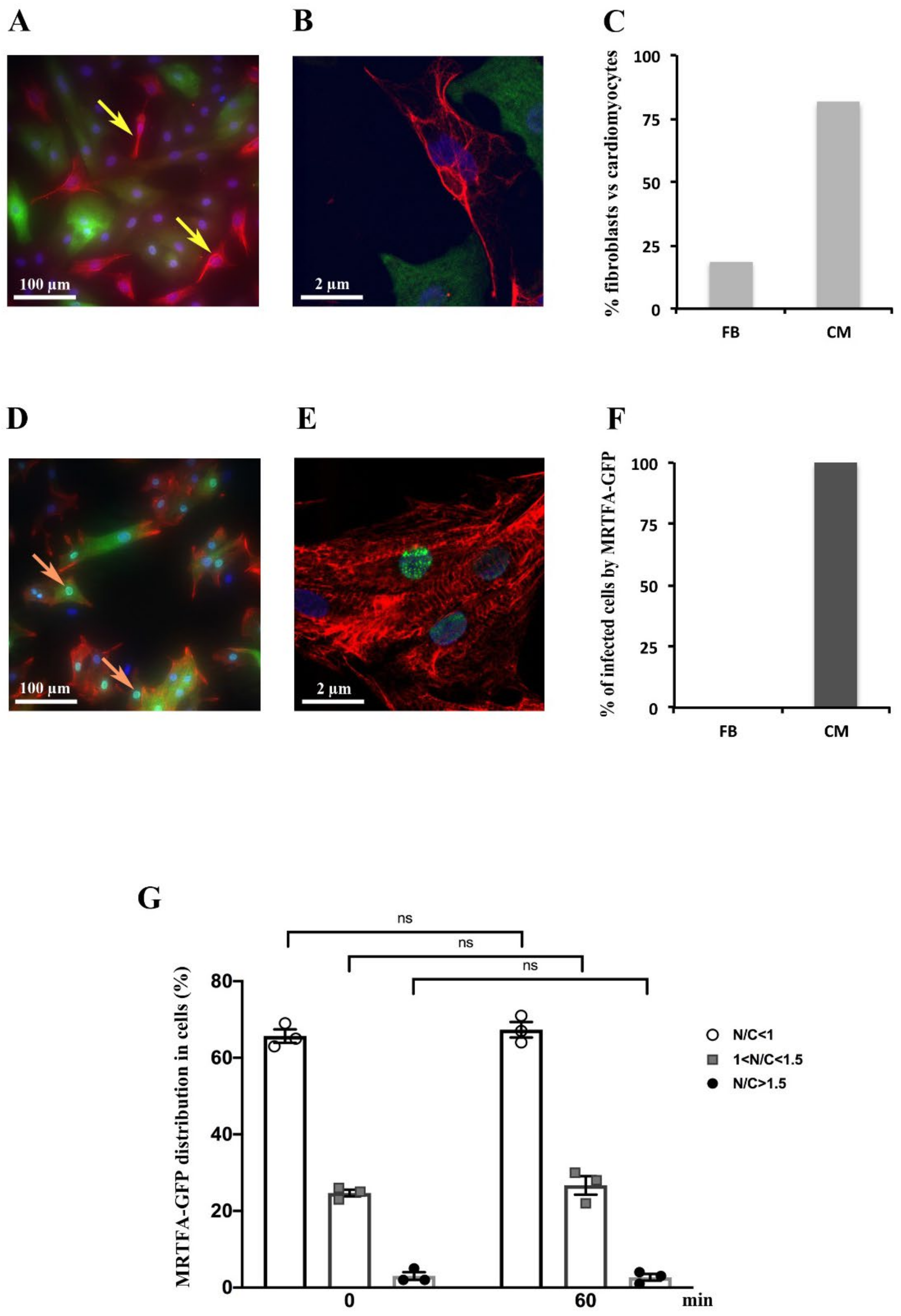
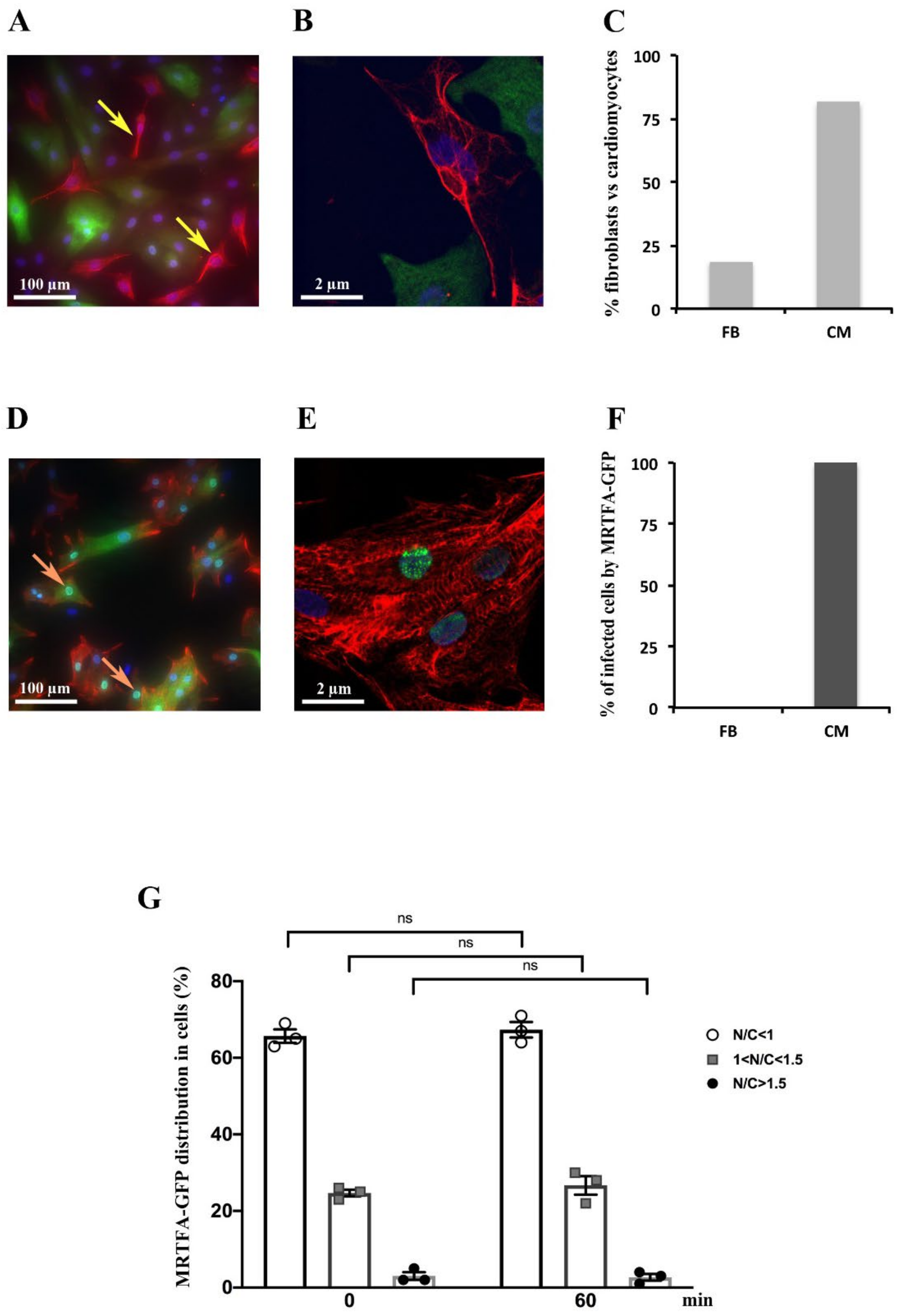
2.1. Detection of MRTFA-GFP in Cardiomyocytes but Not in Fibroblasts
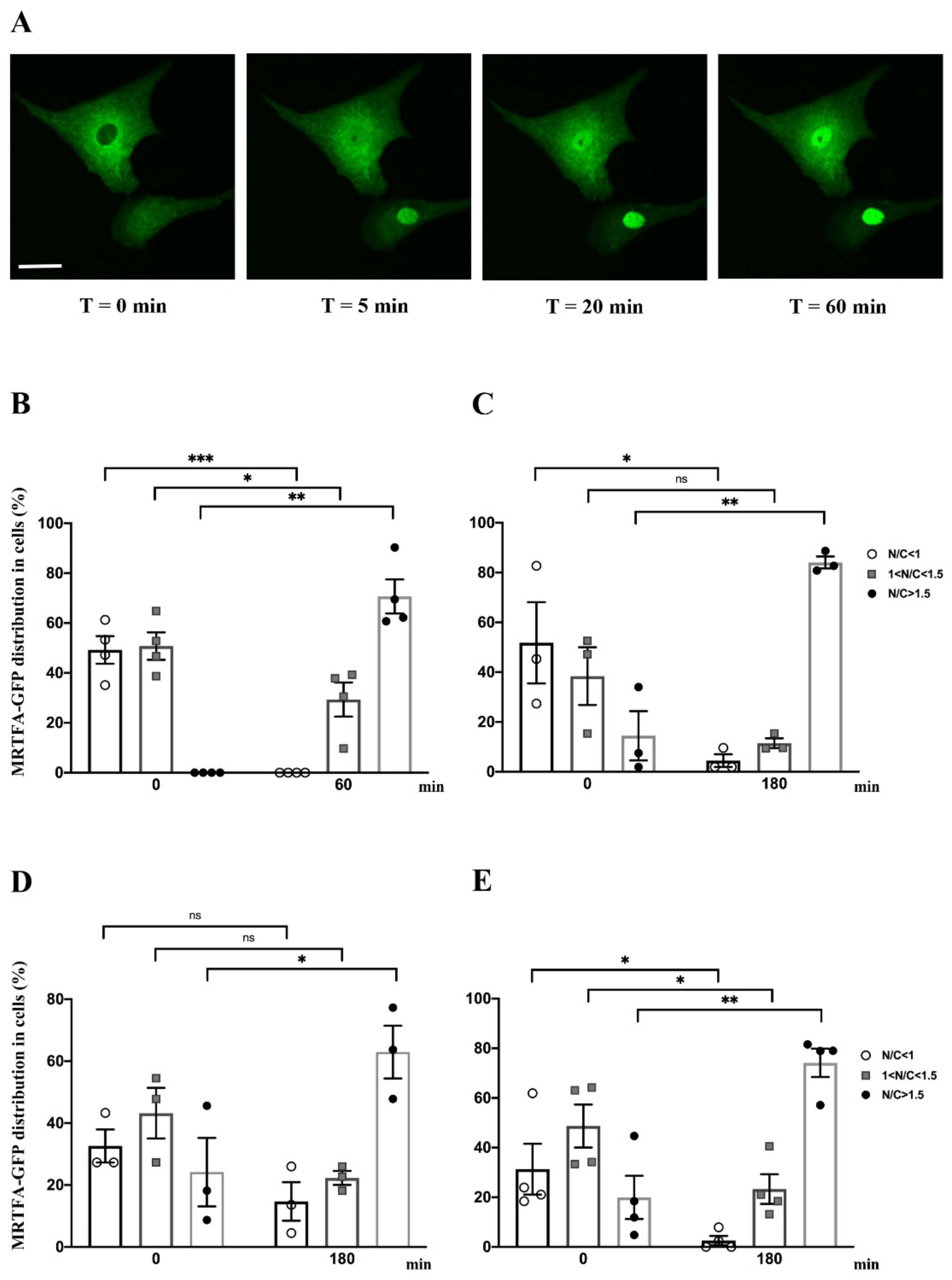
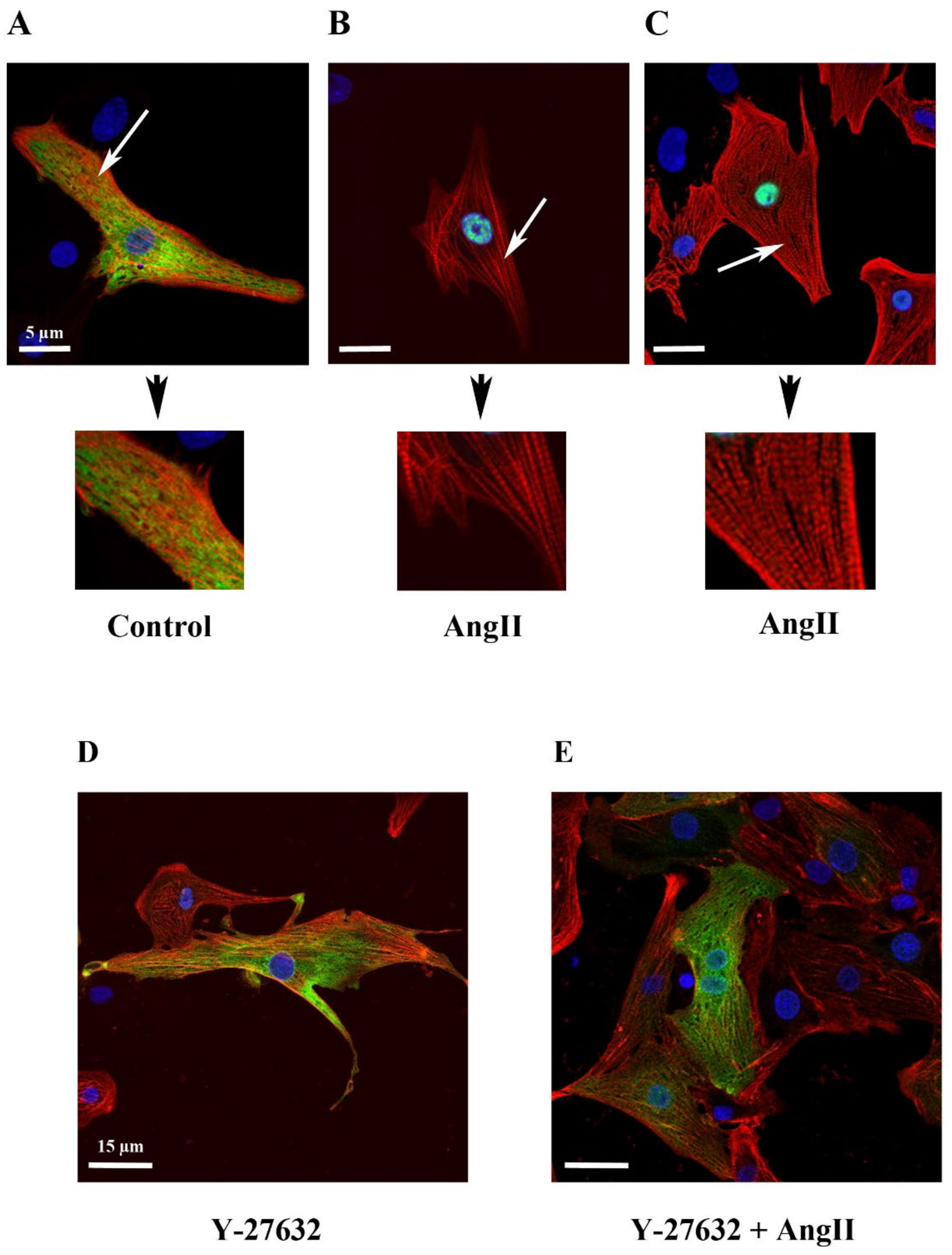
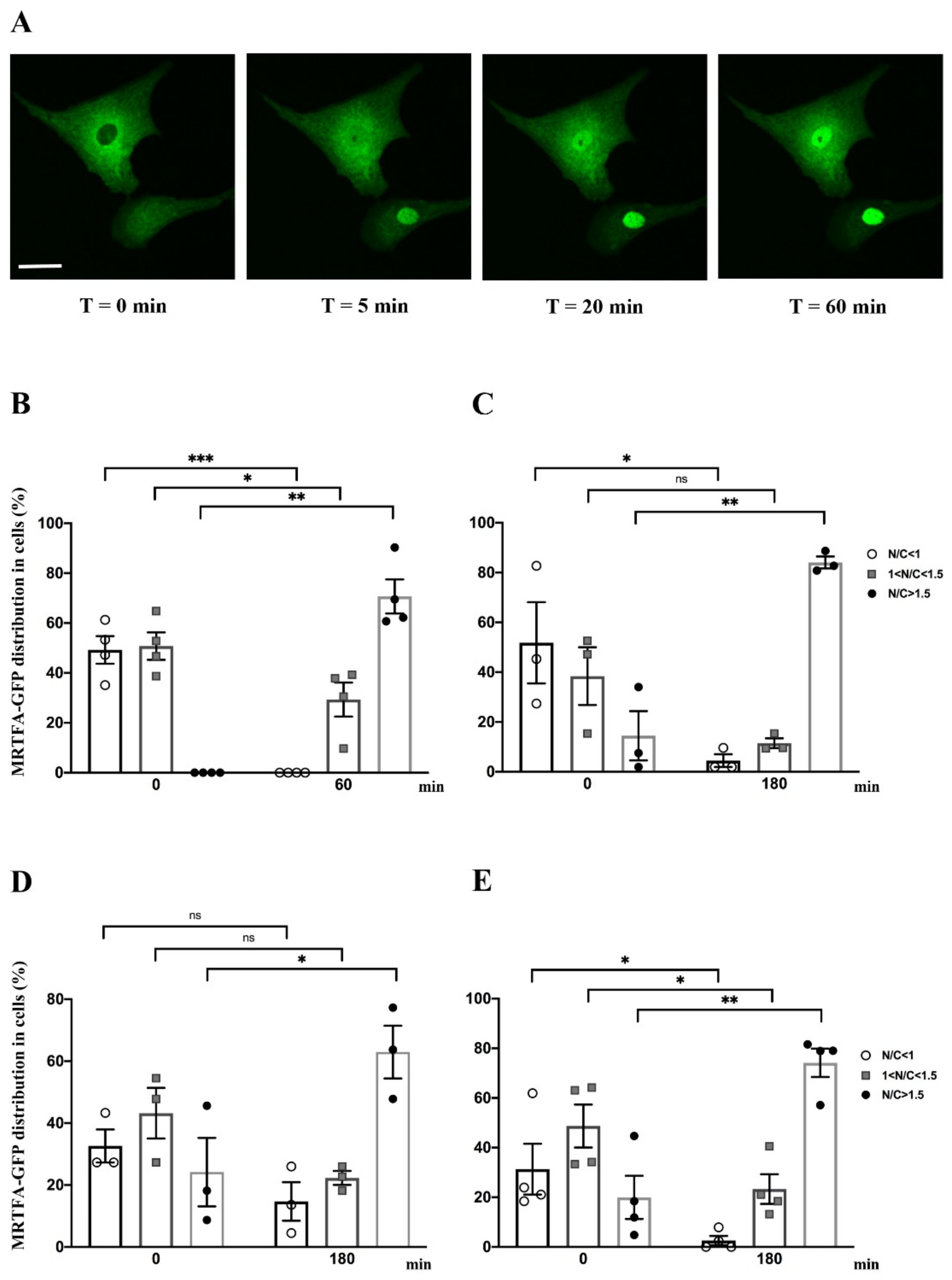
2.2. Effect of Serum, Endothelin-1, Phenylephrine or Angiotensin II on MRTFA Nucleocytoplasmic Shuttling in Cardiac Cells
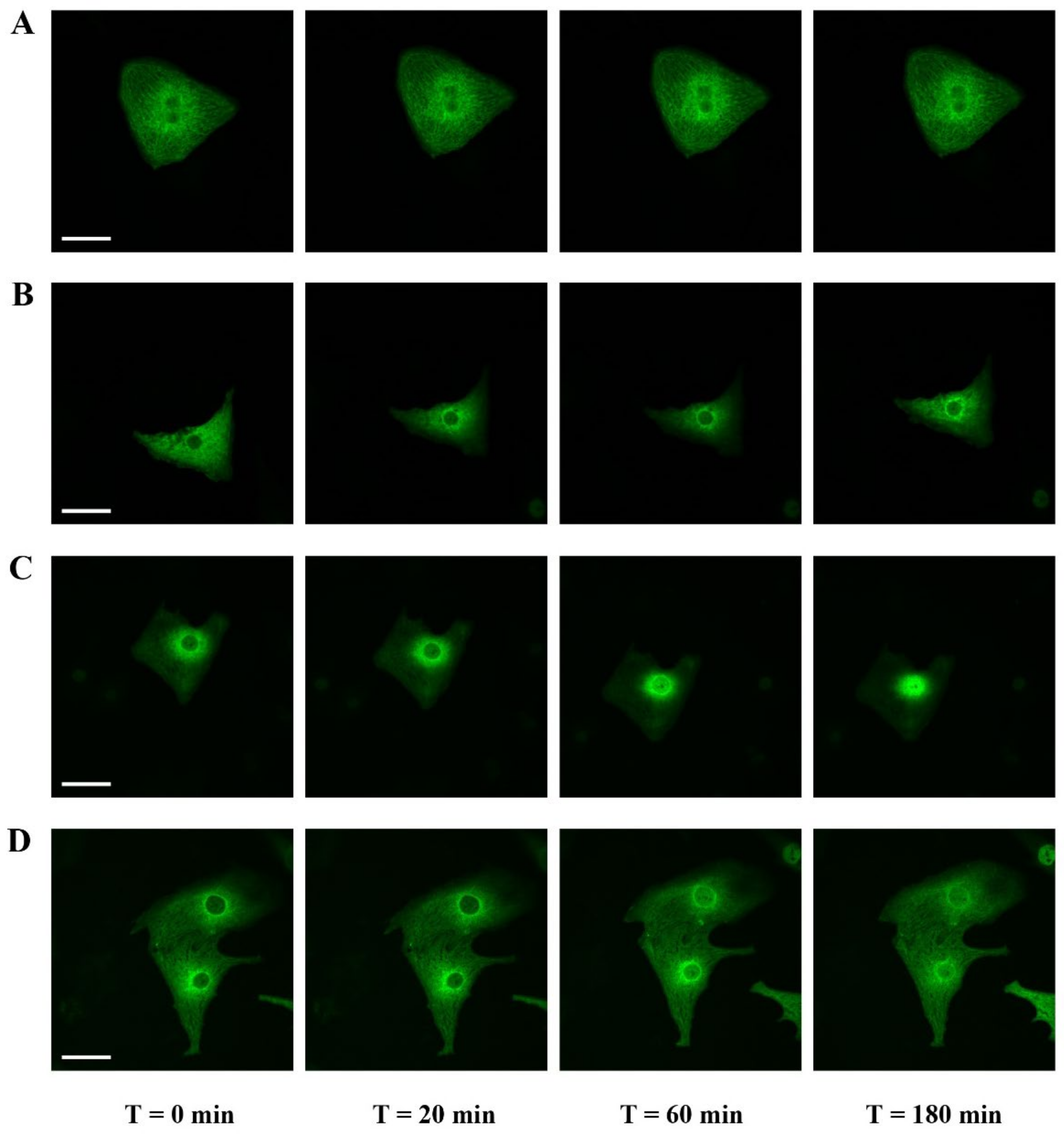
2.3. Effect of Y-27632, a ROCK Inhibitor, on MRTFA Shuttling in Cultured Cardiomyocytes
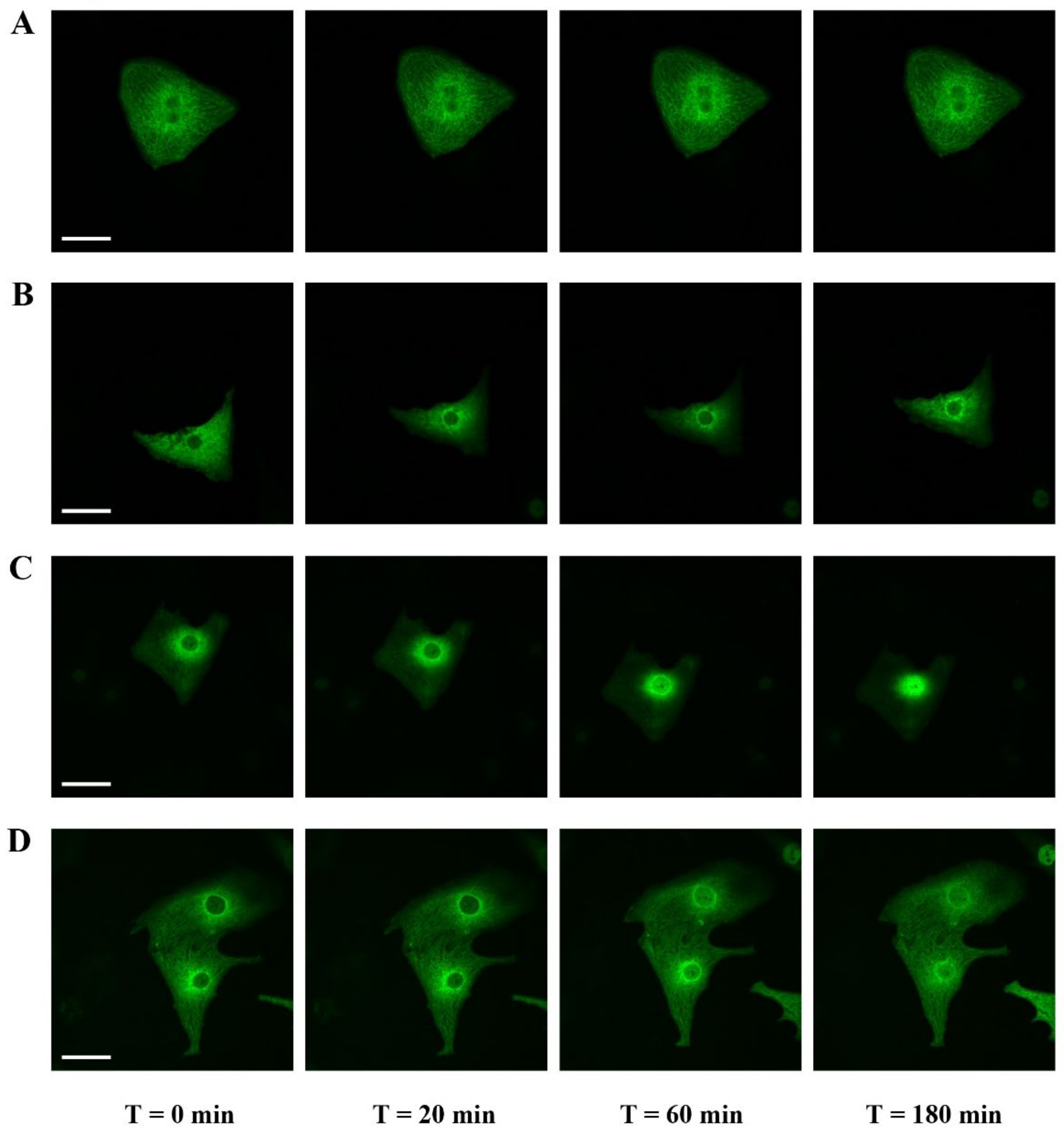
2.4. Filamentous Actin Dynamics in Relation to MRTFA Shuttling in Cultured Cardiomyocytes
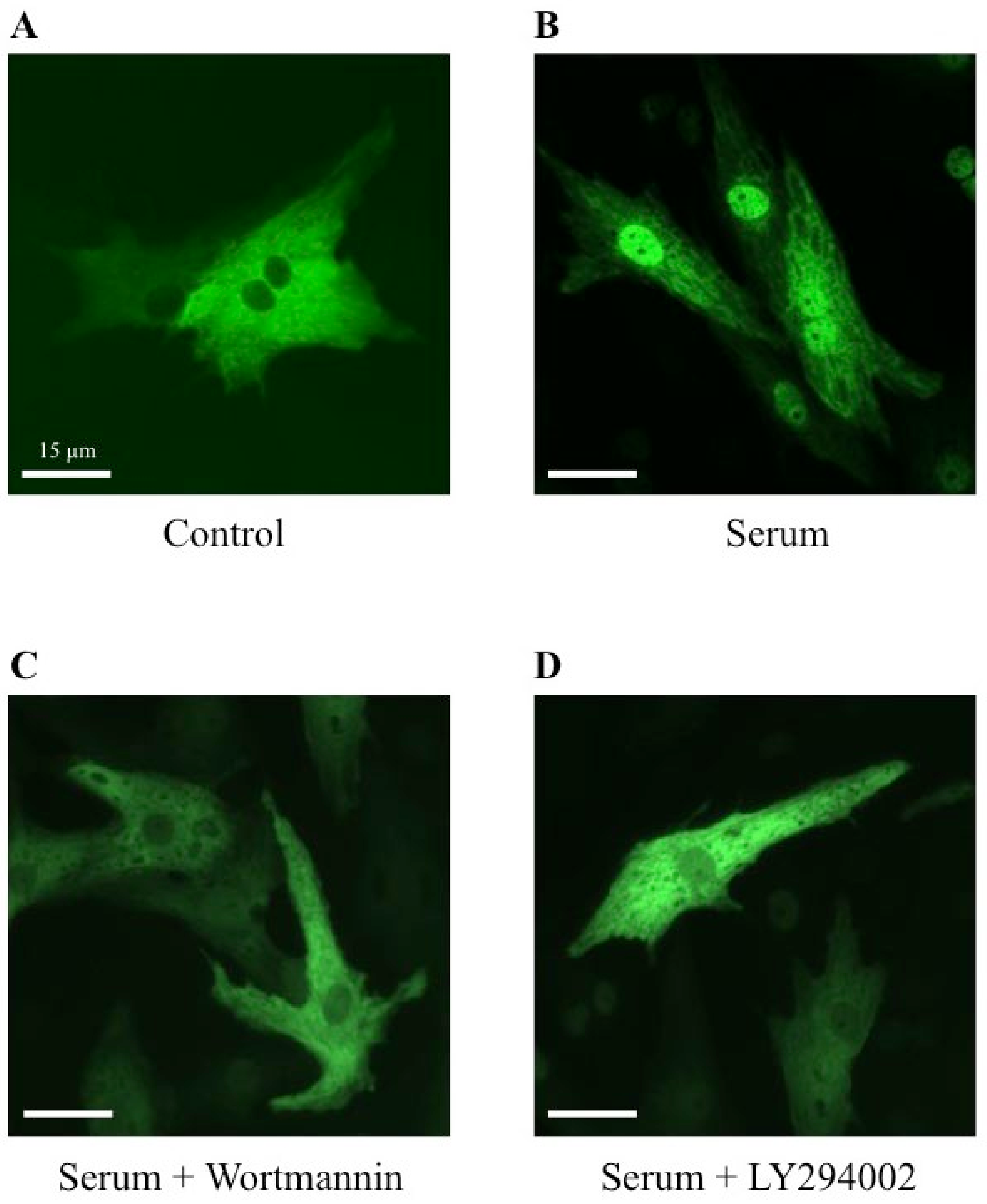
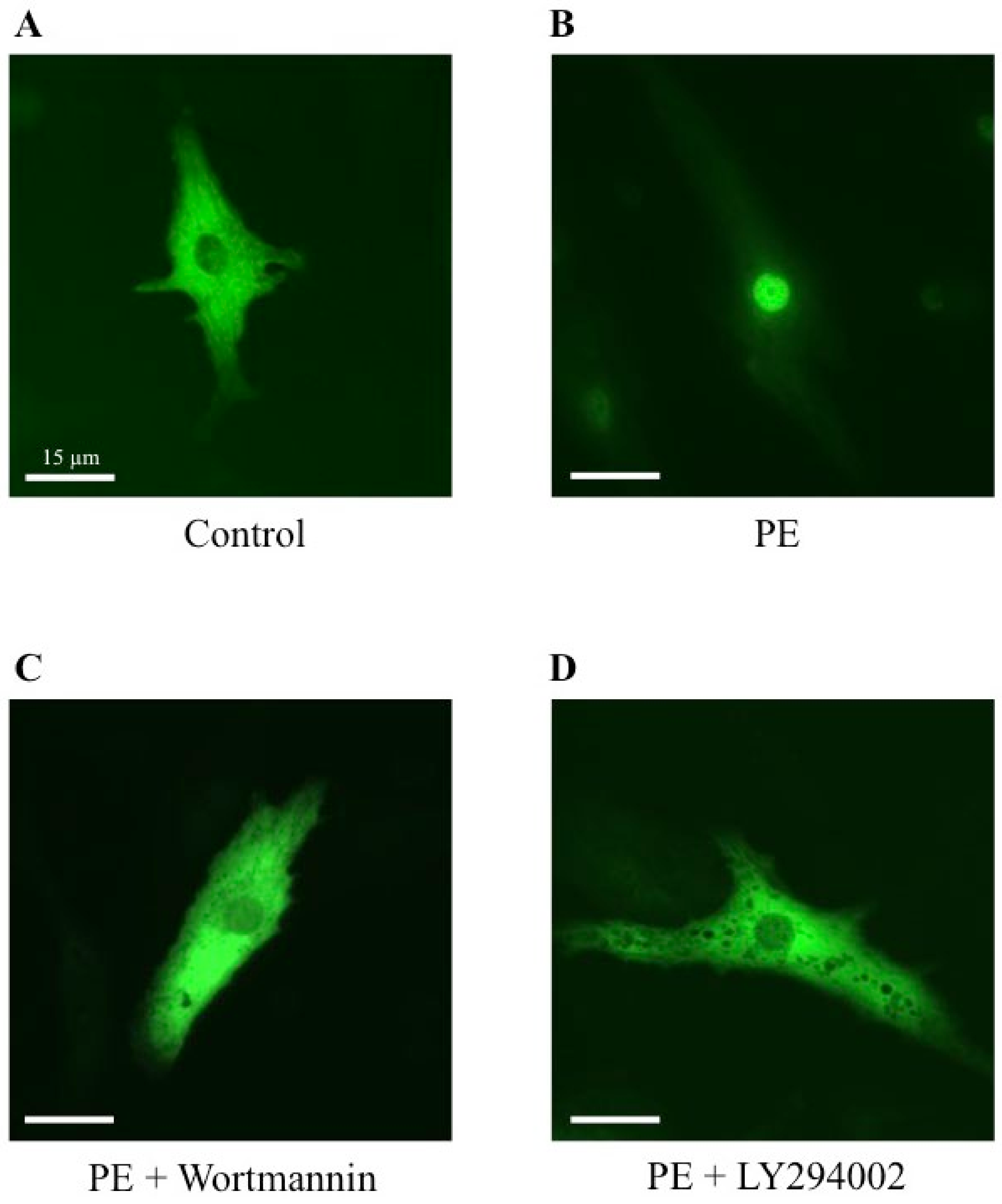
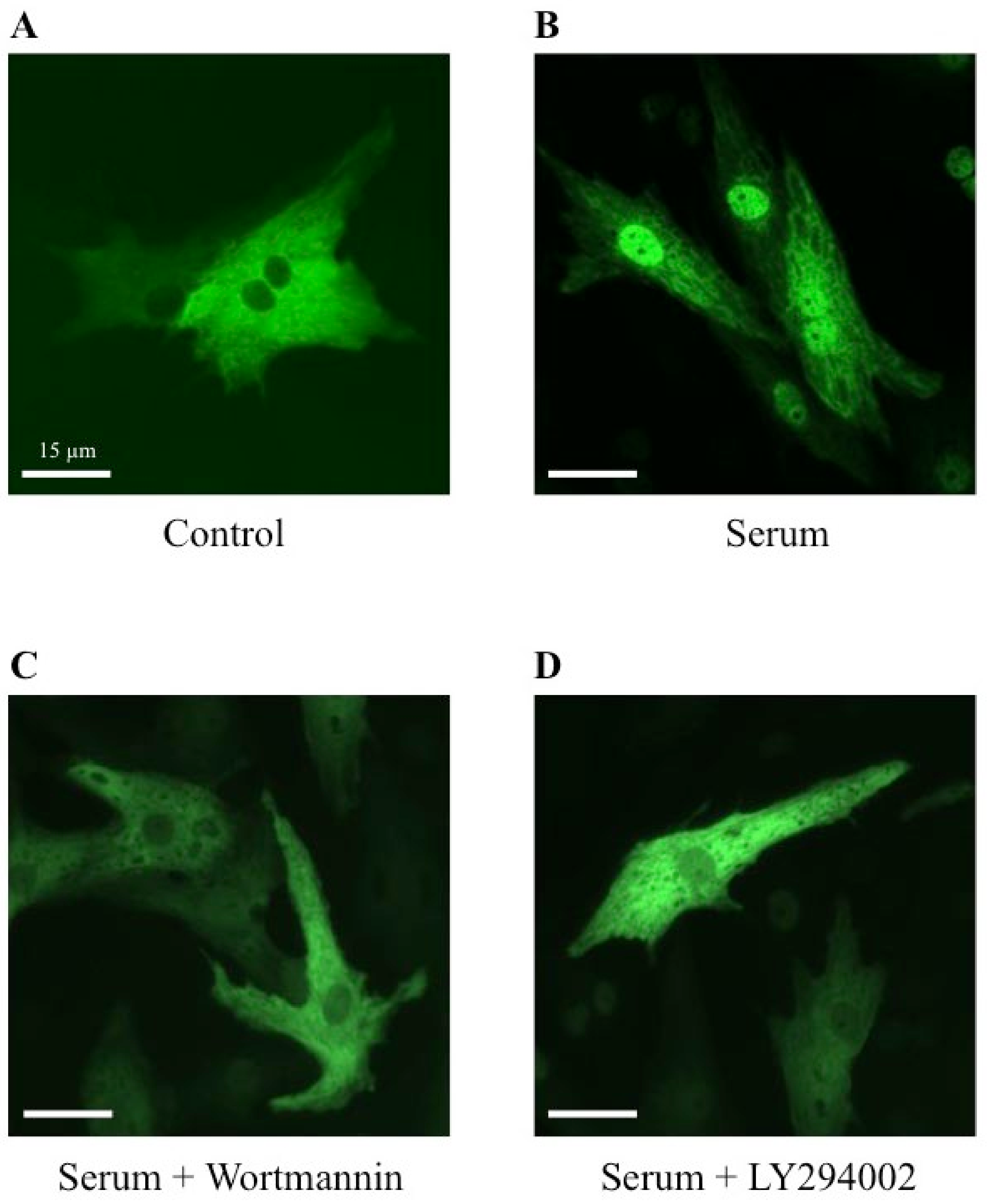
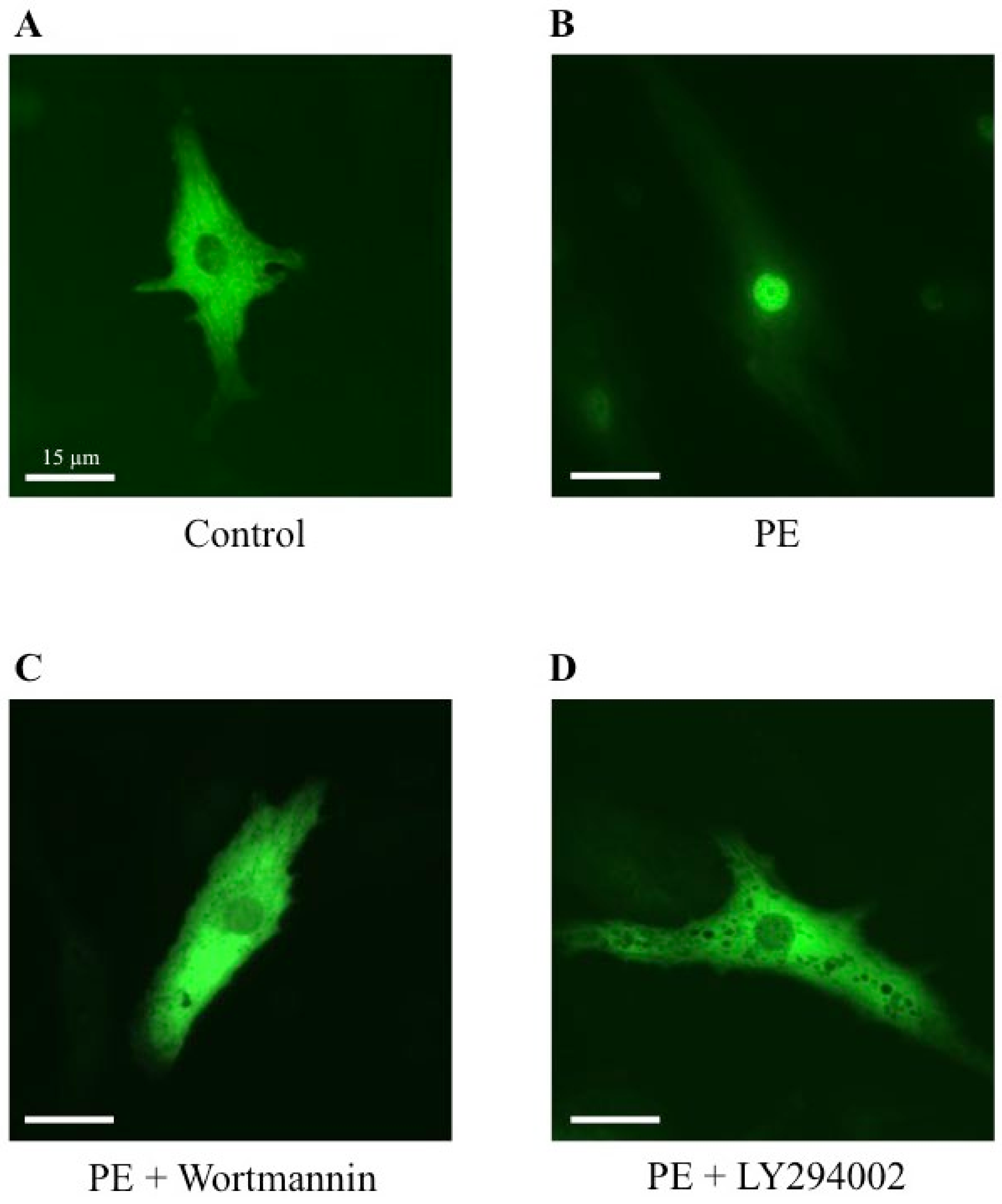
2.5. Effect of Wortmannin and LY294002, Two PI3K Inhibitors, on MRTFA Shuttling in Cultured Cardiomyocytes
3. Discussion
4. Materials and Methods
4.1. Isolation of Neonatal Rat Cardiomyocytes
4.2. Culture Conditions
4.3. Fluorescence Microscopy
4.4. Quantification of MRTFA-GFP Distribution
4.5. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NRCs | Neonatal rat cardiomyocytes |
| MRTFA | Myocardin-related transcription factor-A |
| SRF | Serum response factor |
| Ang II | Angiotensin II |
| PE | Phenylephrine |
| ET-1 | Endothelin-1 |
| ROCK | Rho-associated protein kinase |
