Abstract
Cisplatin (cis-diamminedichloroplatinum (II)) is the oldest known chemotherapeutic agent. Since the identification of its anti-tumour activity, it earned a remarkable place as a treatment of choice for several cancer types. It remains effective against testicular, bladder, lung, head and neck, ovarian, and other cancers. Cisplatin treatment triggers different cellular responses. However, it exerts its cytotoxic effects by generating inter-strand and intra-strand crosslinks in DNA. Tumour cells often develop tolerance mechanisms by effectively repairing cisplatin-induced DNA lesions or tolerate the damage by adopting translesion DNA synthesis. Cisplatin-associated nephrotoxicity is also a huge challenge for effective therapy. Several preclinical and clinical studies attempted to understand the major limitations associated with cisplatin therapy, and so far, there is no definitive solution. As such, a more comprehensive molecular and genetic profiling of patients is needed to identify those individuals that can benefit from platinum therapy. Additionally, the treatment regimen can be improved by combining cisplatin with certain molecular targeted therapies to achieve a balance between tumour toxicity and tolerance mechanisms. In this review, we discuss the importance of various biological processes that contribute to the resistance of cisplatin and its derivatives. We aim to highlight the processes that can be modulated to suppress cisplatin resistance and provide an insight into the role of uptake transporters in enhancing drug efficacy.
1. Background
Cisplatin is one of the most potent anti-cancer agents that is effective against solid tumours. It is also used for treating lymphomas, sarcomas, and germ cell cancers.
Cisplatin was first synthesised by M. Peyrone in 1844 and its chemical structure was reported by Alfred Werner in 1893 [1]. However, it did not receive scientific interest until 1965, when Dr. Rosenberg at Michigan State University discovered that an oxidising agent generated from the electrolysis of platinum mesh electrodes was capable of inhibiting cell division in Escherichia coli in a bacterial chamber [2]. The author proposed that this agent will affect actively dividing cancer cells and suggested using these platinum derivatives in cancer chemotherapy. Platinum has a unique structure as a metallic compound with a square planar geometry (Figure 1A). It has a crystalline powder texture of white or deep yellow in colour and is slightly soluble in water and dimethyl primanide and N,N-dimethylformamide. Cisplatin is stable at room temperature but may transform slowly over time to the trans-isomer rendering it clinically ineffective in DNA crosslinking. Cisplatin has a molecular weight of 301.1 gm/mol, a density of 3.74 g/cm3, a melting point of 270 °C, a log Kow of −2.19 and a water solubility of 2.53 g/L at 25 °C [3].
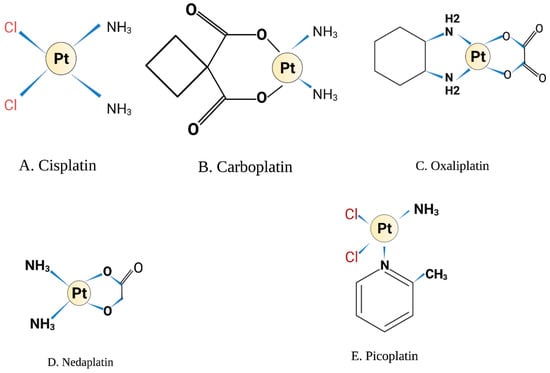
Figure 1.
Chemical structure. Illustrations created by BioRender.com (accessed on 8 May 2022).
Since the identification of cisplatin and its approval by the FDA in 1978, it has proven to have an anti-proliferative effect against different tumour lineages, including sarcomas, bones, muscles, and blood vessels. Although it revolutionised the treatment of solid tumours, a major problem with this agent is the increased nephrotoxicity, which fosters continuous research elucidating other platinum derivatives with equal potency and less toxicity [4].
2. Mechanisms of Cisplatin Toxicity
Cisplatin toxicities were identified as early as the discovery of its anti-tumour functions and it includes gastrointestinal toxicity, myelosuppression, neurotoxicity, vascular and ototoxicity. However, the most severe side effect of cisplatin is nephrotoxicity. The severity of the toxicity is dose-dependent which limits the administration of therapeutic doses to patients [5]. Cisplatin is excreted by glomerular filtration and tubular secretion; it accumulates in the renal parenchymal cells in high concentrations leading to acute kidney injury. Cisplatin undergoes biotransformation in the apical surface of renal cells, where it becomes conjugated to the tripeptide glutathione (glutamate, cysteine and glycine) by glutathione-S-transferase. Inside the kidney, cisplatin-glutathione conjugates are targeted by gamma-glutamyl transpeptidase (GGT), which cleaves the cisplatin glutathione conjugates into cysteinyl-glycine-conjugates that are more prone to enter the proximal tubules. There it is further metabolised into potent nephrotoxic thiols by the enzyme cysteine-S-conjugate β-lyase.
