
Epigenetic and Transcriptomic Programming of HSC Quiescence Signaling in Large for Gestational Age Neonates

 , , , , , and
, , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
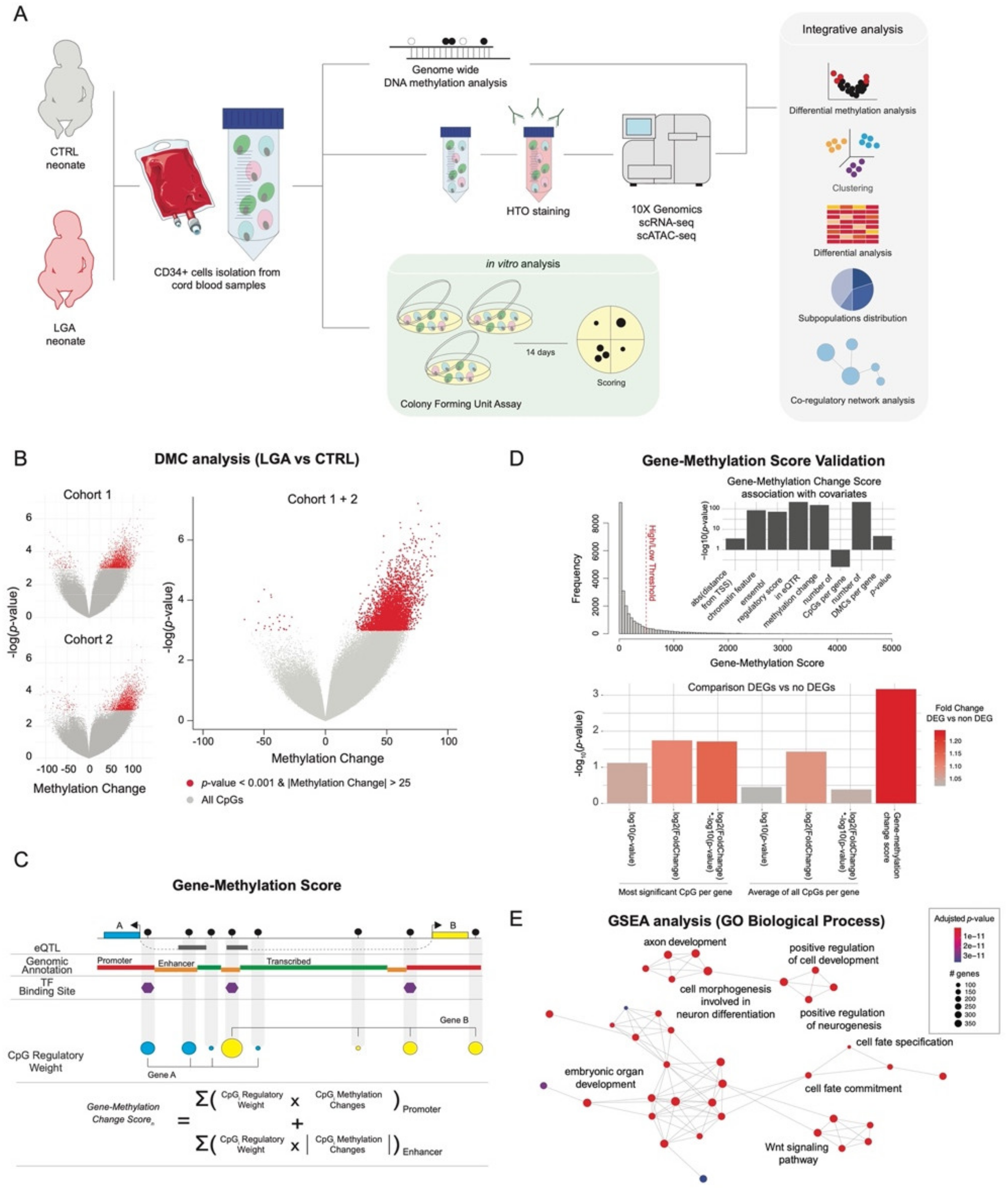
2.1. Optimized Methylation Gene Set Analysis Reveals Association between LGA DNA Hypermethylation and Stem Cell Differentiation Pathways
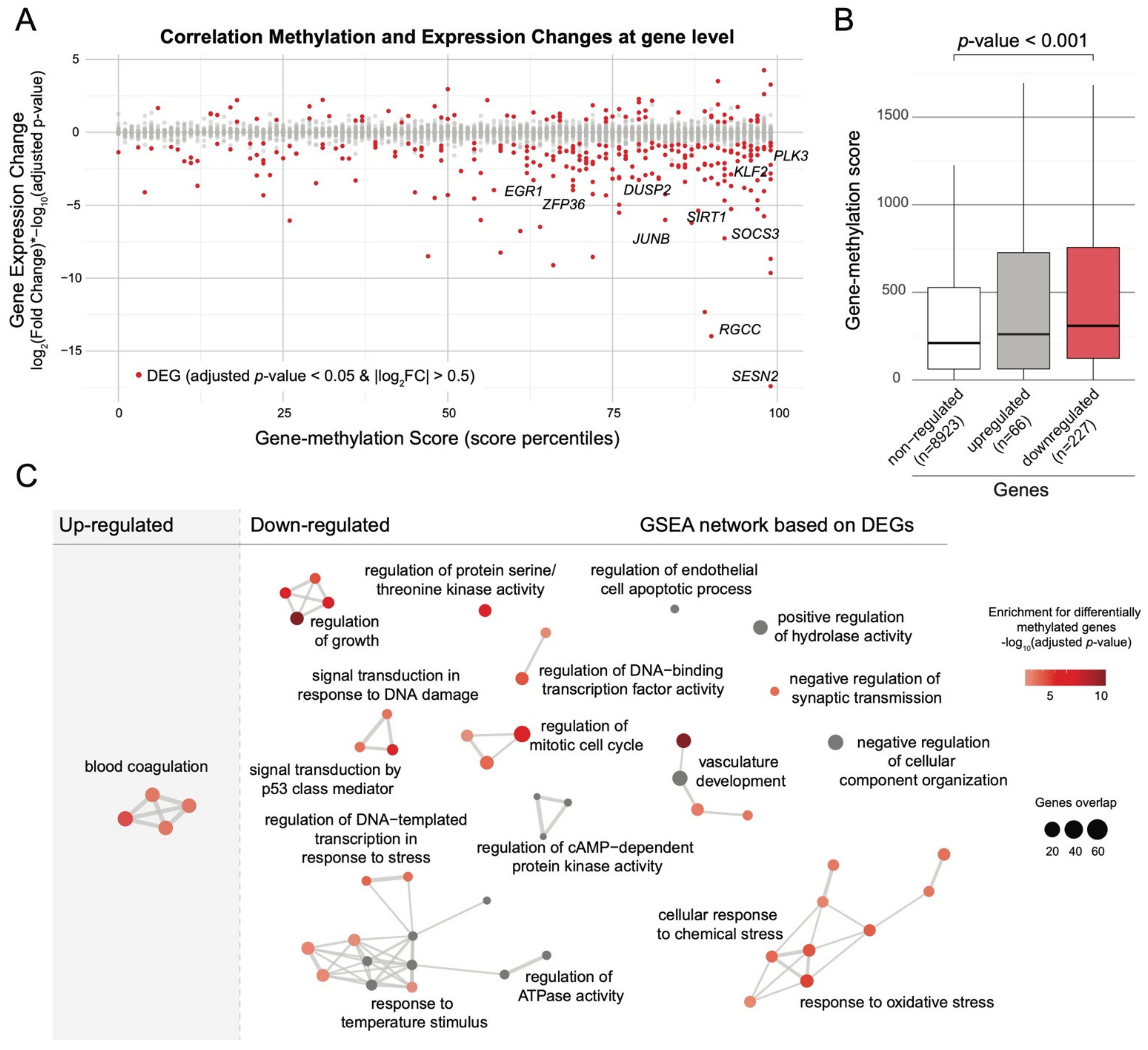
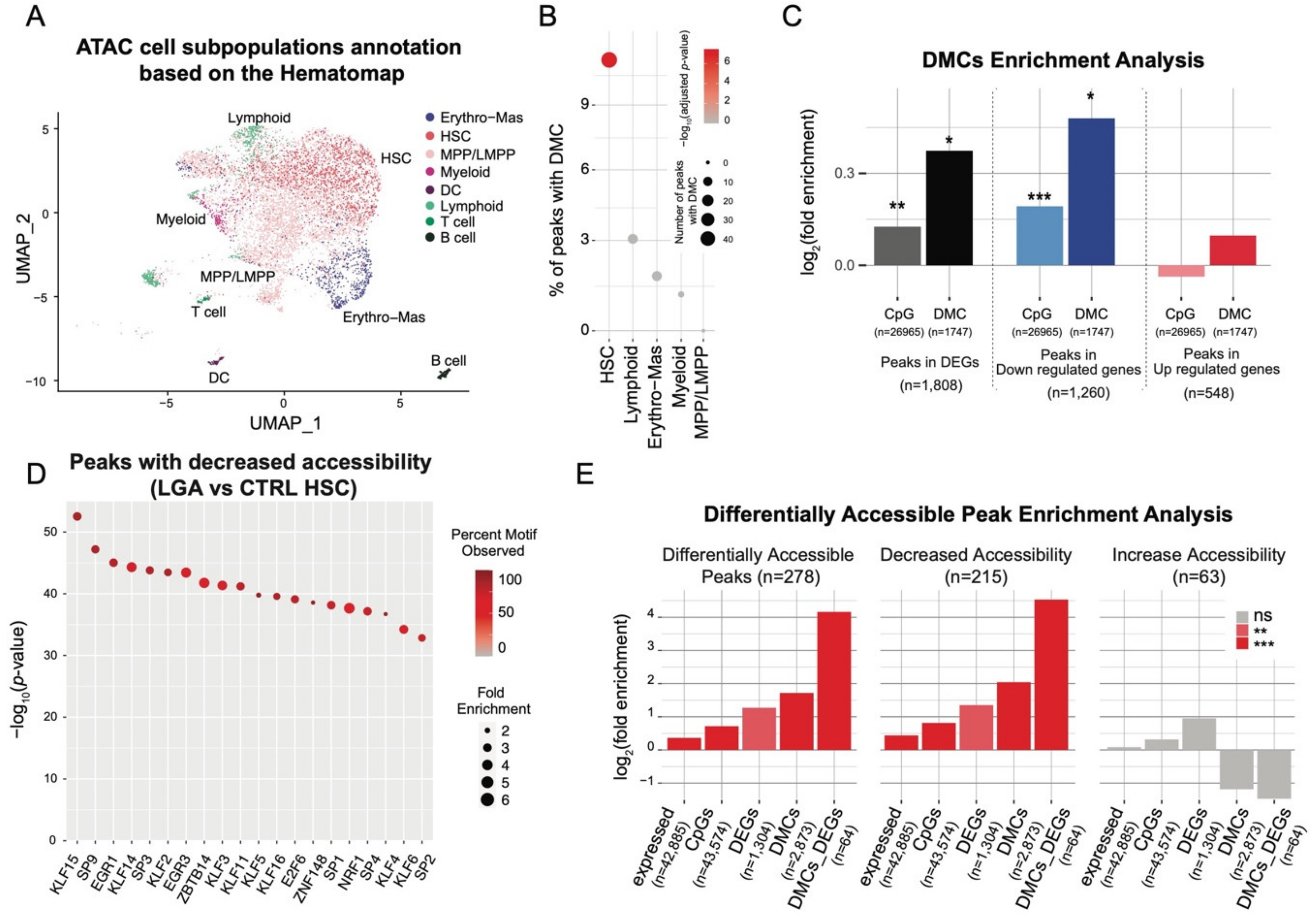
2.2. Single-Cell Transcriptomic Analysis Confirms Alteration of Hyper-Methylated Genes in Pathways Regulating Stem Cell Differentiation among LGA HSCs
2.3. DNA Methylation Changes Occurs in HSCs and DEGs Associated Open Chromatin Regions
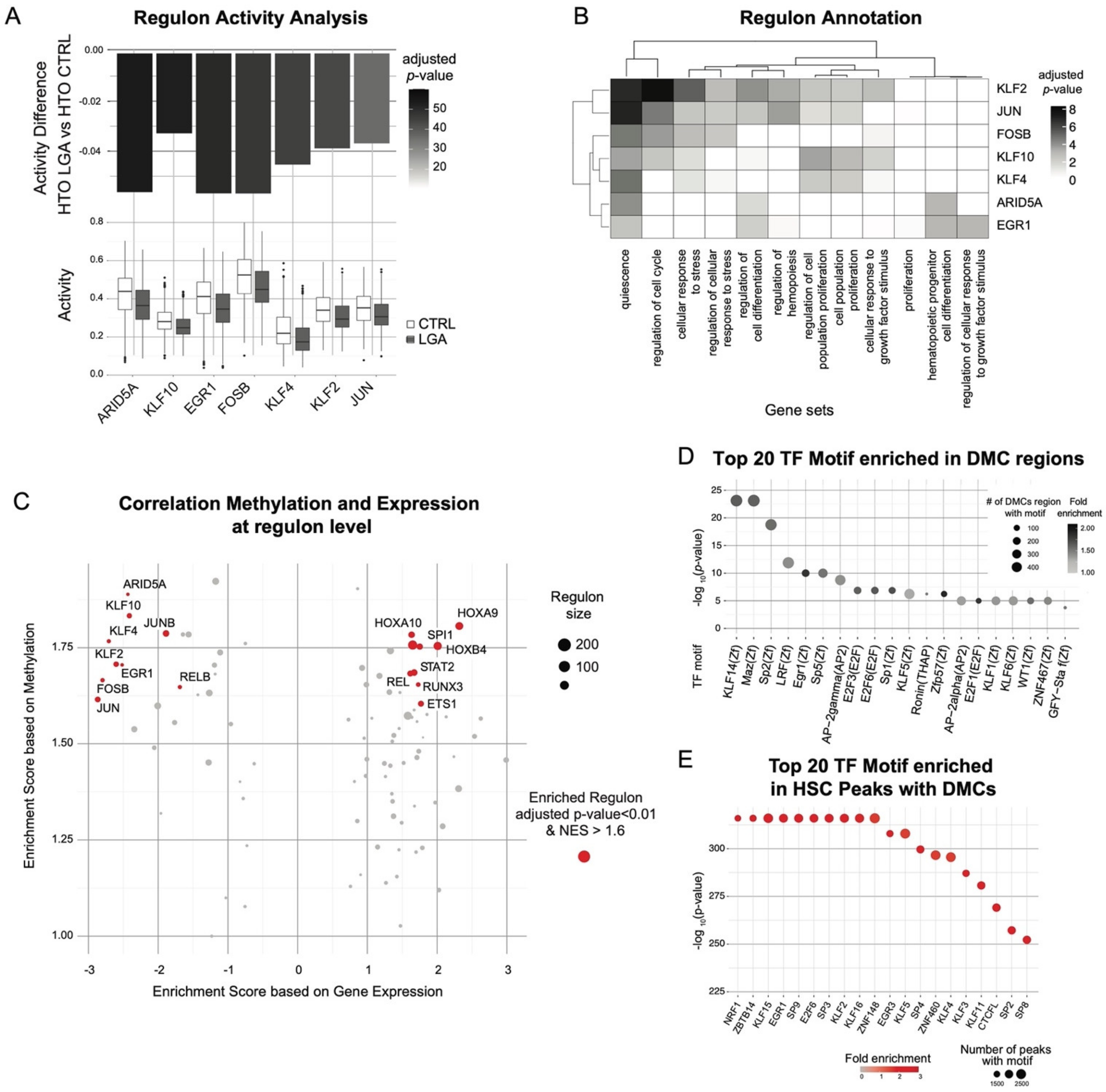
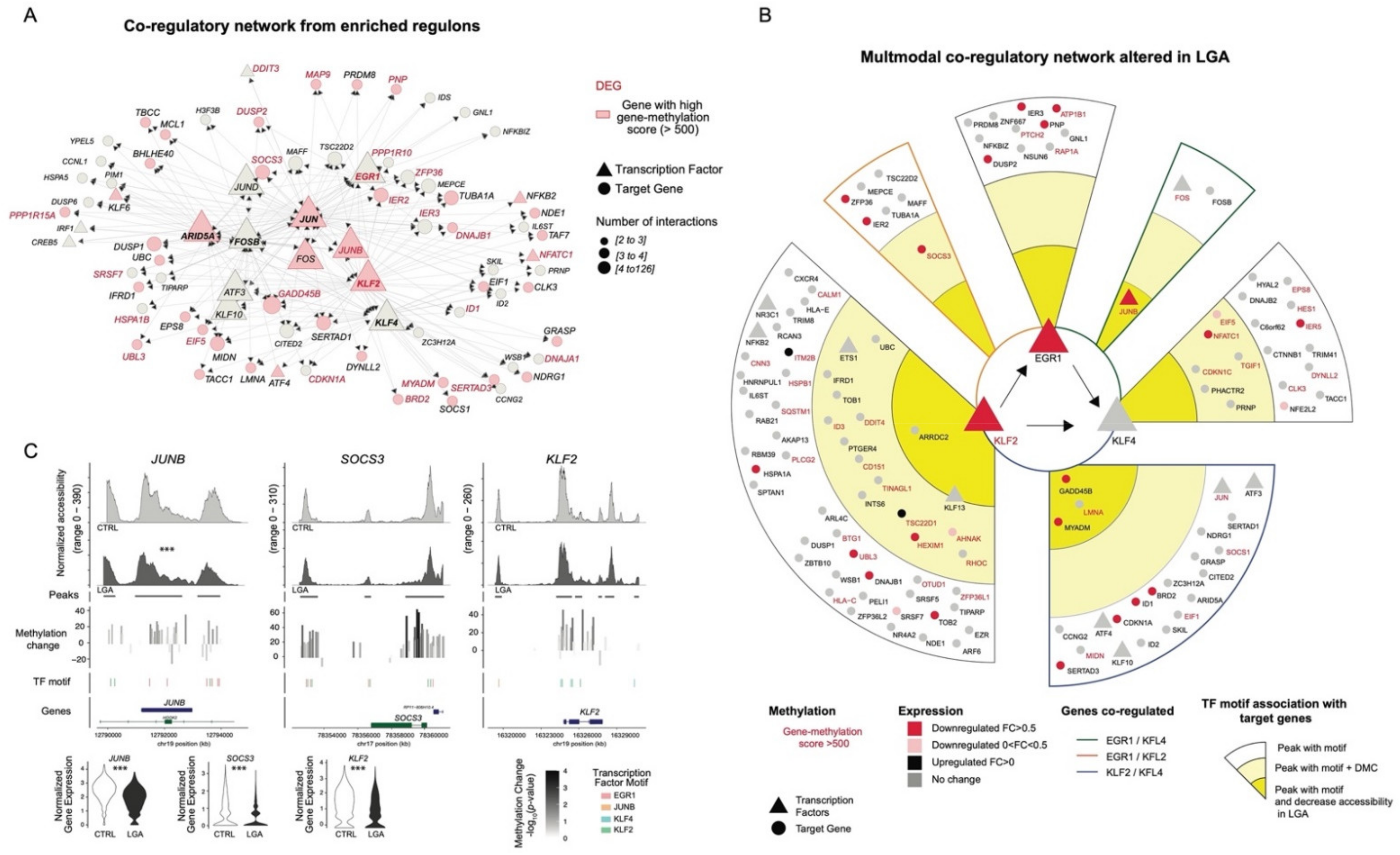
2.4. EGR1, KLF2, and KLF4 Are Key Upstream Regulators Influenced by Early Epigenetic Programming in LGA
2.5. Multimodal Co-Regulatory Network Recapitulating TF-Gene Interactions Influenced by Early Epigenetic Programming in LGA
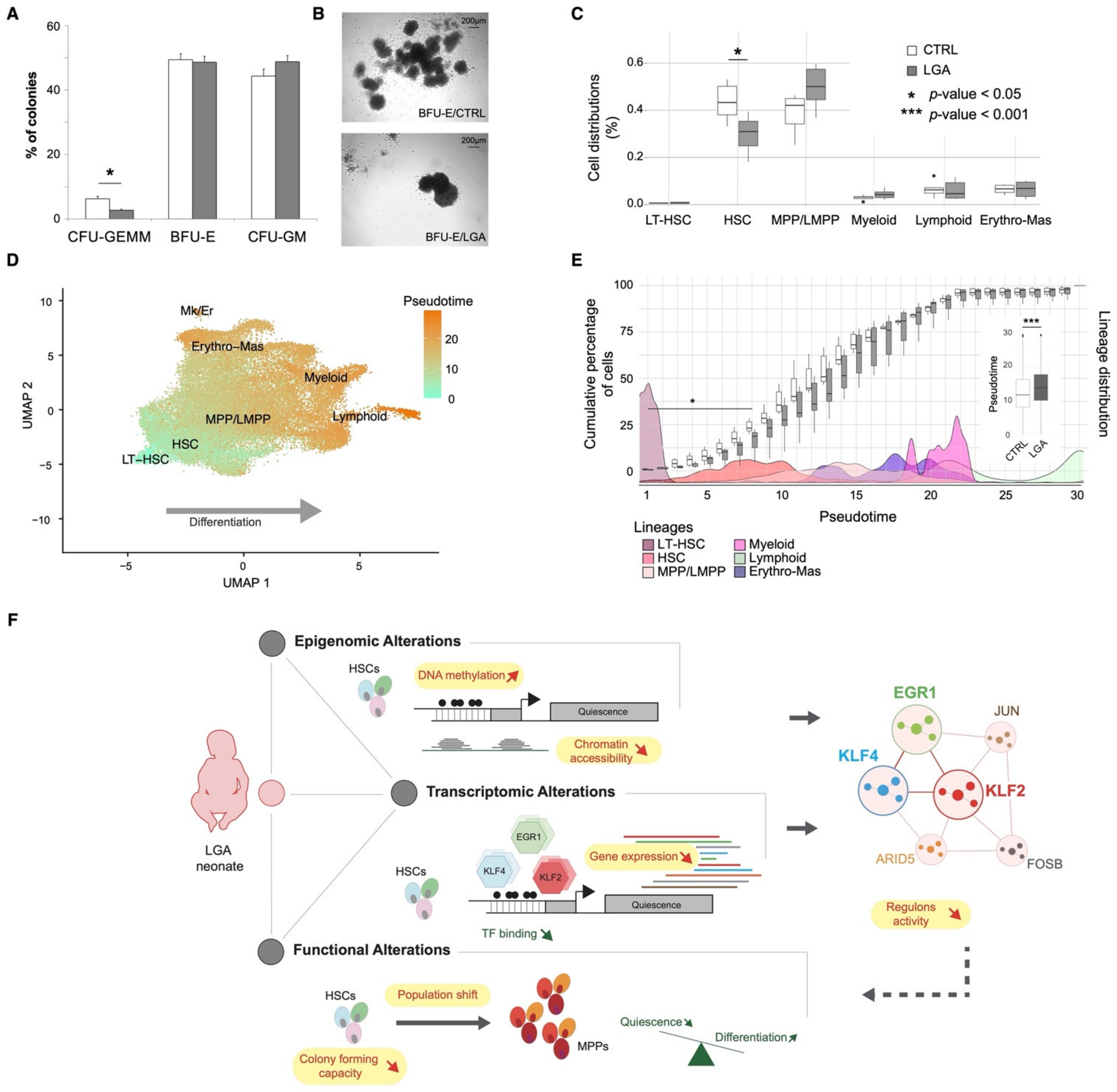
2.6. In Vitro Analysis Confirms the Alteration of HSPCs Differentiation Capacities in LGA
3. Discussion
4. Methods
4.1. Clinical Sample Collection
4.2. Isolation of CD34+ HSPCs
4.3. Genome-Wide DNA Methylation Assay
4.4. Single-Cell RNA Sequencing Libraries Preparation
4.5. Single-Cell ATAC Sequen1cing Libraries Preparation
4.6. HTO Protocol
