
Long-Term Changes in Axon Calibers after Injury: Observations on the Mouse Corticospinal Tract

 , ,
, ,
Abstract
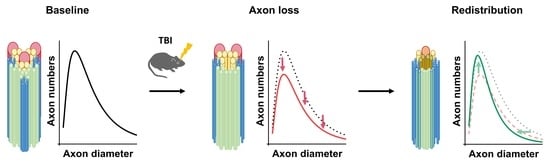
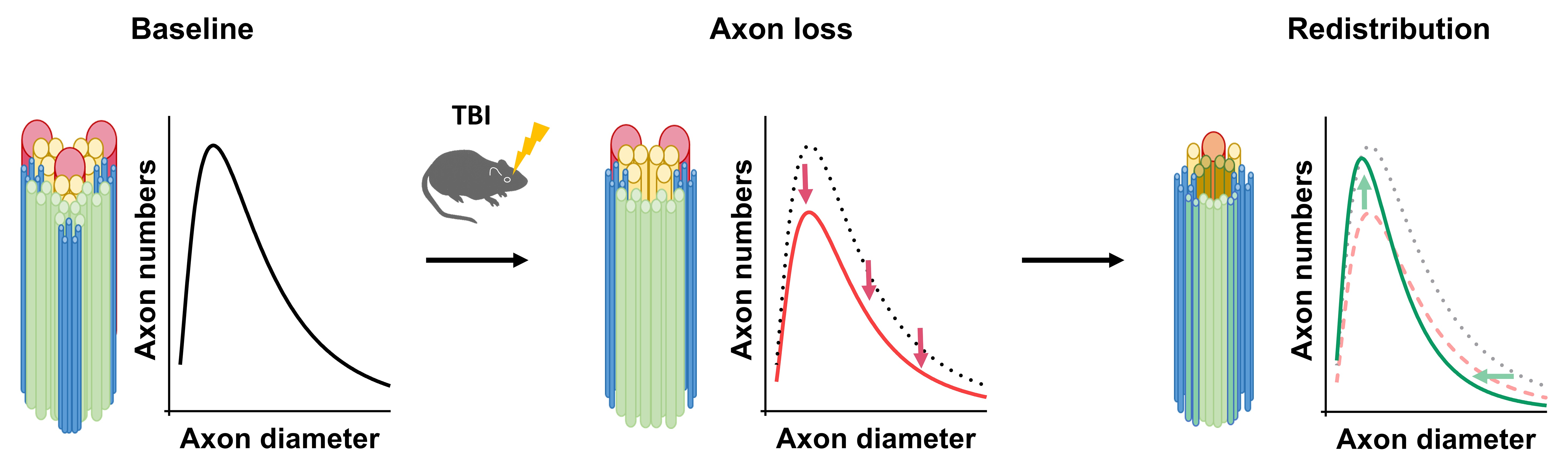
:
1. Introduction
2. Results
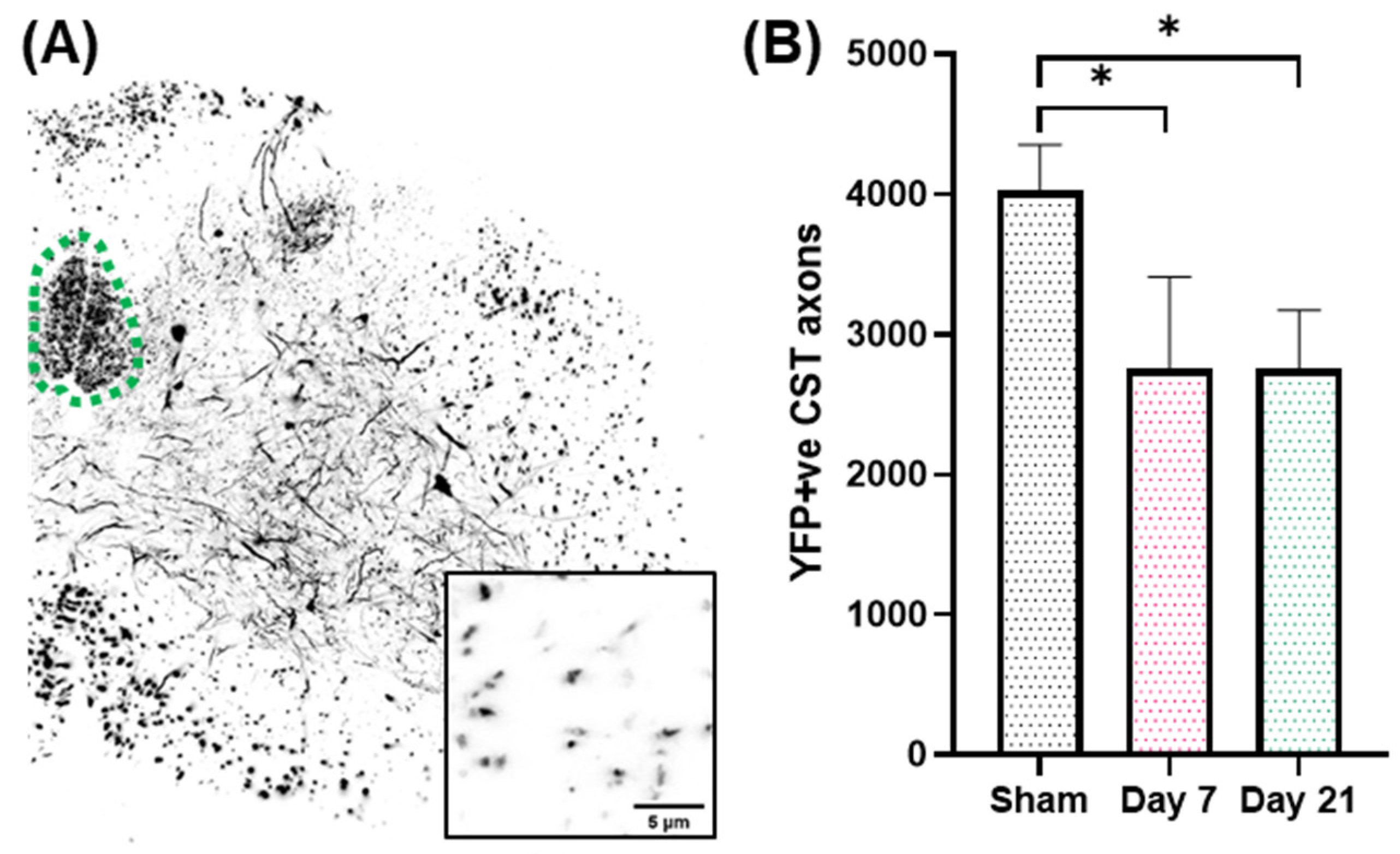
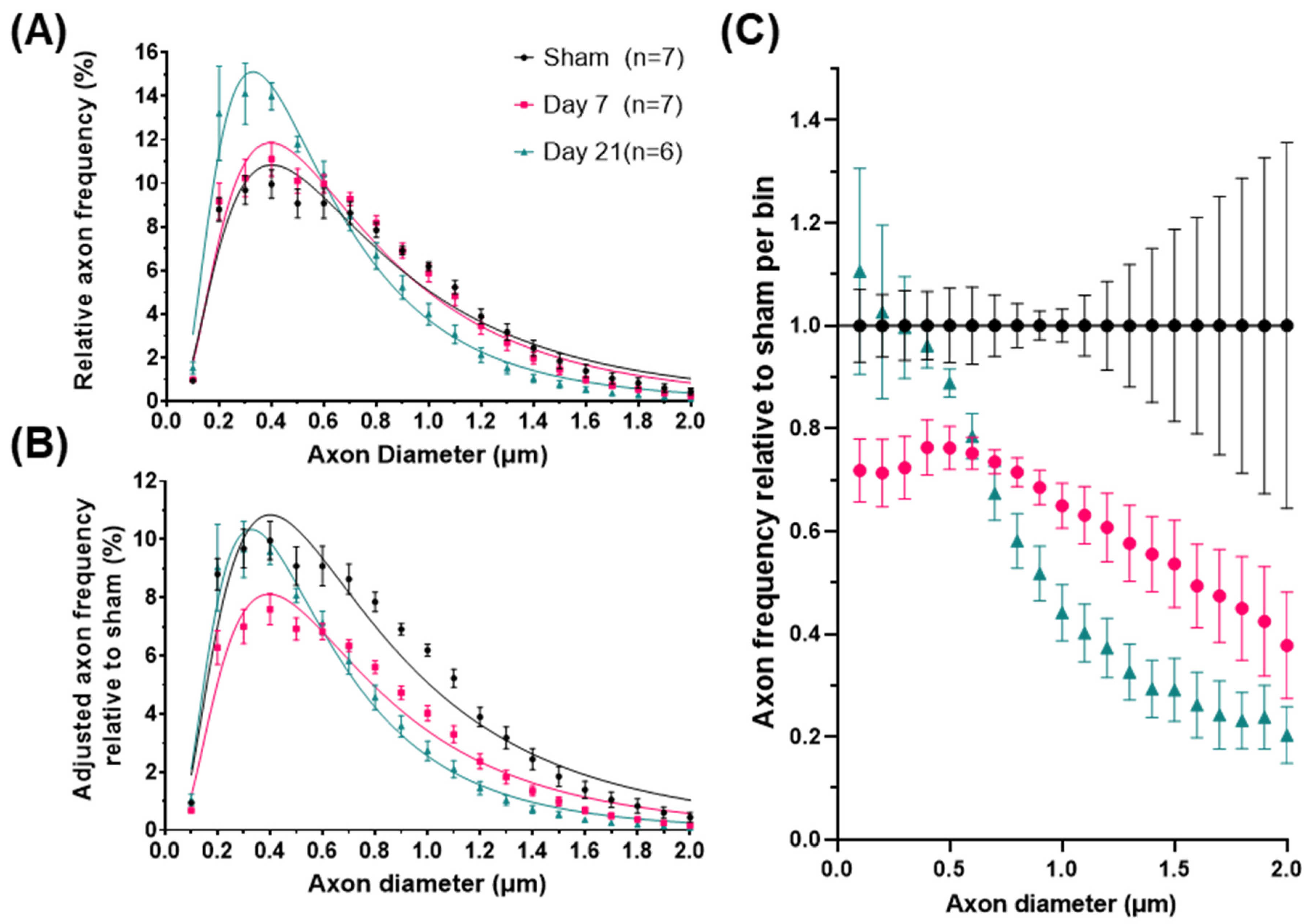
2.1. Impact Acceleration TBI Leads to CST Axon Degeneration in the First Week Post Injury
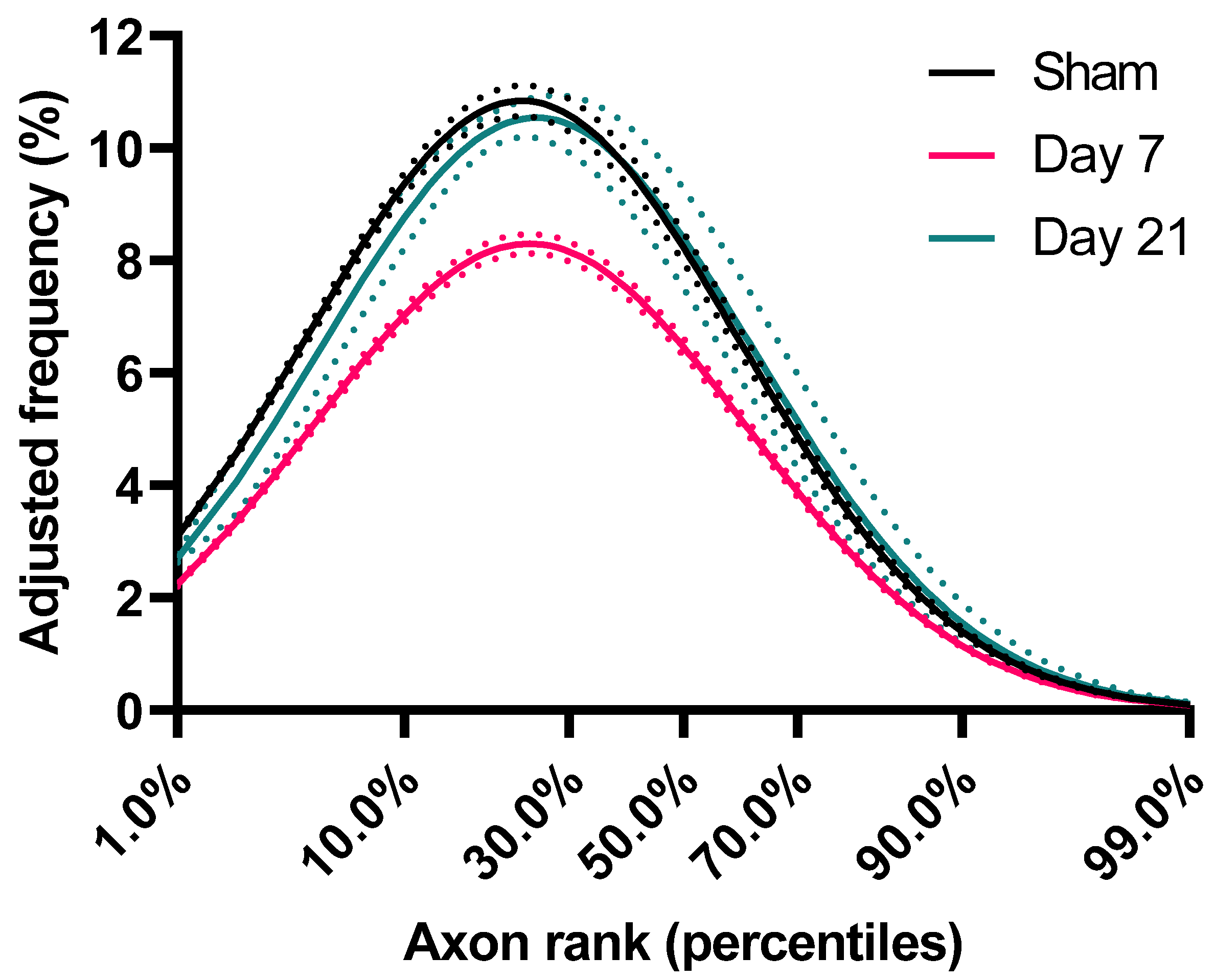
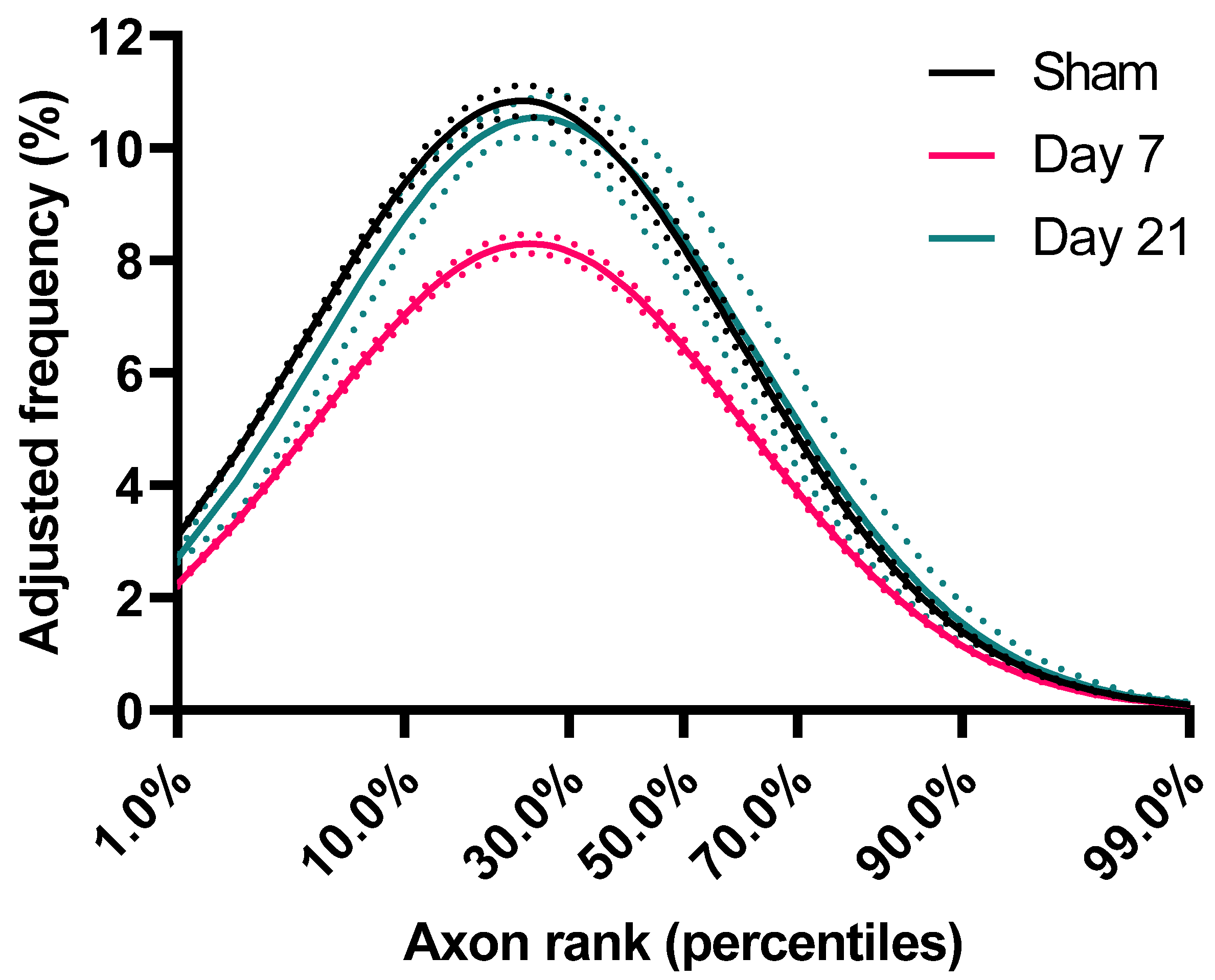
2.2. Impact Acceleration TBI Leads to Significant ADD Changes
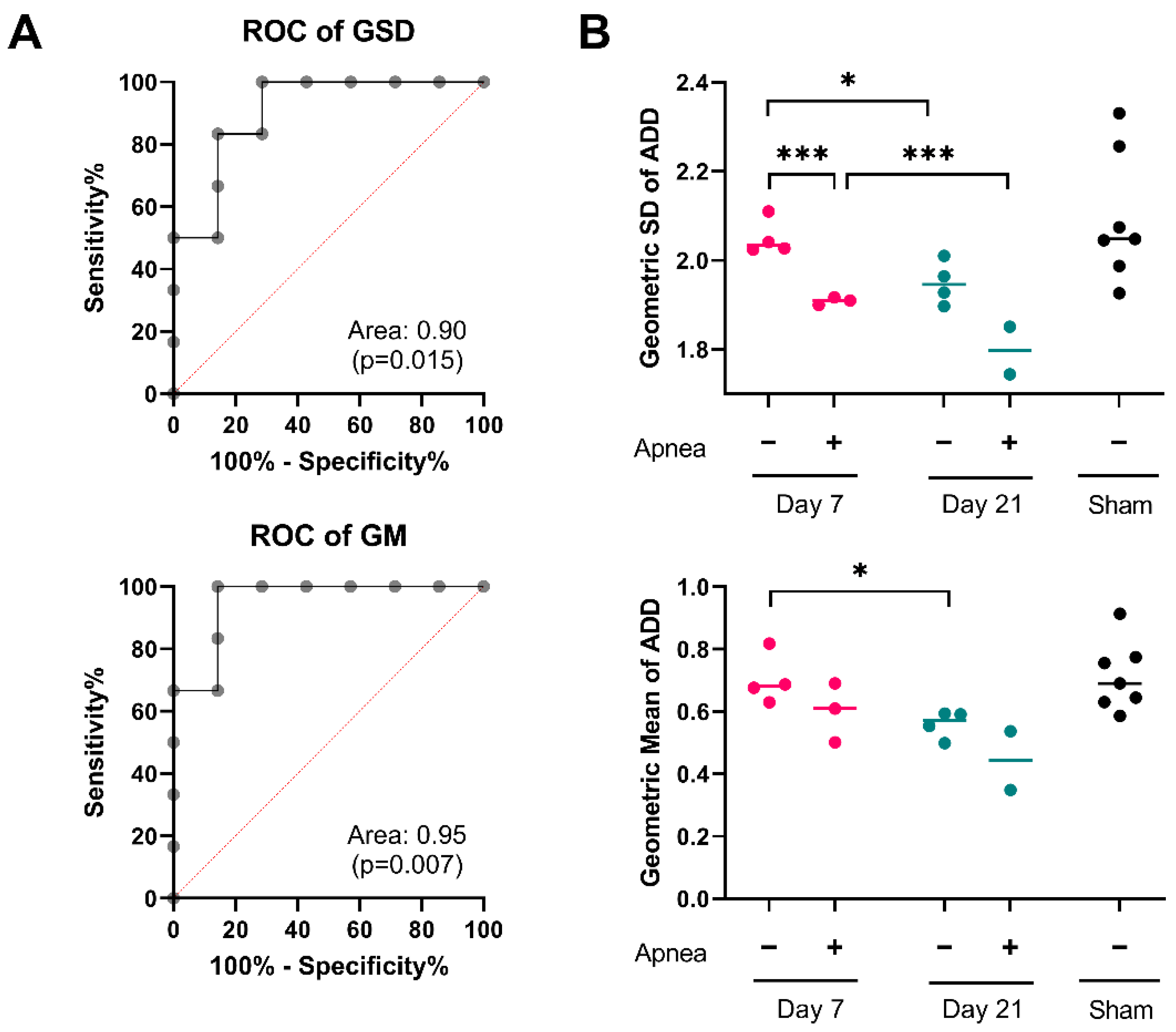
2.3. ADD Injury Signatures in Individual Mice
3. Discussion
3.1. Axon Calibers and Lognormal Distribution
3.2. ADD Changes after Injury
3.3. Potential Mechanisms Underlying ADD Changes
3.4. Clinical Relevance of ADD
4. Materials and Methods
4.1. Experimental Subjects and Impact Acceleration TBI (IA-TBI) Model
4.2. Preparation of Tissues, Imaging and Morphometry
4.3. Axon Diameter Distribution (ADD) Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perge, J.A.; Niven, J.E.; Mugnaini, E.; Balasubramanian, V.; Sterling, P. Why do axons differ in caliber? J. Neurosci. 2012, 32, 626–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegiel, J.; Kaczmarski, W.; Flory, M.; Martinez-Cerdeno, V.; Wisniewski, T.; Nowicki, K.; Kuchna, I.; Wegiel, J. Deficit of corpus callosum axons, reduced axon diameter and decreased area are markers of abnormal development of interhemispheric connections in autistic subjects. Acta Neuropathol. Commun. 2018, 6, 143. [Google Scholar] [CrossRef] [PubMed]
