
Mutation-Specific Differences in Kv7.1 (KCNQ1) and Kv11.1 (KCNH2) Channel Dysfunction and Long QT Syndrome Phenotypes

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
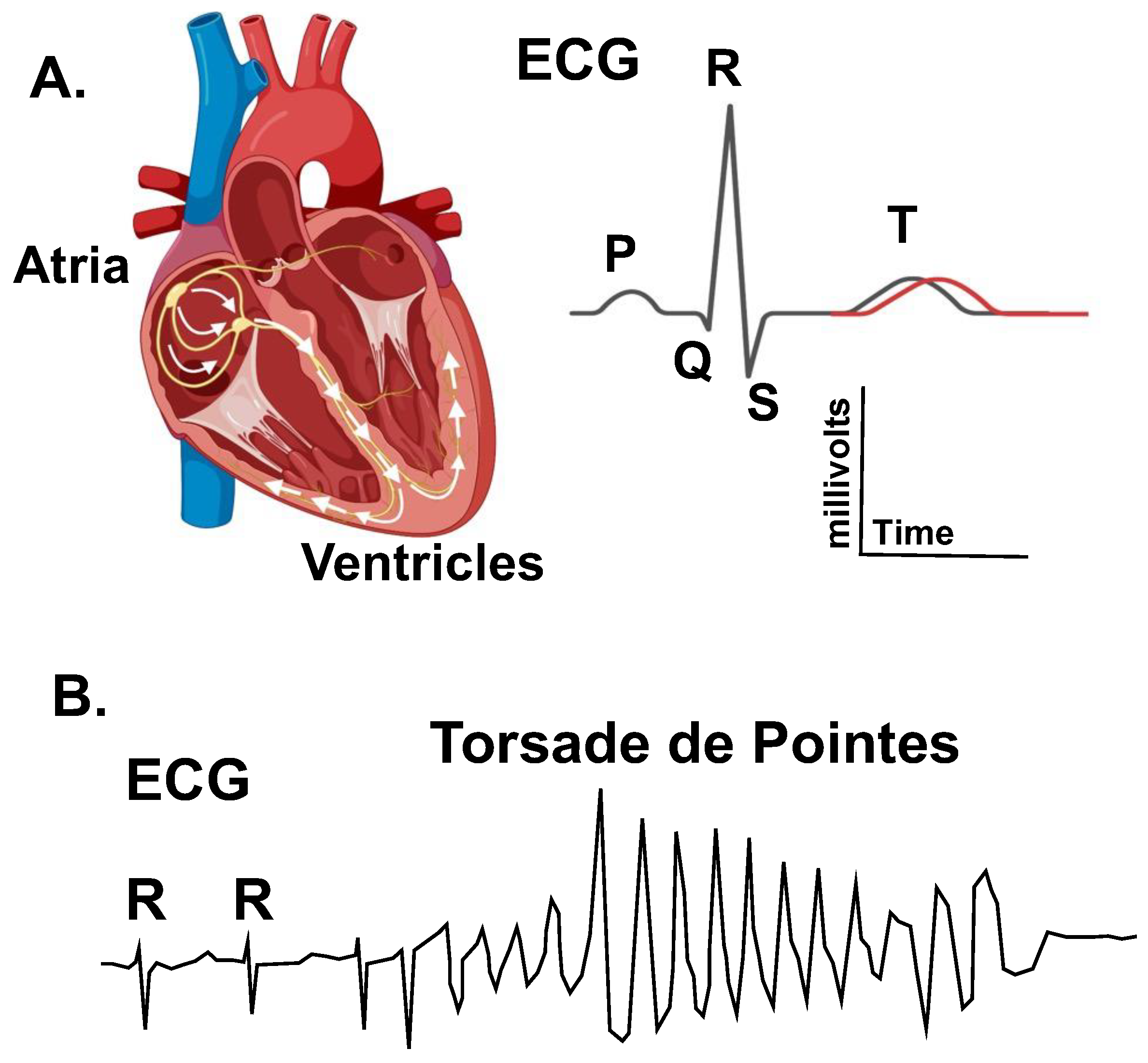
2. The QT-Interval: From Biomarker to Clinical Genetics
“The QT-interval in the present cases is, however, so very prolonged that it must be regarded as pathological by any standard. The E.C.G. changes suggest a metabolic disorder in the myocardium which slows repolarization of the muscle after systole… Undue sensitivity of the myocardium to sympathetic stimulation is postulated. Excessive sympathetic activity may prolong QT-interval and may also contribute to ventricular fibrillation. The clinical observation that the children have improved on a drug which blocks the effect of the sympathetic at the beta receptors in the myocardium might confirm this hypothesis…”.
3. LQT1 and LQT2 Clinical Phenotypes
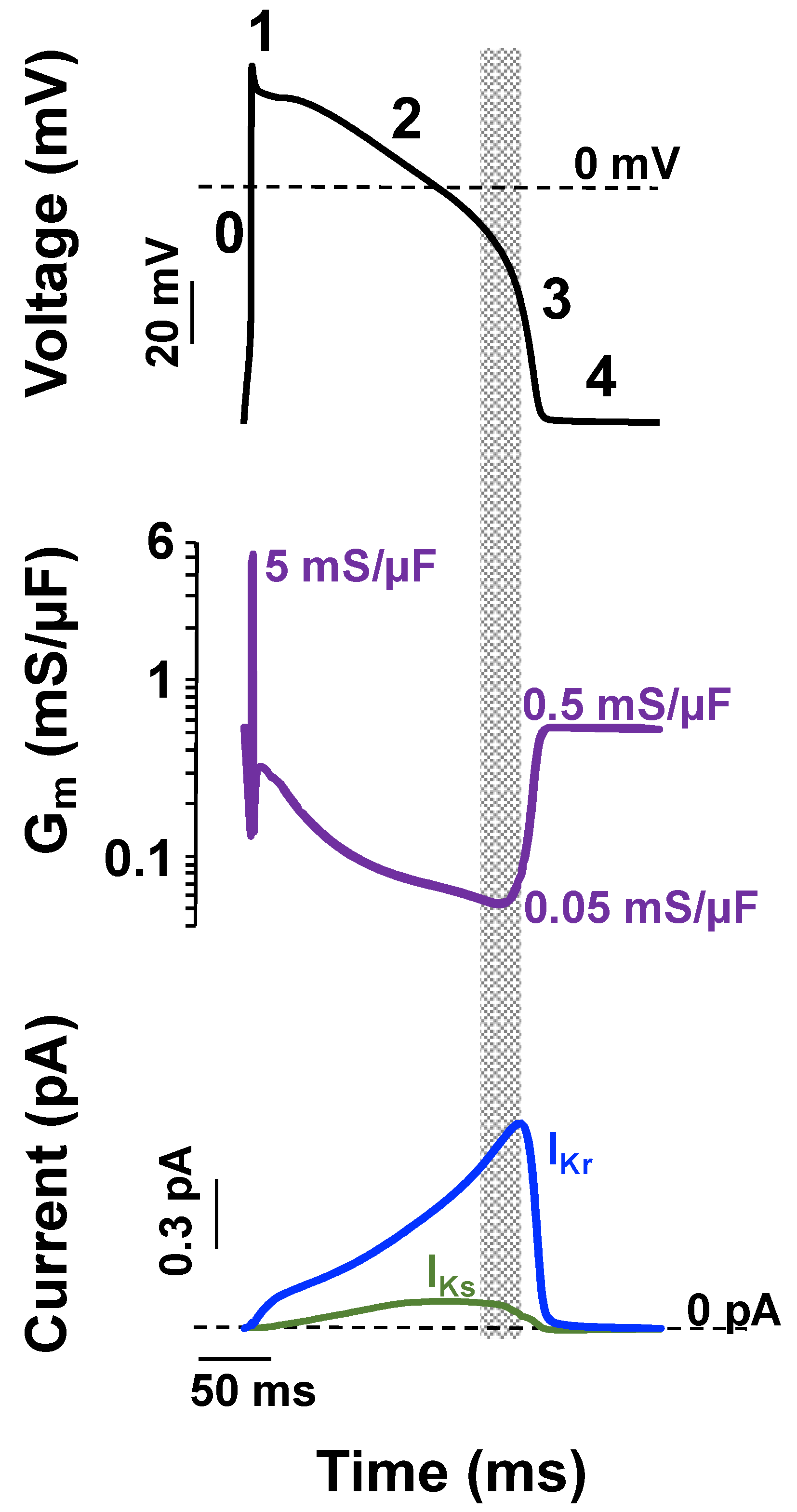
4. Importance of IKs and IKr in Cardiac Action Potential Repolarization
5. Mechanistic Classification of LQT1- and LQT2-Linked Mutations
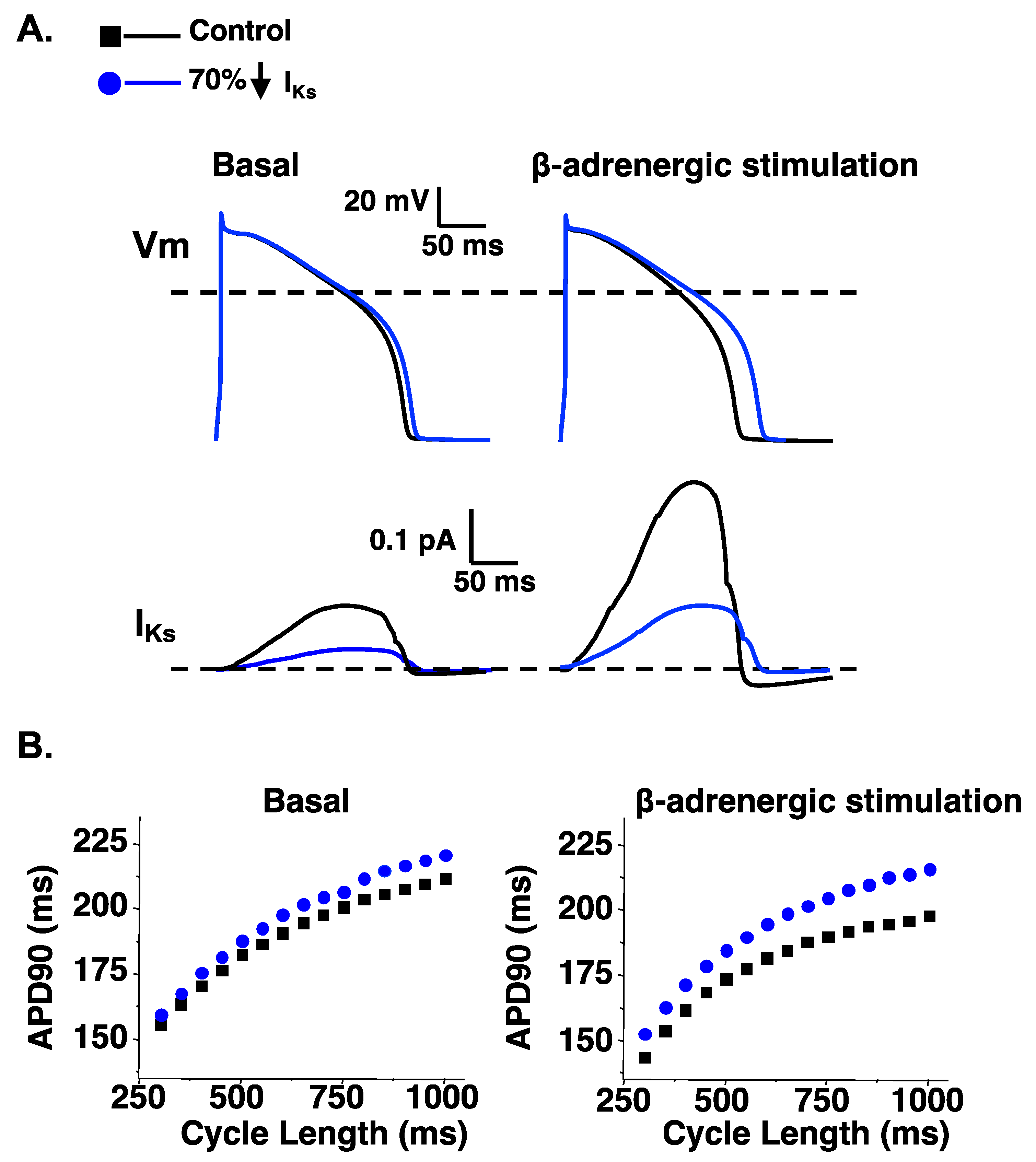
6. LQT1-Linked Mutation Dysfunctional Phenotypes: PKA, CaM, PIP2, and Pleiotropy
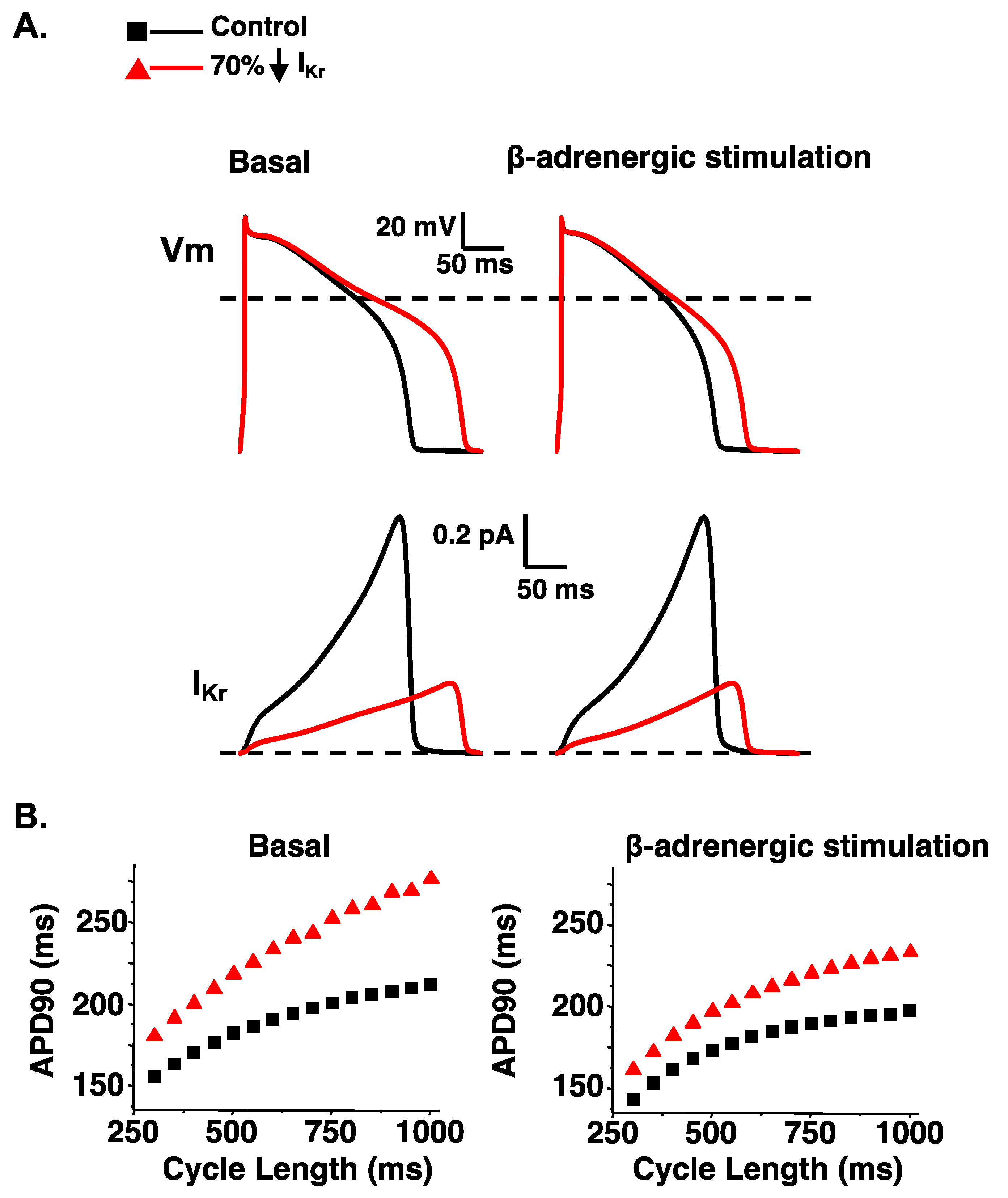
7. LQT2-Linked Mutation Dysfunctional Phenotypes: Trafficking in Heterotetramers
8. Visualizing Mutation-Specific Differences in Kv7.1 and Kv11.1 Channel Structures
9. Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moss, A.J. Long QT Syndrome. JAMA 2003, 289, 2041–2044. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013, 10, 1932–1963. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.; Novelli, V.; Amin, A.S.; Abiusi, E.; Care, M.; Nannenberg, E.A.; Feilotter, H.; Amenta, S.; Mazza, D.; Bikker, H.; et al. An International, Multicentered, Evidence-Based Reappraisal of Genes Reported to Cause Congenital Long QT Syndrome. Circulation 2020, 141, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Liu, F.; Li, Q.; Qing, T.; Zhai, Z.; Xia, Z.; Li, J. The Efficacy of Beta-Blockers in Patients with Long QT Syndrome 1-3 According to Individuals’ Gender, Age, and QTc Intervals: A Network Meta-analysis. Front. Pharm. 2020, 11, 579525. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Amin, A.S.; Postema, P.G. Diagnosis, management and therapeutic strategies for congenital long QT syndrome. Heart 2022, 108, 332–338. [Google Scholar] [CrossRef]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. Executive Summary: HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes. Heart Rhythm Off. J. Heart Rhythm Soc. 2013, 15, 1389–1406. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Crotti, L.; Insolia, R. Long-QT syndrome: From genetics to management. Circ. Arrhythm Electrophysiol. 2012, 5, 868–877. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Q.; Towbin, J.A. Genetics, molecular mechanisms and management of long QT syndrome. Ann. Med. 1998, 30, 58–65. [Google Scholar] [CrossRef]
- Takigawa, M.; Kawamura, M.; Noda, T.; Yamada, Y.; Miyamoto, K.; Okamura, H.; Satomi, K.; Aiba, T.; Kamakura, S.; Sakaguchi, T.; et al. Seasonal and circadian distributions of cardiac events in genotyped patients with congenital long QT syndrome. Circ. J. 2012, 76, 2112–2118. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Priori, S.G.; Spazzolini, C.; Moss, A.J.; Vincent, G.M.; Napolitano, C.; Denjoy, I.; Guicheney, P.; Breithardt, G.; Keating, M.T.; et al. Genotype-phenotype correlation in the long-QT syndrome: Gene-specific triggers for life-threatening arrhythmias. Circulation 2001, 103, 89–95. [Google Scholar] [CrossRef]
- Shimizu, W.; Moss, A.J.; Wilde, A.A.; Towbin, J.A.; Ackerman, M.J.; January, C.T.; Tester, D.J.; Zareba, W.; Robinson, J.L.; Qi, M.; et al. Genotype-phenotype aspects of type 2 long QT syndrome. J. Am. Coll. Cardiol. 2009, 54, 2052–2062. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.; Lopes, C.M.; Barsheshet, A.; Moss, A.J.; Migdalovich, D.; Ouellet, G.; McNitt, S.; Polonsky, S.; Robinson, J.L.; Zareba, W.; et al. Combined assessment of sex- and mutation-specific information for risk stratification in type 1 long QT syndrome. Heart Rhythm 2012, 9, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.; Moss, A.J.; Kaufman, E.S.; Shimizu, W.; Peterson, D.R.; Benhorin, J.; Lopes, C.; Towbin, J.A.; Spazzolini, C.; Crotti, L.; et al. Clinical Aspects of Type 3 Long-QT Syndrome: An International Multicenter Study. Circulation 2016, 134, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.J.; Shimizu, W.; Wilde, A.A.; Towbin, J.A.; Zareba, W.; Robinson, J.L.; Qi, M.; Vincent, G.M.; Ackerman, M.J.; Kaufman, E.S.; et al. Clinical aspects of type-1 long-QT syndrome by location, coding type, and biophysical function of mutations involving the KCNQ1 gene. Circulation 2007, 115, 2481–2489. [Google Scholar] [CrossRef] [PubMed]
- Moss, A.J.; Zareba, W.; Kaufman, E.S.; Gartman, E.; Peterson, D.R.; Benhorin, J.; Towbin, J.A.; Keating, M.T.; Priori, S.G.; Schwartz, P.J.; et al. Increased risk of arrhythmic events in long-QT syndrome with mutations in the pore region of the human ether-a-go-go-related gene potassium channel. Circulation 2002, 105, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Ponce-Balbuena, D.; Deschenes, I. Long QT syndrome—Bench to bedside. Heart Rhythm O2 2021, 2, 89–106. [Google Scholar] [CrossRef]
- Krahn, A.D.; Laksman, Z.; Sy, R.W.; Postema, P.G.; Ackerman, M.J.; Wilde, A.A.M.; Han, H.C. Congenital Long QT Syndrome. JACC Clin. Electrophysiol. 2022, 8, 687–706. [Google Scholar] [CrossRef]