4.7. Colony Forming Unit Assay
4.8. Data Processing and Statistical Analysis
4.9. Gene-Methylation Score
- (1)
- CpG-score
- (2)
- To concatenate CpG-Scores at gene level: gene-methylation score
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guenechea, G.; Gan, O.I.; Dorrell, C.; Dick, J.E. Distinct classes of human stem cells that differ in proliferative and self-renewal potential. Nat. Immunol. 2001, 2, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Kamimae-Lanning, A.N.; Krasnow, S.M.; Goloviznina, N.A.; Zhu, X.; Roth-Carter, Q.R.; Levasseur, P.R.; Jeng, S.; McWeeney, S.K.; Kurre, P.; Marks, D.L. Maternal high-fat diet and obesity compromise fetal hematopoiesis. Mol. Metab. 2015, 4, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Delahaye, F.; Wijetunga, N.A.; Heo, H.J.; Tozour, J.N.; Zhao, Y.M.; Greally, J.M.; Einstein, F.H. Sexual dimorphism in epigenomic responses of stem cells to extreme fetal growth. Nat. Commun. 2014, 5, 5187. [Google Scholar] [CrossRef] [Green Version]
- Wijetunga, N.A.; Delahaye, F.; Zhao, Y.M.; Golden, A.; Mar, J.C.; Einstein, F.H.; Greally, J.M. The meta-epigenomic structure of purified human stem cell populations is defined at cis-regulatory sequences. Nat. Commun. 