- Kjellstrom, C.; Conradi, N.G. Decreased axonal calibres without axonal loss in optic nerve following chronic alcohol feeding in adult rats: A morphometric study. Acta Neuropathol. 1993, 85, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Alcami, P.; El Hady, A. Axonal Computations. Front. Cell. Neurosci. 2019, 13, 413. [Google Scholar] [CrossRef] [PubMed]
- Caminiti, R.; Ghaziri, H.; Galuske, R.; Hof, P.R.; Innocenti, G.M. Evolution amplified processing with temporally dispersed slow neuronal connectivity in primates. Proc. Natl. Acad. Sci. USA 2009, 106, 19551–19556. [Google Scholar] [CrossRef] [Green Version]
- Innocenti, G.M. Network causality, axonal computations, and Poffenberger. Exp. Brain Res. 2017, 235, 2349–2357. [Google Scholar] [CrossRef] [Green Version]
- Chereau, R.; Saraceno, G.E.; Angibaud, J.; Cattaert, D.; Nagerl, U.V. Superresolution imaging reveals activity-dependent plasticity of axon morphology linked to changes in action potential conduction velocity. Proc. Natl. Acad. Sci. USA 2017, 114, 1401–1406. [Google Scholar] [CrossRef] [Green Version]
- Ziogas, N.K.; Koliatsos, V.E. Primary Traumatic Axonopathy in Mice Subjected to Impact Acceleration: A Reappraisal of Pathology and Mechanisms with High-Resolution Anatomical Methods. J. Neurosci. 2018, 38, 4031–4047. [Google Scholar] [CrossRef]