| PI3K | Phosphoinositide 3-kinase |
| GFP | Green fluorescent protein |
| DMEM | Dulbecco’s modified Eagle’ |
| HS | Horse serum |
| FCS | Fetal calf serum |
References
- Parlakian, A.; Charvet, C.; Escoubet, B.; Mericskay, M.; Molkentin, J.D.; Gary-Bobo, G.; De Windt, L.J.; Ludosky, M.A.; Paulin, D.; Daegelen, D.; et al. Temporally controlled onset of dilated cardiomyopathy through disruption of the SRF gene in adult heart. Circulation 2005, 112, 2930–2939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Chai, J.; Azhar, G.; Sheridan, P.; Borras, A.M.; Furr, M.C.; Khrapko, K.; Lawitts, J.; Misra, R.P.; Wei, J.Y. Early postnatal cardiac changes and premature death in transgenic mice overexpressing a mutant form of serum response factor. J. Biol. Chem. 2001, 276, 40033–40040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotiropoulos, A.; Gineitis, D.; Copeland, J.; Treisman, R. Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell 1999, 98, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Vartiainen, M.K.; Guettler, S.; Larijani, B.; Treisman, R. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 2007, 316, 1749–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diguet, N.; Mallat, Y.; Ladouce, R.; Clodic, G.; Prola, A.; Tritsch, E.; Blanc, J.; Larcher, J.C.; Delcayre, C.; Samuel, J.L.; et al. Muscle creatine kinase deficiency triggers both actin