- Pandit, M.; Finn, C.; Tahir, U.; Frishman, W.H. Congenital Long QT Syndrome: A Review of Genetic and Pathophysiologic Etiologies, Phenotypic Subtypes and Clinical Management. Cardiol. Rev. 2022. [Google Scholar] [CrossRef]
- AlGhatrif, M.; Lindsay, J. A brief review: History to understand fundamentals of electrocardiography. J. Commun. Hosp. Intern. Med. Perspect. 2012, 2, 14383. [Google Scholar] [CrossRef]
- Fisch, C. Evolution of the clinical electrocardiogram. J. Am. Coll. Cardiol. 1989, 14, 1127–1138. [Google Scholar] [CrossRef]
- Lewis, T. Report Cxix. Auricular Fibrillation: A Common Clinical Condition. Br. Med. J. 1909, 2, 1528. [Google Scholar] [CrossRef] [PubMed]
- Levine, S.A. Coronary thrombosis: Its various clinical features. Medicine 1929, 8, 245–418. [Google Scholar] [CrossRef]
- RECOMMENDATIONS for standardization of electrocardiographic and vectorcardiographic leads. Circulation 1954, 10, 564–573. [CrossRef]
- Kadish, A.H.; Buxton, A.E.; Kennedy, H.L.; Knight, B.P.; Mason, J.W.; Schuger, C.D.; Tracy, C.M.; Winters, W.L., Jr.; Boone, A.W.; Elnicki, M.; et al. ACC/AHA clinical competence statement on electrocardiography and ambulatory electrocardiography: A report of the ACC/AHA/ACP-ASIM task force on clinical competence (ACC/AHA Committee to develop a clinical competence statement on electrocardiography and ambulatory electrocardiography) endorsed by the International Society for Holter and noninvasive electrocardiology. Circulation 2001, 104, 3169–3178. [Google Scholar] [PubMed]
- Jervell, A.; Lange-Nielsen, F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am. Heart J. 1957, 54, 59–68. [Google Scholar] [CrossRef]
- Romano, C.; Gemme, G.; Pongiglione, R. Rare Cardiac Arrythmias of the Pediatric Age. Ii. Syncopal Attacks Due to Paroxysmal Ventricular Fibrillation (Presentation of 1st Case in Italian Pediatric Literature). Clin. Pediatr. 1963, 45, 656–683. [Google Scholar] [PubMed]
- Ward, O.C. A New Familial Cardiac Syndrome in Children. J. Ir. Med. Assoc. 1964, 54, 103–106. [Google Scholar]
- Schwartz, P.J. 1970-2020: 50 years of research on the long QT syndrome-from almost zero knowledge to precision medicine. Eur. Heart J. 2021, 42, 1063–1072. [Google Scholar] [CrossRef]
- Moss, A.J.; Schwartz, P.J. 25th anniversary of the International Long-QT Syndrome Registry: An ongoing quest to uncover the secrets of long-QT syndrome. Circulation 2005, 111, 1199–1201. [Google Scholar] [CrossRef]
- Schwartz, P.J. Idiopathic long QT syndrome: Progress and questions. Am. Heart J. 1985, 109, 399–411. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Moss, A.J.; Vincent, G.M.; Crampton, R.S. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993, 88, 782–784. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Crotti, L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation 2011, 124, 2181–2184. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.E.; Splawski, I.; Timothy, K.W.; Vincent, G.M.; Green, E.D.; Keating, M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995, 80, 795–803. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Jiang, C.; Curran, M.E.; Keating, M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995, 81, 299–307. [Google Scholar] [CrossRef]
- Trudeau, M.C.; Warmke, J.W.; Ganetzky, B.; Robertson, G.A. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science 1995, 269, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shen, J.; Splawski, I.; Atkinson, D.; Li, Z.; Robinson, J.L.; Moss, A.J.; Towbin, J.A.; Keating, M.T. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995, 80, 805–811. [Google Scholar] [CrossRef]
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; de Jager, T.; et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996, 12, 17–23. [Google Scholar] [CrossRef]
- Wilde, A.A.; Brugada, R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ. Res. 2011, 108, 884–897. [Google Scholar] [CrossRef]
- Abriel, H. Cardiac sodium channel Na(v)1.5 and interacting proteins: Physiology and pathophysiology. J. Mol. Cell Cardiol. 2010, 48, 2–11. [Google Scholar] [CrossRef]
- Remme, C.A.; Wilde, A.A.; Bezzina, C.R. Cardiac sodium channel overlap syndromes: Different faces of SCN5A mutations. Trends Cardiovasc. Med. 2008, 18, 78–87. [Google Scholar] [CrossRef]
- Viskin, S.; Rosovski, U.; Sands, A.J.; Chen, E.; Kistler, P.M.; Kalman, J.M.; Rodriguez Chavez, L.; Iturralde Torres, P.; Cruz, F.F.; Centurion, O.A.; et al. Inaccurate electrocardiographic interpretation of long QT: The majority of physicians cannot recognize a long QT when they see one. Heart Rhythm Off. J. Heart Rhythm Soc. 2005, 2, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.; Lehmann, M.H. Prolonged QT interval diagnosis suppression by a widely used computerized ECG analysis system. Circ. Arrhythm Electrophysiol. 2013, 6, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Garson, A., Jr. How to measure the QT interval—What is normal? Am. J. Cardiol. 1993, 72, 14B–16B. [Google Scholar] [CrossRef]
- Lepeschkin, E.; Surawicz, B. The measurement of the Q-T interval of the electrocardiogram. Circulation 1952, 6, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Postema, P.G.; Wilde, A.A. The measurement of the QT interval. Curr. Cardiol. Rev. 2014, 10, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Taggart, N.W.; Haglund, C.M.; Tester, D.J.; Ackerman, M.J. Diagnostic miscues in congenital long-QT syndrome. Circulation 2007, 115, 2613–2620. [Google Scholar] [CrossRef]