- Alexandris, S.A.; Lee, Y.; Lehar, M.; Alam, Z.; Saminemi, P.; Ryu, J.; Koliatsos, V.E. Traumatic axonopathy in spinal tracts after impact acceleration head injury: Ultrastructural observations and evidence of SARM1-dependent axonal degeneration [Poster presentation]. J. Neurotrauma. 2022, 38, A15–A16. [Google Scholar]
- Luders, E.; Steinmetz, H.; Jancke, L. Brain size and grey matter volume in the healthy human brain. Neuroreport 2002, 13, 2371–2374. [Google Scholar] [CrossRef]
- Hasenstaub, A.; Otte, S.; Callaway, E.; Sejnowski, T.J. Metabolic cost as a unifying principle governing neuronal biophysics. Proc. Natl. Acad. Sci. USA 2010, 107, 12329–12334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limpert, E.; Stahel, W.A.; Abbt, M. Log-normal Distributions across the Sciences: Keys and Clues: On the charms of statistics, and how mechanical models resembling gambling machines offer a link to a handy way to characterize log-normal distributions, which can provide deeper insight into variability and probability—normal or log-normal: That is the question. BioScience 2001, 51, 341–352. [Google Scholar] [CrossRef]
- Buzsaki, G.; Mizuseki, K. The log-dynamic brain: How skewed distributions affect network operations. Nat. Rev. Neurosci. 2014, 15, 264–278. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Shultz, J.R.; Burish, M.J.; Harrison, K.H.; Hof, P.R.; Towns, L.C.; Wagers, M.W.; Wyatt, K.D. Functional trade-offs in white matter axonal scaling. J. Neurosci. 2008, 28, 4047–4056. [Google Scholar] [CrossRef] [Green Version]