2014, 5, 5195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabezas-Wallscheid, N.; Klimmeck, D.; Hansson, J.; Lipka, D.B.; Reyes, A.; Wang, Q.; Weichenhan, D.; Lier, A.; von Paleske, L.; Renders, S.; et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 2014, 15, 507–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamitros, D.; Stoilova, B.; Aboukhalil, Z.; Hamey, F.; Reinisch, A.; Samitsch, M.; Quek, L.; Otto, G.; Repapi, E.; Doondeea, J.; et al. Single-cell analysis reveals the continuum of human lympho-myeloid progenitor cells. Nat. Immunol. 2018, 19, 85–97. [Google Scholar] [CrossRef]
- Stoeckius, M.; Zheng, S.; Houck-Loomis, B.; Hao, S.; Yeung, B.Z.; Mauck, W.M., 3rd; Smibert, P.; Satija, R. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018, 19, 224. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Emerson, S.G. Hematopoietic cytokines, transcription factors and lineage commitment. Oncogene 2002, 21, 3295–3313. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.H.; Hu, Y.; Zhang, Y.; Hu, L.D.; Kong, X. Distinguishing three subtypes of hematopoietic cells based on gene expression profiles using a support vector machine. Biochim. Biophys. Acta. Mol. Basis. Dis. 2018, 1864, 2255–2265. [Google Scholar] [CrossRef]
- Zheng, S.; Papalexi, E.; Butler, A.; Stephenson, W.; Satija, R. Molecular transitions in early progenitors during human cord blood hematopoiesis. Mol. Syst. Biol. 2018, 14, e8041. [Google Scholar] [CrossRef]