Earlier work attempted to reduce nephrotoxicity by inhibiting the cisplatin metabolic pathway in the kidney by targeting GGT and cysteine S-conjugate β-lyase in mice with acivicin and aminooxy acetic acid (AOAA), respectively [6]. However, it has been shown that GGT is essential for cisplatin detoxification through the interaction with cisplatin metabolites [7]. Amifostine is an FDA-approved nephroprotective drug to minimise cisplatin-associated nephrotoxicity, but the overall benefit of using it with cisplatin is not confirmed [8].
3. Cisplatin Derivatives
Other platinum analogues have been developed and investigated in solid tumours as a way to minimise the side effects of cisplatin. Carboplatin was the first attractive platinum derivative in which the two chloride molecules of cisplatin are replaced with cyclobutene-decarboxylate groups and exist in the same planar structure as platinum molecule (Figure 1B) [9]. Carboplatin was believed to have less nephrotoxicity, gastrointestinal and ototoxicity compared to cisplatin [10]. This is due to the slower hydrolysis of decarboxylating ligands in comparison to the labile chloride molecules in the cisplatin structure. However, carboplatin is associated with myelosuppression, predominantly thrombocytopenia which is dose-limiting for carboplatin [11]. The DNA adducts formed by carboplatin are similar to cisplatin, but the cyclobutene-decarboxylate leaving groups require esterase cleavage affecting the rate of adduct formation and calculated to be 10-fold slower compared to cisplatin [9]. As such, 20- to 40-fold higher concentrations of carboplatin are required for effective treatment.
Carboplatin is approved for the treatment of ovarian and testicular cancers [12]. It is also widely used for treating head and neck [13], oesophageal [14], cervical, salivary gland, retinoblastoma [15] and glioblastoma cancers [16]. However, carboplatin is not beneficial for treating cisplatin-resistant tumours as the mechanisms of resistance for both drugs are similar. Carboplatin-resistant cells exhibit alteration in mitotic checkpoints, and the combination of carboplatin and prexasertib (CHEK1 inhibitor) suppressed the growth of carboplatin-resistant triple-negative breast cancers xenografts in mice [17]. Thus, checkpoint inhibitors could prevent the propagation of carboplatin-resistant cancer cells. The availability of nearly 250 antibodies that specifically detect checkpoint proteins that are phosphorylated in response to drug treatment would be useful to pre-screen patients who might be candidates for checkpoint inhibitor therapy in response to cisplatin and its derivatives [18]. Table 1 summarises recent clinical studies involving cisplatin derivatives.

Table 1.
Recent studies on the combination of platinum derivatives with other drugs.
The third generation of platinum derivative developed was oxaliplatin. Its structure differs from cisplatin, in which the amine groups of cisplatin are replaced by diaminocyclohexane (DACH) [28] (Figure 1C). Besides the formation of intra-strand and inter-strand DNA adducts, oxaliplatin exerts higher efficacy than cisplatin through inhibition of protein synthesis and induction of ribosomal biogenesis stress that halts translation machinery and cell division [29]. Interestingly, a recent report compared the effect of both oxaliplatin and cisplatin on DNA damage and nucleolar mechanisms, revealing that oxaliplatin, but not cisplatin, significantly inhibited ribosomal RNA synthesis by Pol I without modulating rRNA processing [30]. Oxaliplatin has earned significant importance in recent clinical trials for the treatment of solid tumours, and as such, the combination of oxaliplatin and 5-fluorouracil has been approved for treating advanced colorectal cancer [28,31].
Nedaplatin is another cisplatin analogue with a different leaving group in which glycolate is bound to platinum through a bidentate ligand (Figure 1D). It has a similar mechanism to cisplatin and carboplatin. However, it causes less nephrotoxicity compared to cisplatin [32]. The main toxicity of nedaplatin is myelosuppression, primarily thrombocytopenia [33]. It has been approved in Japan for the treatment of solid tumours of the lung, ovarian, head and neck [34]. Recent in vitro studies showed that encapsulation of nedaplatin on PEGylated liposomes increased its cytotoxicity and enhanced platinum uptake by cancer cell lines [35].