- Perge, J.A.; Koch, K.; Miller, R.; Sterling, P.; Balasubramanian, V. How the optic nerve allocates space, energy capacity, and information. J. Neurosci. 2009, 29, 7917–7928. [Google Scholar] [CrossRef]
- Tomasi, S.; Caminiti, R.; Innocenti, G.M. Areal differences in diameter and length of corticofugal projections. Cereb. Cortex 2012, 22, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Innocenti, G.M.; Caminiti, R. Axon diameter relates to synaptic bouton size: Structural properties define computationally different types of cortical connections in primates. Brain Struct. Funct. 2017, 222, 1169–1177. [Google Scholar] [CrossRef]
- Bramlett, H.M.; Dietrich, W.D. Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J. Neurotrauma. 2015, 32, 1834–1848. [Google Scholar] [CrossRef]
- Chen, X.H.; Johnson, V.E.; Uryu, K.; Trojanowski, J.Q.; Smith, D.H. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain. Pathol. 2009, 19, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Greer, J.E.; McGinn, M.J.; Povlishock, J.T. Diffuse traumatic axonal injury in the mouse induces atrophy, c-Jun activation, and axonal outgrowth in the axotomized neuronal population. J. Neurosci. 2011, 31, 5089–5105. [Google Scholar] [CrossRef] [Green Version]