- Venezia, T.A.; Merchant, A.A.; Ramos, C.A.; Whitehouse, N.L.; Young, A.S.; Shaw, C.A.; Goodell, M.A. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2004, 2, e301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenen, D.G.; Hromas, R.; Licht, J.D.; Zhang, D.E. Transcription factors, normal myeloid development, and leukemia. Blood 1997, 90, 489–519. [Google Scholar] [CrossRef] [PubMed]
- Antonchuk, J.; Sauvageau, G.; Humphries, R.K. HOXB4-induced expansion of adult hematopoietic stem cells ex vivo. Cell 2002, 109, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhou, B.; Mao, F.; Xu, J.; Miao, H.; Zou, Z.; Phuc Khoa, L.T.; Jang, Y.; Cai, S.; Witkin, M.; et al. HOXA9 Reprograms the Enhancer Landscape to Promote Leukemogenesis. Cancer Cell 2018, 34, 643–658.e5. [Google Scholar] [CrossRef] [Green Version]
- Magnusson, M.; Brun, A.C.; Miyake, N.; Larsson, J.; Ehinger, M.; Bjornsson, J.M.; Wutz, A.; Sigvardsson, M.; Karlsson, S. HOXA10 is a critical regulator for hematopoietic stem cells and erythroid/megakaryocyte development. Blood 2007, 109, 3687–3696. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Hill, A.; Packer, J.; Lin, D.; Ma, Y.A.; Trapnell, C. Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 2017, 14, 309–315. [Google Scholar] [CrossRef]
- Min, I.M.; Pietramaggiori, G.; Kim, F.S.; Passegue, E.; Stevenson, K.E.; Wagers, A.J. The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell 2008, 2, 380–391. [Google Scholar] [CrossRef] [Green Version]