Picoplatin is another platinum-based chemotherapy that was designed to overcome cisplatin and carboplatin resistance [36] (Figure 1E). Picoplatin accumulation has been shown to be high in the cell, and it reached the nucleus despite the presence of high levels of glutathione, which is known to chelate cisplatin. This is due to the steric hindrance structure around the platinum molecule [9]. The in vitro data for picoplatin showed promising anti-tumour activity and overcoming some cisplatin resistance mechanisms in different cancer cell lines. Additionally, early phase I and phase II clinical trials for picoplatin in non-small cell lung cancer and ovarian cancer were well tolerated, although no overall survival benefit or response rate was confirmed over other cytotoxic drugs [37,38]. Importantly, in the SPEAR phase III study conducted on 401 small cell lung cancer patients [39], there was no overall survival benefit for patients treated with picoplatin, which led to the discontinuation of further phase III trials for this agent [33]. However, picoplatin showed some activity on cisplatin- and carboplatin-resistant tumours, and this led to the development of two novel picoplatin derivatives Pt(Oro)(NH3)(2-pic), 1, and Pt(5-FOro)(NH3)(2-pic), which were synthesised by joining a fragment of picoplatin to orotate or 5-fluoroorotate bioactive ligands. In vitro experiments of these derivatives showed lower toxicity to normal cells and more potency over cisplatin and picoplatin. However, additional in vivo studies are needed to confirm the clinical benefit of these analogues [40].
Another promising cisplatin derivative in clinical trials is lipoplatin, which is formulated from cisplatin (9%) and lipids (91%). This novel tumour drug delivery has increased efficacy against tumours with less toxicity to normal tissues compared to cisplatin [41,42]. A meta-analysis study comparing the efficacy and safety of lipoplatin over cisplatin in non-small cell lung cancer and head and neck cancers revealed that lipoplatin offered survival benefits and less tissue toxicity than cisplatin. The meta-analysis included data from five clinical trials and 523 patients [26]. Table 2 and Table 3 summarise ongoing clinical trials on cisplatin and its derivatives in different cancer types.
Table 2.
Recent clinical trials on Cisplatin.
Table 3.
Recent clinical trials on cisplatin derivatives.
Platinum Loaded Nanoparticles
Significant research focused on limiting cisplatin and carboplatin side toxicities caused by high dose requirements due to poor cellular uptake of the drug [9]. To enhance the therapeutic efficacy of the platinated drugs, researchers have altered the drug delivery system. For example, carboplatin prodrug complex with Fe3O4 nanoparticles proved to be more toxic than carboplatin in the ovarian cancer cell lines model [43]. The carboplatin nanoparticles are taken up by the endocytosis process leading to increase drug accumulation that can crosslink with the DNA and cause more toxicity to cancer cells than in normal tissues [43]. Similarly, bovine serum albumin nanoparticles were created that encapsulated carboplatin, which was more potent in cytotoxicity experiments in A2780 ovarian cancer cells with 2-fold lower IC50 compared to carboplatin alone [44]. Others have examined carboplatin liposomal nanoparticles in lung cancer in in vitro environments and showed high drug loading efficiency and retention capability [45]. Thus, improving the delivery system for the platinated drugs is likely to reduce the toxicity of non-targeted tissues significantly.
Indeed, self-assembled nanomedicine for cisplatin and lipoplatin, as well as other chemotherapeutic drugs, is an accelerating field of research that is believed to overcome the major toxicity problem limiting their use. Molecular self-assembly is an interesting process by which molecules exist in a disordered system, either static or dynamic and are set to interact locally without external direction. In static self-assembly, the molecules interact to reach an equilibrium state to reduce their free energy, while in dynamic self-assembly, the pre-existing molecules can self-organise to produce a stable structure [46]. Thus, this can overcome the existing drawback in cisplatin conventional delivery systems, as increased detoxification and high drug loading lead to normal tissue toxicity. Moreover, nanoparticles have a prolonged half-life in vivo, allowing optimum platinum accumulation in solid tumours [47]. Carrier self-assembled nanomedicine are approved for other chemotherapeutic drugs like doxorubicin, irinotecan and paclitaxel [48]. A recent study showed that self-assembled Pt (IV-NPs) from biotin-labelled Pt (IV) prodrug demonstrated specific mitochondrial targeting in cancer cells. The self-assembled drug promoted mitochondrial DNA damage and Pt accumulation by reducing GSH levels in vitro as well as in tumour-bearing animal models [49]. Another designed nanoplatform (PDA-pt-NPs) in which Pt was loaded into polydopamine nanoparticles showed efficient cisplatin release in vivo and optimum anti-cancer activity [50]. While only lipoplatin has made it to clinical trials. It will be interesting to evaluate other self-assembled cisplatin-loaded nanoparticles in a clinical setting.