- Mierzwa, A.J.; Marion, C.M.; Sullivan, G.M.; McDaniel, D.P.; Armstrong, R.C. Components of myelin damage and repair in the progression of white matter pathology after mild traumatic brain injury. J. Neuropathol. Exp. Neurol. 2015, 74, 218–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimura, H.; Lacroix, C.; Said, G. Vulnerability of nerve fibres to ischaemia. A quantitative light and electron microscope study. Brain 1991, 114, Pt 4. 1929–1942. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, M.J.; Mu, E.W.H.; Lavidis, N.A.; Noakes, P.G.; Bellingham, M.C. Size-Dependent Vulnerability of Lumbar Motor Neuron Dendritic Degeneration in SOD1(G93A) Mice. Anat. Rec. 2020, 303, 1455–1471. [Google Scholar] [CrossRef] [PubMed]
- Giguere, N.; Delignat-Lavaud, B.; Herborg, F.; Voisin, A.; Li, Y.; Jacquemet, V.; Anand-Srivastava, M.; Gether, U.; Giros, B.; Trudeau, L.E. Increased vulnerability of nigral dopamine neurons after expansion of their axonal arborization size through D2 dopamine receptor conditional knockout. PLoS Genet. 2019, 15, e1008352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, A.; Rao, M.V.; Veeranna; Nixon, R.A. Neurofilaments and Neurofilament Proteins in Health and Disease. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephan, R.; Goellner, B.; Moreno, E.; Frank, C.A.; Hugenschmidt, T.; Genoud, C.; Aberle, H.; Pielage, J. Hierarchical microtubule organization controls axon caliber and transport and determines synaptic structure and stability. Dev. Cell. 2015, 33, 5–21. [Google Scholar] [CrossRef] [Green Version]