- Cartron, P.F.; Blanquart, C.; Hervouet, E.; Gregoire, M.; Vallette, F.M. HDAC1-mSin3a-NCOR1, Dnmt3b-HDAC1-Egr1 and Dnmt1-PCNA-UHRF1-G9a regulate the NY-ESO1 gene expression. Mol. Oncol. 2013, 7, 452–463. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, M.W.; Wara, A.K.; Cao, Z.; Lebedeva, M.A.; Rosenbauer, F.; Iwasaki, H.; Hirai, H.; Katz, J.P.; Haspel, R.L.; Gray, S.; et al. The Kruppel-like factor KLF4 is a critical regulator of monocyte differentiation. EMBO. J. 2007, 26, 4138–4148. [Google Scholar] [CrossRef] [Green Version]
- Jiang, J.; Chan, Y.S.; Loh, Y.H.; Cai, J.; Tong, G.Q.; Lim, C.A.; Robson, P.; Zhong, S.; Ng, H.H. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat. Cell Biol. 2008, 10, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.C.; McKay, C.; Wang, D.; Topham, D.J.; Parganas, E.; Nakajima, H.; Pendeville, H.; Yasukawa, H.; Sasaki, A.; Yoshimura, A.; et al. SOCS3 is essential in the regulation of fetal liver erythropoiesis. Cell 1999, 98, 617–627. [Google Scholar] [CrossRef] [Green Version]
- Matsui, K.; Ezoe, S.; Oritani, K.; Shibata, M.; Tokunaga, M.; Fujita, N.; Tanimura, A.; Sudo, T.; Tanaka, H.; McBurney, M.W.; et al. NAD-dependent histone deacetylase, SIRT1, plays essential roles in the maintenance of hematopoietic stem cells. Biochem. Biophys. Res. Commun. 