depolymerization and desmin disorganization by advanced glycation end products in dilated cardiomyopathy. J. Biol. Chem. 2011, 286, 35007–35019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokalled, M.H.; Carroll, K.J.; Cenik, B.K.; Chen, B.; Liu, N.; Olson, E.N.; Bassel-Duby, R. Myocardin-related transcription factors are required for cardiac development and function. Dev. Biol. 2015, 406, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Touvron, M.; Escoubet, B.; Mericskay, M.; Angelini, A.; Lamotte, L.; Santini, M.P.; Rosenthal, N.; Daegelen, D.; Tuil, D.; Decaux, J.F. Locally expressed IGF1 propeptide improves mouse heart function in induced dilated cardiomyopathy by blocking myocardial fibrosis and SRF-dependent CTGF induction. Dis. Models Mech. 2012, 5, 481–491. [Google Scholar]
- Wang, D.Z.; Li, S.; Hockemeyer, D.; Sutherland, L.; Wang, Z.; Schratt, G.; Richardson, J.A.; Nordheim, A.; Olson, E.N. Potentiation of serum response factor activity by a family of myocardin-related transcription factors. Proc. Natl. Acad. Sci. USA 2002, 99, 14855–14860. [Google Scholar] [CrossRef] [Green Version]
- Baarlink, C.; Wang, H.; Grosse, R. Nuclear actin network assembly by formins regulates the SRF coactivator MAL. Science 2013, 340, 864–867. [Google Scholar] [CrossRef]