- Kapa, S.; Tester, D.J.; Salisbury, B.A.; Harris-Kerr, C.; Pungliya, M.S.; Alders, M.; Wilde, A.A.; Ackerman, M.J. Genetic testing for long-QT syndrome: Distinguishing pathogenic mutations from benign variants. Circulation 2009, 120, 1752–1760. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Kapplinger, J.D.; Tester, D.J.; Alders, M.; Salisbury, B.A.; Wilde, A.A.; Ackerman, M.J. Phylogenetic and physicochemical analyses enhance the classification of rare nonsynonymous single nucleotide variants in type 1 and 2 long-QT syndrome. Circ. Cardiovasc. Genet. 2012, 5, 519–528. [Google Scholar] [CrossRef]
- Viskin, S.; Postema, P.G.; Bhuiyan, Z.A.; Rosso, R.; Kalman, J.M.; Vohra, J.K.; Guevara-Valdivia, M.E.; Marquez, M.F.; Kogan, E.; Belhassen, B.; et al. The response of the QT interval to the brief tachycardia provoked by standing: A bedside test for diagnosing long QT syndrome. J. Am. Coll Cardiol. 2010, 55, 1955–1961. [Google Scholar] [CrossRef]
- Adler, A.; van der Werf, C.; Postema, P.G.; Rosso, R.; Bhuiyan, Z.A.; Kalman, J.M.; Vohra, J.K.; Guevara-Valdivia, M.E.; Marquez, M.F.; Halkin, A.; et al. The phenomenon of “QT stunning”: The abnormal QT prolongation provoked by standing persists even as the heart rate returns to normal in patients with long QT syndrome. Heart Rhythm 2012, 9, 901–908. [Google Scholar] [CrossRef]
- Sy, R.W.; van der Werf, C.; Chattha, I.S.; Chockalingam, P.; Adler, A.; Healey, J.S.; Perrin, M.; Gollob, M.H.; Skanes, A.C.; Yee, R.; et al. Derivation and validation of a simple exercise-based algorithm for prediction of genetic testing in relatives of LQTS probands. Circulation 2011, 124, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Khositseth, A.; Tester, D.J.; Hejlik, J.B.; Shen, W.K.; Porter, C.B. Epinephrine-induced QT interval prolongation: A gene-specific paradoxical response in congenital long QT syndrome. Mayo Clin. Proc. 2002, 77, 413–421. [Google Scholar] [CrossRef]
- Horner, J.M.; Horner, M.M.; Ackerman, M.J. The diagnostic utility of recovery phase QTc during treadmill exercise stress testing in the evaluation of long QT syndrome. Heart Rhythm 2011, 8, 1698–1704. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, W.; Noda, T.; Takaki, H.; Nagaya, N.; Satomi, K.; Kurita, T.; Suyama, K.; Aihara, N.; Sunagawa, K.; Echigo, S.; et al. Diagnostic value of epinephrine test for genotyping LQT1, LQT2, and LQT3 forms of congenital long QT syndrome. Heart Rhythm Off. J. Heart Rhythm Soc. 2004, 1, 276–283. [Google Scholar] [CrossRef]
- Vyas, H.; Ackerman, M.J. Epinephrine QT stress testing in congenital long QT syndrome. J. Electrocardiol. 2006, 39, S107–S113. [Google Scholar] [CrossRef]
- Peroz, D.; Rodriguez, N.; Choveau, F.; Baro, I.; Merot, J.; Loussouarn, G. Kv7.1 (KCNQ1) properties and channelopathies. J. Physiol. 2008, 586, 1785–1789. [Google Scholar] [CrossRef]
- Wong, J.A.; Gula, L.J.; Klein, G.J.; Yee, R.; Skanes, A.C.; Krahn, A.D. Utility of treadmill testing in identification and genotype prediction in long-QT syndrome. Circ. Arrhythm Electrophysiol. 2010, 3, 120–125. [Google Scholar] [CrossRef]
- Krahn, A.D.; Klein, G.J.; Yee, R. Hysteresis of the RT interval with exercise: A new marker for the long-QT syndrome? Circulation 1997, 96, 1551–1556. [Google Scholar] [CrossRef]
- Bos, J.M.; Attia, Z.I.; Albert, D.E.; Noseworthy, P.A.; Friedman, P.A.; Ackerman, M.J. Use of Artificial Intelligence and Deep Neural Networks in Evaluation of Patients With Electrocardiographically Concealed Long QT Syndrome From the Surface 12-Lead Electrocardiogram. JAMA Cardiol. 2021, 6, 532–538. [Google Scholar] [CrossRef]
- Aufiero, S.; Bleijendaal, H.; Robyns, T.; Vandenberk, B.; Krijger, C.; Bezzina, C.; Zwinderman, A.H.; Wilde, A.A.M.; Pinto, Y.M. A deep learning approach identifies new ECG features in congenital long QT syndrome. BMC Med. 2022, 20, 162. [Google Scholar] [CrossRef]
- Roden, D.M. Taking the “idio” out of “idiosyncratic”: Predicting torsades de pointes. Pacing Clin. Electrophysiol. 1998, 21, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M. Repolarization reserve: A moving target. Circulation 2008, 118, 981–982. [Google Scholar] [CrossRef] [PubMed]
- Soltis, A.R.; Saucerman, J.J. Synergy between CaMKII substrates and beta-adrenergic signaling in regulation of cardiac myocyte Ca(2+) handling. Biophys. J. 2010, 99, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Kurokawa, J.; Reiken, S.; Motoike, H.; D’Armiento, J.; Marks, A.R.; Kass, R.S. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 2002, 295, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Zipes, D.P.; Jalife, J.; Stevenson, W.G. Cardiac Electrophysiology: From Cell to Bedside; Elsevier Health Science: Philadelphia, PA, USA, 2018. [Google Scholar]
- Bartos, D.C.; Giudicessi, J.R.; Tester, D.J.; Ackerman, M.J.; Ohno, S.; Horie, M.; Gollob, M.H.; Burgess, D.E.; Delisle, B.P. A KCNQ1 mutation contributes to the concealed type 1 long QT phenotype by limiting the Kv7.1 channel conformational changes associated with protein kinase A phosphorylation. Heart Rhythm 2014, 11, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Anderson, C.L.; Burgess, D.E.; Elayi, C.S.; January, C.T.; Delisle, B.P. Molecular pathogenesis of long QT syndrome type 2. J. Arrhythm 2016, 32, 373–380. [Google Scholar] [CrossRef]
- Anderson, C.L.; Kuzmicki, C.E.; Childs, R.R.; Hintz, C.J.; Delisle, B.P.; January, C.T. Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat. Commun. 2014, 5, 5535. [Google Scholar] [CrossRef]
- Napolitano, C.; Priori, S.G.; Schwartz, P.J.; Bloise, R.; Ronchetti, E.; Nastoli, J.; Bottelli, G.; Cerrone, M.; Leonardi, S. Genetic testing in the long QT syndrome: Development and validation of an efficient approach to genotyping in clinical practice. JAMA 2005, 294, 2975–2980. [Google Scholar] [CrossRef]