2012, 418, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Parmigiani, A.; Nourbakhsh, A.; Ding, B.; Wang, W.; Kim, Y.C.; Akopiants, K.; Guan, K.L.; Karin, M.; Budanov, A.V. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 2014, 9, 1281–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaul, P.; Savu, A.; Yeung, R.O.; Ryan, E.A. Association between maternal glucose and large for gestational outcomes: Real-world evidence to support Hyperglycaemia and Adverse Pregnancy Outcomes (HAPO) study findings. Diabet. Med. 2022, 39, e14786. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Moore, D.; Subramanian, A.; Cheng, K.K.; Toulis, K.A.; Qiu, X.; Saravanan, P.; Price, M.J.; Nirantharakumar, K. Gestational dyslipidaemia and adverse birthweight outcomes: A systematic review and meta-analysis. Obes. Rev. 2018, 19, 1256–1268. [Google Scholar] [CrossRef]
- Chen, K.Y.; Lin, S.Y.; Lee, C.N.; Wu, H.T.; Kuo, C.H.; Kuo, H.C.; Chuang, C.C.; Kuo, C.H.; Chen, S.C.; Fan, K.C.; et al. Maternal Plasma Lipids During Pregnancy, Insulin-like Growth Factor-1, and Excess Fetal Growth. J. Clin. Endocrinol. Metab. 2021, 106, e3461–e3472. [Google Scholar] [CrossRef]
- Hay, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., III; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Barker, D.J. In utero programming of chronic disease. Clin. Sci. 1998, 95, 115–128. [Google Scholar] [CrossRef]