- Scheler, G. Logarithmic distributions prove that intrinsic learning is Hebbian. F1000Res 2017, 6, 1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilliam, M.; Ziogas, N.; Koliatsos, V.E. Department of Pathology, Johns Hopkins School of Medicine, Baltimore, MD 21205, USA. 2021; manuscript in preparation. [Google Scholar]
- Jones, D.K.; Alexander, D.C.; Bowtell, R.; Cercignani, M.; Dell’Acqua, F.; McHugh, D.J.; Miller, K.L.; Palombo, M.; Parker, G.J.M.; Rudrapatna, U.S.; et al. Microstructural imaging of the human brain with a ‘super-scanner’: 10 key advantages of ultra-strong gradients for diffusion MRI. Neuroimage 2018, 182, 8–38. [Google Scholar] [CrossRef] [Green Version]
- Barazany, D.; Basser, P.J.; Assaf, Y. In vivo measurement of axon diameter distribution in the corpus callosum of rat brain. Brain 2009, 132, 1210–1220. [Google Scholar] [CrossRef] [Green Version]
- Barazany, D.; Jones, D.; Assaf, Y. AxCaliber 3D. In Proceedings of the International Society of Magnetic Resonance in Medicine. 19th Annual ISMRM Scientific Meeting and Exhibition 2011, Montreal, QC, Canada, 7–13 May 2011. [Google Scholar]
- Zhang, H.; Hubbard, P.L.; Parker, G.J.M.; Alexander, D.C. Axon diameter mapping in the presence of orientation dispersion with diffusion MRI. NeuroImage 2011, 56, 1301–1315. [Google Scholar] [CrossRef]
- Xu, J.; Li, H.; Harkins, K.D.; Jiang, X.; Xie, J.; Kang, H.; Does, M.D.; Gore, J.C. Mapping mean axon diameter and axonal volume fraction by MRI using temporal diffusion spectroscopy. Neuroimage 2014, 103, 10–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harkins, K.D.; Beaulieu, C.; Xu, J.; Gore, J.C.; Does, M.D. A simple estimate of axon size with diffusion MRI. Neuroimage 2021, 227, 117619. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Aravind, A.; Pfister, B.J.; Chandra, N.; Haorah, J. Animal Models of Traumatic Brain Injury and Assessment of Injury Severity. Mol. Neurobiol. 2019, 56, 5332–5345. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, C.N.; Roberts, K.N.; Higgins, E.K.; Bachstetter, A.D. A Systematic Review of Closed Head Injury Models of Mild Traumatic Brain Injury in Mice and Rats. J. Neurotrauma 2019, 36, 1683–1706. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods. 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Zaimi, A.; Wabartha, M.; Herman, V.; Antonsanti, P.L.; Perone, C.S.; Cohen-Adad, J. AxonDeepSeg: Automatic axon and myelin segmentation from microscopy data using convolutional neural networks. Sci. Rep. 2018, 8, 3816. [Google Scholar] [CrossRef] [Green Version]
- Efron, B.; Stein, C. The Jackknife Estimate of Variance. Ann. Stat. 1981, 9, 586–596. [Google Scholar] [CrossRef]
- Ernst, M.D. Permutation Methods: A Basis for Exact Inference. Stat. Sci. 2004, 19, 676–685. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADD Features | Sham (n = 7) | Day 7 (n = 7) | Day 21 (n = 6) |
|---|---|---|---|
| Geometric mean, GM (CI95%) | 0.69 (0.62–0.77) | 0.65 (0.58–0.72) | 0.51 (0.43–0.59) |
| Geometric standard deviation, GSD (CI95%) | 2.10 (1.99–2.21) | 2.03 (1.97–2.09) | 1.97 (1.92–2.02) |