- Anderson, C.L.; Delisle, B.P.; Anson, B.D.; Kilby, J.A.; Will, M.L.; Tester, D.J.; Gong, Q.; Zhou, Z.; Ackerman, M.J.; January, C.T. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation 2006, 113, 365–373. [Google Scholar] [CrossRef]
- Burgess, D.E.; Bartos, D.C.; Reloj, A.R.; Campbell, K.S.; Johnson, J.N.; Tester, D.J.; Ackerman, M.J.; Fressart, V.; Denjoy, I.; Guicheney, P.; et al. High-risk long QT syndrome mutations in the Kv7.1 (KCNQ1) pore disrupt the molecular basis for rapid K(+) permeation. Biochemistry 2012, 51, 9076–9085. [Google Scholar] [CrossRef]
- Delisle, B.P.; Anson, B.D.; Rajamani, S.; January, C.T. Biology of cardiac arrhythmias: Ion channel protein trafficking. Circ. Res. 2004, 94, 1418–1428. [Google Scholar] [CrossRef]
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed.; Sinauer Associates, Inc.: Sunderland, MA, USA, 2001; p. 814. [Google Scholar]
- Gong, Q.; Zhang, L.; Vincent, G.M.; Horne, B.D.; Zhou, Z. Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense-mediated mRNA decay in human long-QT syndrome. Circulation 2007, 116, 17–24. [Google Scholar] [CrossRef]
- Dausse, E.; Berthet, M.; Denjoy, I.; Andre-Fouet, X.; Cruaud, C.; Bennaceur, M.; Faure, S.; Coumel, P.; Schwartz, K.; Guicheney, P. A mutation in HERG associated with notched T waves in long QT syndrome. J. Mol. Cell Cardiol. 1996, 28, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Curran, M.E.; Spector, P.S.; Keating, M.T. Spectrum of HERG K+-channel dysfunction in an inherited cardiac arrhythmia. Proc. Natl. Acad. Sci. USA 1996, 93, 2208–2212. [Google Scholar] [CrossRef] [PubMed]
- Satler, C.A.; Walsh, E.P.; Vesely, M.R.; Plummer, M.H.; Ginsburg, G.S.; Jacob, H.J. Novel missense mutation in the cyclic nucleotide-binding domain of HERG causes long QT syndrome. Am. J. Med. Genet. 1996, 65, 27–35. [Google Scholar] [CrossRef]
- Li, X.; Xu, J.; Li, M. The human delta1261 mutation of the HERG potassium channel results in a truncated protein that contains a subunit interaction domain and decreases the channel expression. J. Biol. Chem. 1997, 272, 705–708. [Google Scholar] [CrossRef]
- Nakajima, T.; Furukawa, T.; Tanaka, T.; Katayama, Y.; Nagai, R.; Nakamura, Y.; Hiraoka, M. Novel mechanism of HERG current suppression in LQT2: Shift in voltage dependence of HERG inactivation. Circ. Res. 1998, 83, 415–422. [Google Scholar] [CrossRef]
- Zhou, Z.; Gong, Q.; Epstein, M.L.; January, C.T. HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. J. Biol. Chem. 1998, 273, 21061–21066. [Google Scholar] [CrossRef]
- Chen, J.; Zou, A.; Splawski, I.; Keating, M.T.; Sanguinetti, M.C. Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. J. Biol. Chem. 1999, 274, 10113–10118. [Google Scholar] [CrossRef]
- Yoshida, H.; Horie, M.; Otani, H.; Takano, M.; Tsuji, K.; Kubota, T.; Fukunami, M.; Sasayama, S. Characterization of a novel missense mutation in the pore of HERG in a patient with long QT syndrome. J. Cardiovasc. Electrophysiol. 1999, 10, 1262–1270. [Google Scholar] [CrossRef]
- Stump, M.R.; Gong, Q.; Zhou, Z. LQT2 nonsense mutations generate trafficking defective NH2-terminally truncated channels by the reinitiation of translation. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1397–H1404. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bianchi, L.; Priori, S.G.; Napolitano, C.; Surewicz, K.A.; Dennis, A.T.; Memmi, M.; Schwartz, P.J.; Brown, A.M. Mechanisms of I(Ks) suppression in LQT1 mutants. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H3003–H3011. [Google Scholar] [CrossRef] [PubMed]
- Chouabe, C.; Neyroud, N.; Guicheney, P.; Lazdunski, M.; Romey, G.; Barhanin, J. Properties of KvLQT1 K+ channel mutations in Romano-Ward and Jervell and Lange-Nielsen inherited cardiac arrhythmias. EMBO J. 1997, 16, 5472–5479. [Google Scholar] [CrossRef] [PubMed]
- Shalaby, F.Y.; Levesque, P.C.; Yang, W.P.; Little, W.A.; Conder, M.L.; Jenkins-West, T.; Blanar, M.A. Dominant-negative KvLQT1 mutations underlie the LQT1 form of long QT syndrome. Circulation 1997, 96, 1733–1736. [Google Scholar] [CrossRef]
- Murray, A.; Donger, C.; Fenske, C.; Spillman, I.; Richard, P.; Dong, Y.B.; Neyroud, N.; Chevalier, P.; Denjoy, I.; Carter, N.; et al. Splicing mutations in KCNQ1: A mutation hot spot at codon 344 that produces in frame transcripts. Circulation 1999, 100, 1077–1084. [Google Scholar] [CrossRef]
- Franqueza, L.; Lin, M.; Shen, J.; Splawski, I.; Keating, M.T.; Sanguinetti, M.C. Long QT syndrome-associated mutations in the S4-S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J. Biol. Chem. 1999, 274, 21063–21070. [Google Scholar] [CrossRef]
- Huang, H.; Kuenze, G.; Smith, J.A.; Taylor, K.C.; Duran, A.M.; Hadziselimovic, A.; Meiler, J.; Vanoye, C.G.; George, A.L., Jr.; Sanders, C.R. Mechanisms of KCNQ1 channel dysfunction in long QT syndrome involving voltage sensor domain mutations. Sci. Adv. 2018, 4, eaar2631. [Google Scholar] [CrossRef]
- Thompson, E.; Eldstrom, J.; Westhoff, M.; McAfee, D.; Balse, E.; Fedida, D. cAMP-dependent regulation of IKs single-channel kinetics. J. Gen. Physiol. 2017, 149, 781–798. [Google Scholar] [CrossRef]
- Lopes, C.M.; Remon, J.I.; Matavel, A.; Sui, J.L.; Keselman, I.; Medei, E.; Shen, Y.; Rosenhouse-Dantsker, A.; Rohacs, T.; Logothetis, D.E. Protein kinase A modulates PLC-dependent regulation and PIP2-sensitivity of K+ channels. Channels 2007, 1, 124–134. [Google Scholar] [CrossRef]
- Lundby, A.; Andersen, M.N.; Steffensen, A.B.; Horn, H.; Kelstrup, C.D.; Francavilla, C.; Jensen, L.J.; Schmitt, N.; Thomsen, M.B.; Olsen, J.V. In vivo phosphoproteomics analysis reveals the cardiac targets of beta-adrenergic receptor signaling. Sci. Signal 2013, 6, rs11. [Google Scholar] [CrossRef]