- Wilson, A.; Laurenti, E.; Trumpp, A. Balancing dormant and self-renewing hematopoietic stem cells. Curr. Opin. Genet. Dev. 2009, 19, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Kotowski, M.; Safranow, K.; Kawa, M.P.; Lewandowska, J.; Klos, P.; Dziedziejko, V.; Paczkowska, E.; Czajka, R.; Celewicz, Z.; Rudnicki, J.; et al. Circulating hematopoietic stem cell count is a valuable predictor of prematurity complications in preterm newborns. BMC. Pediatr. 2012, 12, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, V.W.C.; Yusuf, R.Z.; Oki, T.; Wu, J.; Saez, B.; Wang, X.; Cook, C.; Baryawno, N.; Ziller, M.J.; Lee, E.; et al. Epigenetic Memory Underlies Cell-Autonomous Heterogeneous Behavior of Hematopoietic Stem Cells. Cell 2017, 168, 944–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Jing, Q.; Lia, D.; Pascual, M.; McLellan, A.; Greally, J.M. Optimized design and data analysis of tag-based cytosine methylation assays. Genome Biol. 2010, 11, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic. Acids. Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [Green Version]
- Aibar, S.; Gonzalez-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Wilder, S.P.; Johnson, N.; Juettemann, T.; Flicek, P.R. The ensembl regulatory build. Genome Biol. 2015, 16, 56. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelletier, A.; Carrier, A.; Zhao, Y.; Canouil, M.; Derhourhi, M.; Durand, E.; Berberian-Ferrato, L.; Greally, J.; Hughes, F.; Froguel, P.; et al. Epigenetic and Transcriptomic Programming of HSC Quiescence Signaling in Large for Gestational Age Neonates. Int. J. Mol. Sci. 2022, 23, 7323. https://doi.org/10.3390/ijms23137323
Pelletier A, Carrier A, Zhao Y, Canouil M, Derhourhi M, Durand E, Berberian-Ferrato L, Greally J, Hughes F, Froguel P, et al. Epigenetic and Transcriptomic Programming of HSC Quiescence Signaling in Large for Gestational Age Neonates. International Journal of Molecular Sciences. 2022; 23(13):7323. https://doi.org/10.3390/ijms23137323
Chicago/Turabian StylePelletier, Alexandre, Arnaud Carrier, Yongmei Zhao, Mickaël Canouil, Mehdi Derhourhi, Emmanuelle Durand, Lionel Berberian-Ferrato, John Greally, Francine Hughes, Philippe Froguel, and et al. 2022. "Epigenetic and Transcriptomic Programming of HSC Quiescence Signaling in Large for Gestational Age Neonates" International Journal of Molecular Sciences 23, no. 13: 7323. https://doi.org/10.3390/ijms23137323
APA StylePelletier, A., Carrier, A., Zhao, Y., Canouil, M., Derhourhi, M., Durand, E., Berberian-Ferrato, L., Greally, J., Hughes, F., Froguel, P., Bonnefond, A., & Delahaye, F. (2022). Epigenetic and Transcriptomic Programming of HSC Quiescence Signaling in Large for Gestational Age Neonates. International Journal of Molecular Sciences, 23(13), 7323. https://doi.org/10.3390/ijms23137323