- Miralles, F.; Posern, G.; Zaromytidou, A.I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Olson, E.N.; Nordheim, A. Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 2010, 11, 353–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouilleron, S.; Guettler, S.; Langer, C.A.; Treisman, R.; McDonald, N.Q. Molecular basis for G-actin binding to RPEL motifs from the serum response factor coactivator MAL. EMBO J. 2008, 27, 3198–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staus, D.P.; Weise-Cross, L.; Mangum, K.D.; Medlin, M.D.; Mangiante, L.; Taylor, J.M.; Mack, C.P. Nuclear RhoA signaling regulates MRTF-dependent SMC-specific transcription. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H379–H390. [Google Scholar] [CrossRef] [PubMed]
- Blajecka, K.; Marinov, M.; Leitner, L.; Uth, K.; Posern, G.; Arcaro, A. Phosphoinositide 3-Kinase C2β Regulates RhoA and the Actin Cytoskeleton through an Interaction with Dbl. PLoS ONE 2012, 7, e44945. [Google Scholar] [CrossRef]
- Brachmann, S.M.; Yballe, C.M.; Innocenti, M.; Deane, J.A.; Fruman, D.A.; Thomas, S.M.; Cantley, L.C. Role of phosphoinositide 3-kinase regulatory isoforms in development and actin rearrangement. Mol. Cell Biol. 2005, 25, 2593–2606. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, C.; Portela, R.A.; Mellado, M.; Rodríguez-Frade, J.M.; Collard, J.; Serrano, A.; Martínez-A, C.; Avila, J.; Carrera, A.C. Role of the PI3K regulatory subunit in the control of actin organization and cell migration. J. Cell Biol. 