- Barhanin, J.; Lesage, F.; Guillemare, E.; Fink, M.; Lazdunski, M.; Romey, G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 1996, 384, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Curran, M.E.; Zou, A.; Shen, J.; Spector, P.S.; Atkinson, D.L.; Keating, M.T. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 1996, 384, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, J.; Motoike, H.K.; Rao, J.; Kass, R.S. Regulatory actions of the A-kinase anchoring protein Yotiao on a heart potassium channel downstream of PKA phosphorylation. Proc. Natl. Acad. Sci. USA 2004, 101, 16374–16378. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Marquardt, M.L.; Tester, D.J.; Sampson, K.J.; Ackerman, M.J.; Kass, R.S. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc. Natl. Acad. Sci. USA 2007, 104, 20990–20995. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Vanoli, E.; Crotti, L.; Spazzolini, C.; Ferrandi, C.; Goosen, A.; Hedley, P.; Heradien, M.; Bacchini, S.; Turco, A.; et al. Neural control of heart rate is an arrhythmia risk modifier in long QT syndrome. J. Am. Coll. Cardiol. 2008, 51, 920–929. [Google Scholar] [CrossRef]
- Thomas, D.; Khalil, M.; Alter, M.; Schweizer, P.A.; Karle, C.A.; Wimmer, A.B.; Licka, M.; Katus, H.A.; Koenen, M.; Ulmer, H.E.; et al. Biophysical characterization of KCNQ1 P320 mutations linked to long QT syndrome 1. J. Mol. Cell. Cardiol. 2010, 48, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Brink, P.A.; Crotti, L.; Corfield, V.; Goosen, A.; Durrheim, G.; Hedley, P.; Heradien, M.; Geldenhuys, G.; Vanoli, E.; Bacchini, S.; et al. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation 2005, 112, 2602–2610. [Google Scholar] [CrossRef]
- Heijman, J.; Spatjens, R.L.; Seyen, S.R.; Lentink, V.; Kuijpers, H.J.; Boulet, I.R.; de Windt, L.J.; David, M.; Volders, P.G. Dominant-negative control of cAMP-dependent IKs upregulation in human long-QT syndrome type 1. Circ. Res. 2012, 110, 211–219. [Google Scholar] [CrossRef]
- Barsheshet, A.; Goldenberg, I.; O-Uchi, J.; Moss, A.J.; Jons, C.; Shimizu, W.; Wilde, A.A.; McNitt, S.; Peterson, D.R.; Zareba, W.; et al. Mutations in cytoplasmic loops of the KCNQ1 channel and the risk of life-threatening events: Implications for mutation-specific response to beta-blocker therapy in type 1 long-QT syndrome. Circulation 2012, 125, 1988–1996. [Google Scholar] [CrossRef]
- Wu, J.; Naiki, N.; Ding, W.G.; Ohno, S.; Kato, K.; Zang, W.J.; Delisle, B.P.; Matsuura, H.; Horie, M. A molecular mechanism for adrenergic-induced long QT syndrome. J. Am. Coll Cardiol. 2014, 63, 819–827. [Google Scholar] [CrossRef]
- Policarova, M.; Novotny, T.; Bebarova, M. Impaired Adrenergic/Protein Kinase A Response of Slow Delayed Rectifier Potassium Channels as a Long QT Syndrome Motif: Importance and Unknowns. Can. J. Cardiol. 2019, 35, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Nunziato, D.A.; Pitt, G.S. KCNQ1 assembly and function is blocked by long-QT syndrome mutations that disrupt interaction with calmodulin. Circ. Res. 2006, 98, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Shamgar, L.; Ma, L.; Schmitt, N.; Haitin, Y.; Peretz, A.; Wiener, R.; Hirsch, J.; Pongs, O.; Attali, B. Calmodulin is essential for cardiac IKS channel gating and assembly: Impaired function in long-QT mutations. Circ. Res. 2006, 98, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Loussouarn, G.; Park, K.H.; Bellocq, C.; Baro, I.; Charpentier, F.; Escande, D. Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: A functional homology between voltage-gated and inward rectifier K+ channels. EMBO J. 2003, 22, 5412–5421. [Google Scholar] [CrossRef]
- Zaydman, M.A.; Silva, J.R.; Delaloye, K.; Li, Y.; Liang, H.; Larsson, H.P.; Shi, J.; Cui, J. Kv7.1 ion channels require a lipid to couple voltage sensing to pore opening. Proc. Natl. Acad. Sci. USA 2013, 110, 13180–13185. [Google Scholar] [CrossRef]
- Park, K.H.; Piron, J.; Dahimene, S.; Merot, J.; Baro, I.; Escande, D.; Loussouarn, G. Impaired KCNQ1-KCNE1 and phosphatidylinositol-4,5-bisphosphate interaction underlies the long QT syndrome. Circ. Res. 2005, 96, 730–739. [Google Scholar] [CrossRef]
- Matavel, A.; Medei, E.; Lopes, C.M. PKA and PKC partially rescue long QT type 1 phenotype by restoring channel-PIP2 interactions. Channels 2010, 4, 3–11. [Google Scholar] [CrossRef]
- Chen, Y.H.; Xu, S.J.; Bendahhou, S.; Wang, X.L.; Wang, Y.; Xu, W.Y.; Jin, H.W.; Sun, H.; Su, X.Y.; Zhuang, Q.N.; et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 2003, 299, 251–254. [Google Scholar] [CrossRef]
- Hong, K.; Piper, D.R.; Diaz-Valdecantos, A.; Brugada, J.; Oliva, A.; Burashnikov, E.; Santos-de-Soto, J.; Grueso-Montero, J.; Diaz-Enfante, E.; Brugada, P.; et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc. Res. 2005, 68, 433–440. [Google Scholar] [CrossRef]
- Bartos, D.C.; Duchatelet, S.; Burgess, D.E.; Klug, D.; Denjoy, I.; Peat, R.; Lupoglazoff, J.M.; Fressart, V.; Berthet, M.; Ackerman, M.J.; et al. R231C mutation in KCNQ1 causes long QT syndrome type 1 and familial atrial fibrillation. Heart Rhythm Off. J. Heart Rhythm Soc. 2011, 8, 48–55. [Google Scholar] [CrossRef]