4. Cisplatin-Induced DNA Damage
Cisplatin targets a wide range of cellular components, including membrane phospholipids and thiol-containing peptides [51]. It can bind to some proteins as well as the pre-transcription of RNA. However, DNA remains the most critical target for cisplatin [52]. Once inside the cell, cisplatin exchanges one or two of its chlorine molecules with H2O causing different types of irreversible DNA lesions [4]. One of the most critical lesions is the intra-strand crosslink that the platinum atom forms covalently with two guanosine nucleotides (GG) or with adenosine-guanosine nucleotides (AG) [52]. Most of the adducts occur as a result of cisplatin binding to purine bases on the same strand of the DNA double helix (Figure 2). As estimated by DNA renaturation studies and alkaline elution, the 1,2 GG intra-strand adduct is the most abundant cisplatin DNA adduct accounting for nearly 60% of the platinum lesions. The next abundant lesion is 1,3 GG intra-strand adduct and GG inter-strand adduct (ICL), which accounts for 1% of the total platinated lesions. It has been suggested that cisplatin tumour cytotoxicity is based on the formation of ICLs, which can prevent DNA synthesis and RNA transcription elongation, and the mechanisms of resistance to the drug stem from tumour cells exploiting multiple DNA repair pathways to process ICL lesions [53].
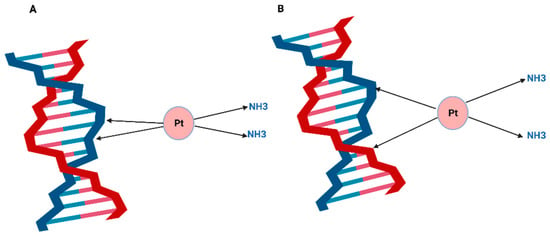
Figure 2.
Cisplatin DNA adducts are formed when cisplatin exchange one or two of its chloride molecules for water and bind covalently to the purines at the N7 position to form (A) Cisplatin intra-strand adduct or (B) Cisplatin inter-strand crosslink. Illustrations created by BioRender.com (accessed on 8 May 2022).
4.1. Nucleotide Excision Repair Pathway
The nucleotide excision repair pathway (NER) plays a key role in repairing cisplatin-induced DNA adducts [54]. NER can be subdivided into two pathways: the transcription-coupled repair (TCR), which mainly processes lesions at the active transcription site in the DNA, and the global genome repair (GGR), which recognises the damage across the majority of the non-transcribing genome. If the adducts occurred at the active transcription site, the lesions would be recognised by RNA polymerase II followed by stalling of transcription elongation and recruitment of several proteins, including the Cockayne syndrome proteins CSA and CSB, the TFIIH complex with XPB and XPD helicases that will unwind the DNA double helix around the lesion. The lesion is then removed by the XPF/ERCC1 and XPG endonucleases that cleave the damaged strand on either side of the lesion to leave a gap of at least 28 to 20 nucleotides [51,52]. The resulting gap is resynthesised by the DNA polymerase ε. It is noteworthy that in the GGR by the NER pathway, the recognition step is initiated by sensor proteins that scan the genomic DNA for chemical distortions such as cisplatin adducts. The protein complex XPC-HR23B together with XPE first recognises the DNA damage, followed by the recruitment of the TFIIH complex to aid in the repair of the lesion by TCR [51,54]. Thus, defects in any proteins in the NER pathway are likely to alter the resistance to cisplatin.
4.2. Mismatch Repair Pathway
The mismatch repair pathway (MMR) plays a role in the recognition of DNA damage induced by cisplatin [55]. The MutSα complex consisting of the MSH2-MSH6 heterodimer can recognise ICLs (1–2 bases) caused by cisplatin. In case of more than two bases mispairing, the MutSb complex (MSH2-MSH3) can be activated. Recognition of the mismatched bases by the MutS complexes helps recruit MutLa (MLH1·PMS2 heterodimer), which, in turn, activates EXO1 excision activity to initiate DNA synthesis by polymerase δ followed by the action of DNA ligase I to seal the nick [56,57].
Although MMR is involved in recognising the cisplatin-generated DNA adducts, it cannot repair the lesions because MMR can only replace mispairing opposite the cisplatin adduct. Eventually, unrepaired cisplatin-induced DNA lesion will lead to the formation of DNA double-strand breaks (DSBs) [58]. However, it seems that activation of MMR towards cisplatin adducts could be a rate-limiting step regulating platinum sensitivity and independently of the canonical MMR pathway [4]. MSH3 is a key component in the recognition of the mismatch bases by both MutSα and MutSβ complexes, and its downregulation was shown to sensitise colorectal cancer cells to cisplatin, oxaliplatin, and poly (ADP-ribose) polymerase (PARP) inhibitors [57].