2000, 151, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Komuro, I.; Katoh, Y.; Kaida, T.; Shibazaki, Y.; Kurabayashi, M.; Takaku, F.; Yazaki, Y. Mechanical loading stimulates cell hypertrophy and specific gene expression in cultured rat cardiac myocytes. Possible role of protein kinase C activation. J. Biol. Chem. 1990, 266, 1265–1268. [Google Scholar] [CrossRef]
- Rockman, H.A.; Kock, W.J.; Lefkowitz, R.J. Seven-transmembrane-spanning receptors and heart function. Nature 2002, 415, 206–212. [Google Scholar] [CrossRef]
- Simpson, P.C. Norepinephrine-stimulated hypertrophy of cultured rat myocardial cells in an alpha1-adrenergic response. J. Clin. Investig. 1983, 72, 732–738. [Google Scholar] [CrossRef] [Green Version]
- Olson, M.F. Applications for ROCK kinase inhibition. Curr. Opin. Cell Biol. 2008, 20, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Small, E.M.; Thatcher, J.E.; Sutherland, L.B.; Kinoshita, H.; Gerard, R.D.; Richardson, J.A.; Dimaio, J.M.; Sadek, H.; Kuwahara, K.; Olson, E.N. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ. Res. 2010, 107, 294–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedl, J.; Crevenna, A.H.; Kessenbrock, K.; Yu, J.H.; Neukirchen, D.; Bista, M.; Bradke, F.; Jenne, D.; Holak, T.A.; Werb, Z.; et al. Lifeact: A versatile marker to visualize F-actin. Nat. Methods 2008, 5, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Ui, M.; Okada, T.; Hazeki, K.; Hazeki, O. Wortmannin as a unique probe for an intracellular signalling protein, phosphoinositide 3-kinase. Trends Biochem. Sci. 1995, 20, 303–307. [Google Scholar] [CrossRef]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 2000, 6, 909–919. [Google Scholar] [CrossRef]
- Vlahos, C.J.; Matter, W.F.; Hui, K.Y.; Brown, R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 1994, 269, 5241–5248. [Google Scholar] [CrossRef]
- Posern, G.; Miralles, F.; Guettler, S.; Treisman, R. Mutant actins that stabilise F-actin use distinct mechanisms to activate the SRF coactivator MAL. Embo J. 2004, 23, 3973–3983. [Google Scholar] [CrossRef] [Green Version]
- Miano, J.M. Serum response factor: Toggling between disparate programs of gene expression. J. Mol. Cell. Cardiol. 2003, 35, 577–593. [Google Scholar] [CrossRef]
- Miano, J.M.; Long, X.; Fujiwara, K. Serum response factor: Master regulator of the actin cytoskeleton and contractile apparatus. Am. J. Physiol. Cell Physiol. 2007, 292, C70–C81. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, G.; Streb, J.W.; Long, X.; Yang, Y.; Stoeckert, C.J., Jr.; Miano, J.M. Defining the mammalian CArGome. Genome Res. 2006, 16, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Treisman, R. The serum response element. Trends Biochem. Sci. 1992, 17, 423–426. [Google Scholar] [CrossRef]