- Bartos, D.C.; Anderson, J.B.; Bastiaenen, R.; Johnson, J.N.; Gollob, M.H.; Tester, D.J.; Burgess, D.E.; Homfray, T.; Behr, E.R.; Ackerman, M.J.; et al. A KCNQ1 Mutation Causes a High Penetrance for Familial Atrial Fibrillation. J. Cardiovasc. Electrophysiol. 2013, 24, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Makino, S.; Melman, Y.F.; Shea, M.A.; Goyal, S.B.; Rosenzweig, A.; Macrae, C.A.; Ellinor, P.T. Mutation in the S3 segment of KCNQ1 results in familial lone atrial fibrillation. Heart Rhythm Off. J. Heart Rhythm Soc. 2009, 6, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Ohno, S.; Ashihara, T.; Itoh, H.; Ding, W.G.; Toyoda, F.; Makiyama, T.; Aoki, H.; Nakamura, Y.; Delisle, B.P.; et al. A novel KCNQ1 missense mutation identified in a patient with juvenile-onset atrial fibrillation causes constitutively open IKs channels. Heart Rhythm 2014, 11, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Henrion, U.; Zumhagen, S.; Steinke, K.; Strutz-Seebohm, N.; Stallmeyer, B.; Lang, F.; Schulze-Bahr, E.; Seebohm, G. Overlapping cardiac phenotype associated with a familial mutation in the voltage sensor of the KCNQ1 channel. Cell Physiol. Biochem. 2012, 29, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Knoche, J.W.; Orland, K.M.; January, C.T.; Maginot, K.R. Atrial Fibrillation and Long QT Syndrome Presenting in a 12-Year-Old Girl. Case Rep. Pediatr. 2012, 2012, 124838. [Google Scholar] [CrossRef]
- Tamargo, M.; Espinosa, M.Á.; Gómez-Carrillo, V.; Juárez, M.; Fernández-Avilés, F.; Yotti, R. Sudden Death in a Young Patient with Atrial Fibrillation. Cardiogenetics 2017, 7, 18–21. [Google Scholar] [CrossRef][Green Version]
- Kharche, S.; Adeniran, I.; Stott, J.; Law, P.; Boyett, M.R.; Hancox, J.C.; Zhang, H. Pro-arrhythmogenic effects of the S140G KCNQ1 mutation in human atrial fibrillation—Insights from modelling. J. Physiol. 2012, 590, 4501–4514. [Google Scholar] [CrossRef]
- Jonsson, M.K.; van der Heyden, M.A.; van Veen, T.A. Deciphering hERG channels: Molecular basis of the rapid component of the delayed rectifier potassium current. J. Mol. Cell Cardiol. 2012, 53, 369–374. [Google Scholar] [CrossRef]
- Abbott, G.W.; Sesti, F.; Splawski, I.; Buck, M.E.; Lehmann, M.H.; Timothy, K.W.; Keating, M.T.; Goldstein, S.A. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell 1999, 97, 175–187. [Google Scholar] [CrossRef]
- Ficker, E.; Thomas, D.; Viswanathan, P.C.; Dennis, A.T.; Priori, S.G.; Napolitano, C.; Memmi, M.; Wible, B.A.; Kaufman, E.S.; Iyengar, S.; et al. Novel characteristics of a misprocessed mutant HERG channel linked to hereditary long QT syndrome. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H1748–H1756. [Google Scholar] [CrossRef]
- Ono, M.; Burgess, D.E.; Schroder, E.A.; Elayi, C.S.; Anderson, C.L.; January, C.T.; Sun, B.; Immadisetty, K.; Kekenes-Huskey, P.M.; Delisle, B.P. Long QT Syndrome Type 2: Emerging Strategies for Correcting Class 2 KCNH2 (hERG) Mutations and Identifying New Patients. Biomolecules 2020, 10, 1144. [Google Scholar] [CrossRef] [PubMed]
- Ficker, E.; Obejero-Paz, C.A.; Zhao, S.; Brown, A.M. The binding site for channel blockers that rescue misprocessed human long QT syndrome type 2 ether-a-gogo-related gene (HERG) mutations. J. Biol. Chem. 2002, 277, 4989–4998. [Google Scholar] [CrossRef] [PubMed]
- Ficker, E.; Dennis, A.T.; Wang, L.; Brown, A.M. Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ. Res. 2003, 92, e87–e100. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.; Wan, X.; Ramirez-Navarro, A.; Tesar, P.J.; Kaufman, E.S.; Ficker, E.; George, A.L., Jr.; Deschenes, I. Physiological genomics identifies genetic modifiers of long QT syndrome type 2 severity. J. Clin. Investig. 2018, 128, 1043–1056. [Google Scholar] [CrossRef]
- Hall, A.R.; Anderson, C.L.; Smith, J.L.; Mirshahi, T.; Elayi, C.S.; January, C.T.; Delisle, B.P. Visualizing Mutation-Specific Differences in the Trafficking-Deficient Phenotype of Kv11.1 Proteins Linked to Long QT Syndrome Type 2. Front. Physiol. 2018, 9, 584. [Google Scholar] [CrossRef]
- Jones, E.M.; Roti Roti, E.C.; Wang, J.; Delfosse, S.A.; Robertson, G.A. Cardiac IKr channels minimally comprise hERG 1a and 1b subunits. J. Biol. Chem. 2004, 279, 44690–44694. [Google Scholar] [CrossRef]
- McNally, B.A.; Pendon, Z.D.; Trudeau, M.C. hERG1a and hERG1b potassium channel subunits directly interact and preferentially form heteromeric channels. J. Biol. Chem. 2017, 292, 21548–21557. [Google Scholar] [CrossRef]
- London, B.; Trudeau, M.C.; Newton, K.P.; Beyer, A.K.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Satler, C.A.; Robertson, G.A. Two isoforms of the mouse ether-a-go-go-related gene coassemble to form channels with properties similar to the rapidly activating component of the cardiac delayed rectifier K+ current. Circ. Res. 1997, 81, 870–878. [Google Scholar] [CrossRef]
- Feng, L.; Zhang, J.; Lee, C.; Kim, G.; Liu, F.; Petersen, A.J.; Lim, E.; Anderson, C.L.; Orland, K.M.; Robertson, G.A.; et al. Long QT Syndrome KCNH2 Variant Induces hERG1a/1b Subunit Imbalance in Patient-Specific Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Arrhythm Electrophysiol. 2021, 14, e009343. [Google Scholar] [CrossRef]
- Yang, P.C.; Perissinotti, L.L.; Lopez-Redondo, F.; Wang, Y.; DeMarco, K.R.; Jeng, M.T.; Vorobyov, I.; Harvey, R.D.; Kurokawa, J.; Noskov, S.Y.; et al. A multiscale computational modelling approach predicts mechanisms of female sex risk in the setting of arousal-induced arrhythmias. J. Physiol. 2017, 595, 4695–4723. [Google Scholar] [CrossRef]
- Meregalli, P.G.; Westendorp, I.C.; Tan, H.L.; Elsman, P.; Kok, W.E.; Wilde, A.A. Pregnancy and the risk of torsades de pointes in congenital long-QT syndrome. Neth. Heart J. 2008, 16, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Gao, M.; Fang, S.; Zheng, R.; Peng, D.; Luo, Q.; Yu, B. L51P, a novel mutation in the PAS domain of hERG channel, confers long QT syndrome by impairing channel activation. Am. J. Transl. Res. 2020, 12, 8040–8049. [Google Scholar] [PubMed]