| Skewness (CI95%) | 3.21 (2.79–3.62) | 2.94 (2.72–3.16) | 2.72 (2.55–2.88) |
| 90th Percentile (CI95%) | 1.79 (1.49–2.09) | 1.61 (1.4–1.81) | 1.22 (1.02–1.41) |
| Mode (CI95%) | 0.40 (0.37–0.43) | 0.39 (0.35–0.44) | 0.32 (0.2–0.38) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexandris, A.S.; Wang, Y.; Frangakis, C.E.; Lee, Y.; Ryu, J.; Alam, Z.; Koliatsos, V.E. Long-Term Changes in Axon Calibers after Injury: Observations on the Mouse Corticospinal Tract. Int. J. Mol. Sci. 2022, 23, 7391. https://doi.org/10.3390/ijms23137391
Alexandris AS, Wang Y, Frangakis CE, Lee Y, Ryu J, Alam Z, Koliatsos VE. Long-Term Changes in Axon Calibers after Injury: Observations on the Mouse Corticospinal Tract. International Journal of Molecular Sciences. 2022; 23(13):7391. https://doi.org/10.3390/ijms23137391
Chicago/Turabian StyleAlexandris, Athanasios S., Yiqing Wang, Constantine E. Frangakis, Youngrim Lee, Jiwon Ryu, Zahra Alam, and Vassilis E. Koliatsos. 2022. "Long-Term Changes in Axon Calibers after Injury: Observations on the Mouse Corticospinal Tract" International Journal of Molecular Sciences 23, no. 13: 7391. https://doi.org/10.3390/ijms23137391
APA StyleAlexandris, A. S., Wang, Y., Frangakis, C. E., Lee, Y., Ryu, J., Alam, Z., & Koliatsos, V. E. (2022). Long-Term Changes in Axon Calibers after Injury: Observations on the Mouse Corticospinal Tract. International Journal of Molecular Sciences, 23(13), 7391. https://doi.org/10.3390/ijms23137391