- Nakamura, S.; Hayashi, K.; Iwasaki, K.; Fujioka, T.; Egusa, H.; Yatani, H.; Sobue, K. Nuclear import mechanism for myocardin family members and their correlation with vascular smooth muscle cell phenotype. J. Biol. Chem. 2010, 285, 37314–37323. [Google Scholar] [CrossRef] [Green Version]
- Shioi, T.; Kang, P.M.; Douglas, P.S.; Hampe, J.; Yballe, C.M.; Lawitts, J.; Cantley, L.C.; Izumo, S. The conserved phosphoinositide 3-kinase pathway determines heart size in mice. Embo J. 2000, 19, 2537–2548. [Google Scholar] [CrossRef] [Green Version]
- McMullen, J.R.; Amirahmadi, F.; Woodcock, E.A.; Schinke-Braun, M.; Bouwman, R.D.; Hewitt, K.A.; Mollica, J.P.; Zhang, L.; Zhang, Y.; Shioi, T.; et al. Protective effects of exercise and phosphoinositide 3-kinase (p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc. Natl. Acad. Sci. USA 2007, 104, 612–617. [Google Scholar] [CrossRef] [Green Version]
- Pretorius, L.; Owen, K.L.; McMullen, J.R. Role of phosphoinositide 3-kinases in regulating cardiac function. Front. Biosci. 2009, 14, 2221–2229. [Google Scholar] [CrossRef]
- Weeks, K.L.; Gao, X.M.; Du, X.J.; Boey, E.J.H.; Matsumoto, A.; Bernardo, B.C.; Kiriazis, H.; Cemerlang, N.; Tan, J.W.; Tham, Y.K.; et al. Phosphoinositide 3-Kinase p110α is a master regulator of exercise-induced cardioprotection and PI3K gene therapy rescues cardiac dysfunction. Circ. Heart Fail. 2012, 5, 523–534. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, H.N.; Soriano, P. SRF regulates craniofacial development through selective recruitment of MRTF cofactors by PDGF signaling. Dev. Cell 2014, 31, 332–344. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorey, M.-A.; Mericskay, M.; Li, Z.; Decaux, J.-F. Interrelation between α-Cardiac Actin Treadmilling and Myocardin-Related Transcription Factor-A Nuclear Shuttling in Cardiomyocytes. Int. J. Mol. Sci. 2022, 23, 7394. https://doi.org/10.3390/ijms23137394
Gorey M-A, Mericskay M, Li Z, Decaux J-F. Interrelation between α-Cardiac Actin Treadmilling and Myocardin-Related Transcription Factor-A Nuclear Shuttling in Cardiomyocytes. International Journal of Molecular Sciences. 2022; 23(13):7394. https://doi.org/10.3390/ijms23137394
Chicago/Turabian StyleGorey, Mark-Alexander, Mathias Mericskay, Zhenlin Li, and Jean-François Decaux. 2022. "Interrelation between α-Cardiac Actin Treadmilling and Myocardin-Related Transcription Factor-A Nuclear Shuttling in Cardiomyocytes" International Journal of Molecular Sciences 23, no. 13: 7394. https://doi.org/10.3390/ijms23137394
APA StyleGorey, M.-A., Mericskay, M., Li, Z., & Decaux, J.-F. (2022). Interrelation between α-Cardiac Actin Treadmilling and Myocardin-Related Transcription Factor-A Nuclear Shuttling in Cardiomyocytes. International Journal of Molecular Sciences, 23(13), 7394. https://doi.org/10.3390/ijms23137394