- Miranda, W.E.; DeMarco, K.R.; Guo, J.; Duff, H.J.; Vorobyov, I.; Clancy, C.E.; Noskov, S.Y. Selectivity filter modalities and rapid inactivation of the hERG1 channel. Proc. Natl. Acad. Sci. USA 2020, 117, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Kroncke, B.M.; Yang, T.; Kannankeril, P.; Shoemaker, M.B.; Roden, D.M. Exploiting ion channel structure to assess rare variant pathogenicity. Heart Rhythm 2018, 15, 890–894. [Google Scholar] [CrossRef] [PubMed]
- Brewer, K.R.; Kuenze, G.; Vanoye, C.G.; George, A.L., Jr.; Meiler, J.; Sanders, C.R. Structures Illuminate Cardiac Ion Channel Functions in Health and in Long QT Syndrome. Front. Pharm. 2020, 11, 550. [Google Scholar] [CrossRef] [PubMed]
- DeMarco, K.R.; Bekker, S.; Vorobyov, I. Challenges and advances in atomistic simulations of potassium and sodium ion channel gating and permeation. J. Physiol. 2019, 597, 679–698. [Google Scholar] [CrossRef] [PubMed]
- Paquin, A.; Ye, D.; Tester, D.J.; Kapplinger, J.D.; Zimmermann, M.T.; Ackerman, M.J. Even pore-localizing missense variants at highly conserved sites in KCNQ1-encoded Kv7.1 channels may have wild-type function and not cause type 1 long QT syndrome: Do not rely solely on the genetic test company’s interpretation. Heart Rhythm Case Rep. 2018, 4, 37–44. [Google Scholar] [CrossRef]
- Agudelo, W.A.; Gil-Quinones, S.R.; Fonseca, A.; Arenas, A.; Castro, L.; Sierra-Diaz, D.C.; Patarroyo, M.A.; Laissue, P.; Suarez, C.F.; Cabrera, R. Structural Modelling of KCNQ1 and KCNH2 Double Mutant Proteins, Identified in Two Severe Long QT Syndrome Cases, Reveals New Insights into Cardiac Channelopathies. Int. J. Mol. Sci. 2021, 22, 12861. [Google Scholar] [CrossRef]
- Paquet, E.; Viktor, H.L. Molecular dynamics, monte carlo simulations, and langevin dynamics: A computational review. Biomed. Res. Int. 2015, 2015, 183918. [Google Scholar] [CrossRef]
- Costa, F.; Guardiani, C.; Giacomello, A. Molecular dynamics simulations suggest possible activation and deactivation pathways in the hERG channel. Commun. Biol. 2022, 5, 165. [Google Scholar] [CrossRef]
- Kuenze, G.; Duran, A.M.; Woods, H.; Brewer, K.R.; McDonald, E.F.; Vanoye, C.G.; George, A.L., Jr.; Sanders, C.R.; Meiler, J. Upgraded molecular models of the human KCNQ1 potassium channel. PLoS ONE 2019, 14, e0220415. [Google Scholar] [CrossRef] [PubMed]
- Tobelaim, W.S.; Dvir, M.; Lebel, G.; Cui, M.; Buki, T.; Peretz, A.; Marom, M.; Haitin, Y.; Logothetis, D.E.; Hirsch, J.A.; et al. Competition of calcified calmodulin N lobe and PIP2 to an LQT mutation site in Kv7.1 channel. Proc. Natl. Acad. Sci. USA 2017, 114, E869–E878. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; MacKinnon, R. Cryo-EM Structure of the Open Human Ether-a-go-go-Related K(+) Channel hERG. Cell 2017, 169, 422–430.e410. [Google Scholar] [CrossRef] [PubMed]
- van Gunsteren, W.F.; Daura, X.; Fuchs, P.F.J.; Hansen, N.; Horta, B.A.C.; Hunenberger, P.H.; Mark, A.E.; Pechlaner, M.; Riniker, S.; Oostenbrink, C. On the Effect of the Various Assumptions and Approximations used in Molecular Simulations on the Properties of Bio-Molecular Systems: Overview and Perspective on Issues. Chemphyschem 2021, 22, 264–282. [Google Scholar] [CrossRef] [PubMed]
- Meier, K.; Choutko, A.; Dolenc, J.; Eichenberger, A.P.; Riniker, S.; van Gunsteren, W.F. Multi-resolution simulation of biomolecular systems: A review of methodological issues. Angew. Chem. Int. Ed. Engl. 2013, 52, 2820–2834. [Google Scholar] [CrossRef]
- Takada, S. Go model revisited. Biophys. Phys. 2019, 16, 248–255. [Google Scholar] [CrossRef]
- Torrisi, M.; Pollastri, G.; Le, Q. Deep learning methods in protein structure prediction. Comput. Struct. Biotechnol. J. 2020, 18, 1301–1310. [Google Scholar] [CrossRef]
- Pandurangan, A.P.; Blundell, T.L. Prediction of impacts of mutations on protein structure and interactions: SDM, a statistical approach, and mCSM, using machine learning. Protein Sci. 2020, 29, 247–257. [Google Scholar] [CrossRef]
- Li, B.; Mendenhall, J.L.; Kroncke, B.M.; Taylor, K.C.; Huang, H.; Smith, D.K.; Vanoye, C.G.; Blume, J.D.; George, A.L., Jr.; Sanders, C.R.; et al. Predicting the Functional Impact of KCNQ1 Variants of Unknown Significance. Circ. Cardiovasc. Genet. 2017, 10, e001754. [Google Scholar] [CrossRef]
- Gepp, M.M.; Hutter, M.C. Determination of hERG channel blockers using a decision tree. Bioorg. Med. Chem. 2006, 14, 5325–5332. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, J.; Wang, Y.; Fan, Y.; Zhu, L.; Yang, Y.; Chen, X.; Lu, T.; Chen, Y.; Liu, H. Prediction of hERG K+ channel blockage using deep neural networks. Chem. Biol. Drug Des. 2019, 94, 1973–1985. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Yu, M.S.; Kazmi, S.R.; Oh, S.Y.; Rhee, K.H.; Bae, M.A.; Lee, B.H.; Shin, D.S.; Oh, K.S.; Ceong, H.; et al. Computational determination of hERG-related cardiotoxicity of drug candidates. BMC Bioinform. 2019, 20, 250. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, L.; Li, S.; Yang, T.; Liu, L.; Zhao, J.; Liu, H. Prediction of hERG potassium channel blockage using ensemble learning methods and molecular fingerprints. Toxicol. Lett 2020, 332, 88–96. [Google Scholar] [CrossRef]
- Draelos, R.L.; Ezekian, J.E.; Zhuang, F.; Moya-Mendez, M.E.; Zhang, Z.; Rosamilia, M.B.; Manivannan, P.K.R.; Henao, R.; Landstrom, A.P. GENESIS: Gene-Specific Machine Learning Models for Variants of Uncertain Significance Found in Catecholaminergic Polymorphic Ventricular Tachycardia and Long QT Syndrome-Associated Genes. Circ. Arrhythm Electrophysiol. 2022, 15, e010326. [Google Scholar] [CrossRef] [PubMed]
- Oliveira-Mendes, B.; Feliciangeli, S.; Menard, M.; Chatelain, F.; Alameh, M.; Montnach, J.; Nicolas, S.; Ollivier, B.; Barc, J.; Baro, I.; et al. A standardised hERG phenotyping pipeline to evaluate KCNH2 genetic variant pathogenicity. Clin. Transl. Med. 2021, 11, e609. [Google Scholar] [CrossRef] [PubMed]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzym. 2011, 487, 545–574. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5–6. [Google Scholar] [CrossRef]
- Jisna, V.A.; Jayaraj, P.B. Protein Structure Prediction: Conventional and Deep Learning Perspectives. Protein. J. 2021, 40, 522–544. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- AlQuraishi, M. ProteinNet: A standardized data set for machine learning of protein structure. BMC Bioinform. 2019, 20, 311. [Google Scholar] [CrossRef] [PubMed]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of protein structure prediction (CASP)-Round XIII. Proteins 2019, 87, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Meinen, B.A.; Bahl, C.D. Breakthroughs in computational design methods open up new frontiers for de novo protein engineering. Protein Eng. Des. Sel. 2021, 34, gzab007. [Google Scholar] [CrossRef]
- Sruthi, C.K.; Balaram, H.; Prakash, M.K. Toward Developing Intuitive Rules for Protein Variant Effect Prediction Using Deep Mutational Scanning Data. ACS Omega 2020, 5, 29667–29677. [Google Scholar] [CrossRef]
- Thusberg, J.; Vihinen, M. Pathogenic or not? And if so, then how? Studying the effects of missense mutations using bioinformatics methods. Hum. Mutat. 2009, 30, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Coskuner-Weber, O.; Uversky, V.N. Insights into the Molecular Mechanisms of Alzheimer’s and Parkinson’s Diseases with Molecular Simulations: Understanding the Roles of Artificial and Pathological Missense Mutations in Intrinsically Disordered Proteins Related to Pathology. Int. J. Mol. Sci. 2018, 19, 336. [Google Scholar] [CrossRef]
- Sagar, A.; Xue, B. Recent Advances in Machine Learning Based Prediction of RNA-protein Interactions. Protein Pept. Lett. 2019, 26, 601–619. [Google Scholar] [CrossRef]
- Terayama, K.; Shinobu, A.; Tsuda, K.; Takemura, K.; Kitao, A. evERdock BAI: Machine-learning-guided selection of protein-protein complex structure. J. Chem. Phys. 2019, 151, 215104. [Google Scholar] [CrossRef]
- Bitencourt-Ferreira, G.; de Azevedo, W.F., Jr. Machine Learning to Predict Binding Affinity. Methods Mol. Biol. 2019, 2053, 251–273. [Google Scholar] [CrossRef]
- Wang, D.D.; Ou-Yang, L.; Xie, H.; Zhu, M.; Yan, H. Predicting the impacts of mutations on protein-ligand binding affinity based on molecular dynamics simulations and machine learning methods. Comput. Struct. Biotechnol. J. 2020, 18, 439–454. [Google Scholar] [CrossRef]
- Pavlova, A.; Zhang, Z.; Acharya, A.; Lynch, D.L.; Pang, Y.T.; Mou, Z.; Parks, J.M.; Chipot, C.; Gumbart, J.C. Machine Learning Reveals the Critical Interactions for SARS-CoV-2 Spike Protein Binding to ACE2. J. Phys. Chem. Lett. 2021, 12, 5494–5502. [Google Scholar] [CrossRef] [PubMed]
- Gurunathan, V.; Hamre, J., 3rd; Klimov, D.K.; Jafri, M.S. Data Mining of Molecular Simulations Suggest Key Amino Acid Residues for Aggregation, Signaling and Drug Action. Biomolecules 2021, 11, 1541. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Feig, M. High-accuracy protein structures by combining machine-learning with physics-based refinement. Proteins 2020, 88, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Qin, Y.; Fontaine, N.T.; Ng Fuk Chong, M.; Maria-Solano, M.A.; Feixas, F.; Cadet, X.F.; Pandjaitan, R.; Garcia-Borras, M.; Cadet, F.; et al. Machine Learning Enables Selection of Epistatic Enzyme Mutants for Stability Against Unfolding and Detrimental Aggregation. Chembiochem 2021, 22, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Babbitt, G.A.; Fokoue, E.P.; Evans, J.R.; Diller, K.I.; Adams, L.E. DROIDS 3.0-Detecting Genetic and Drug Class Variant Impact on Conserved Protein Binding Dynamics. Biophys. J. 2020, 118, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Ponzoni, L.; Nguyen, N.H.; Bahar, I.; Brodsky, J.L. Complementary computational and experimental evaluation of missense variants in the ROMK potassium channel. PLoS Comput. Biol. 2020, 16, e1007749. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kekenes-Huskey, P.M.; Burgess, D.E.; Sun, B.; Bartos, D.C.; Rozmus, E.R.; Anderson, C.L.; January, C.T.; Eckhardt, L.L.; Delisle, B.P. Mutation-Specific Differences in Kv7.1 (KCNQ1) and Kv11.1 (KCNH2) Channel Dysfunction and Long QT Syndrome Phenotypes. Int. J. Mol. Sci. 2022, 23, 7389. https://doi.org/10.3390/ijms23137389
Kekenes-Huskey PM, Burgess DE, Sun B, Bartos DC, Rozmus ER, Anderson CL, January CT, Eckhardt LL, Delisle BP. Mutation-Specific Differences in Kv7.1 (KCNQ1) and Kv11.1 (KCNH2) Channel Dysfunction and Long QT Syndrome Phenotypes. International Journal of Molecular Sciences. 2022; 23(13):7389. https://doi.org/10.3390/ijms23137389
Chicago/Turabian StyleKekenes-Huskey, Peter M., Don E. Burgess, Bin Sun, Daniel C. Bartos, Ezekiel R. Rozmus, Corey L. Anderson, Craig T. January, Lee L. Eckhardt, and Brian P. Delisle. 2022. "Mutation-Specific Differences in Kv7.1 (KCNQ1) and Kv11.1 (KCNH2) Channel Dysfunction and Long QT Syndrome Phenotypes" International Journal of Molecular Sciences 23, no. 13: 7389. https://doi.org/10.3390/ijms23137389
APA StyleKekenes-Huskey, P. M., Burgess, D. E., Sun, B., Bartos, D. C., Rozmus, E. R., Anderson, C. L., January, C. T., Eckhardt, L. L., & Delisle, B. P. (2022). Mutation-Specific Differences in Kv7.1 (KCNQ1) and Kv11.1 (KCNH2) Channel Dysfunction and Long QT Syndrome Phenotypes. International Journal of Molecular Sciences, 23(13), 7389. https://doi.org/10.3390/ijms23137389