Hypermethylation of the CpG islands of the MLH1 promoter can lead to the accumulation of mismatched nucleotides and the generation of microsatellite instability [59] which have been linked to the poor prognosis of several tumour types but predominantly in colorectal cancers and endometrial cancers [60,61]. Epigenetic silencing of MLH1 is shown to be associated with aggressive tumours and cisplatin resistance in endometrial cancer [62,63] as well as in testicular germ cell tumours [64,65].
5. Role of DSB Pathways in Repairing Cisplatin-Induced DNA Lesions
While cisplatin DNA adducts are more likely to be sensed and processed by single-strand break repair mechanisms such as NER and MMR, these lesions can also trigger double-strand breaks (DSB) [4]. At an active transcriptional site, the TCR pathway can attempt to repair the damage; however, during the DNA incision steps, double-strand breaks could be produced [66]. Nonetheless, cisplatin DNA damage does not appear to trigger an efficient DSB response. Cisplatin combined with ionising radiation is sensed by the Non-Homologous End-Joining (NHEJ) repair pathway [67]. Cisplatin treatment prior to or concurrent with ionising radiation therapies increased cancer cells radiosensitisation in ovarian, head and neck, as well as cervical carcinomas [67]. The exact mechanism for this synergistic effect is not very clear, although it seems to be dependent on the platinum dosage as well as the combination regimen that creates multiple types of DNA lesions. Moreover, cisplatin lesions can inhibit DNA-PK activation lowing its kinase activity and preventing it from binding to the Ku70/80 heterodimer in the NHEJ pathway. In addition, the rate of Ku translocation at the DNA double-strand break is also inhibited in the presence of cisplatin adducts [68]. These findings implicate that the synergism activity between cisplatin and ionising radiation is dependent on cisplatin adducts impairing the NHEJ processing from processing the radiation-induced DNA damage [69,70].
Similarly, cisplatin maximum toxicity is observed in NER deficient tumours such as testicular cancers that have lower expression of XPA and ERCC1, which are key drivers in the NER pathway [71]. In vitro depletion of XPF and ERCC1 sensitised NSCLC to cisplatin [72]. Thus, optimal cisplatin anti-tumour activity could be achieved in patients with known DNA repair defects.
BRCA Mutations and Cisplatin Sensitivity
Mutations in the breast cancer susceptibility genes BRCA1 and BRCA2 confer a high predisposition to breast cancers and other tumour types, including ovarian, pancreatic, and colorectal [73]. BRCA1 colocalises with BARD1, RAD51, and the proliferating cell nuclear antigen (PCNA) during replication. It promotes the 5′ to 3′ resection of the break site leaving behind a 3′ overhang during the S-phase of the cell cycle, whereas BRCA2 binds to RAD51 and regulates RAD51 filament formation. BRCA1/BRCA2 mutations lead to genomic instability because of stalling replication fork during DNA double-strand break repair [74]. A recent report highlighted that ssDNA replication gaps rather than defects in homologous recombination underlie the hypersensitivity of BRCA-deficient cancer to chemotherapy [75]. BRCA1/2 mutated tumours are hypersensitive to ionising radiation and other DNA damaging agents, including platinum derivatives. Breast and ovarian cancers with BRCA mutations are particularly sensitive to platinum. Thus, cisplatin treatment of these tumours showed a high success rate. However, acquired resistance to cisplatin often occurs in recurrent tumours [76,77]. A well-known resistance mechanism occurs through secondary intragenic mutations in BRCA1/2 that restore the wild-type open reading frame and recover homologous recombination [78]. Interestingly, refractory tumours due to BRCA reactivation are as also resistant to the PARP inhibitor olaparib as a monotherapy [79]. Implying similarity in the resistant mechanisms to cisplatin and PARP inhibitors. Loss of MED12, a component of the mediator transcription regulation complex, promotes resistance of BRCA 1/2 deficient cells to cisplatin and PARP inhibitors. MED12 depletion activates the TGF-β pathway independently of the mediator complex and restores HR repair to mediate cisplatin resistance [80].
Emerging studies identify biomarkers of cisplatin resistance and aim to restore tumour sensitivity to platinum derivatives. Mutations in the ATM, RB1 and FANCC genes in muscle-invasive bladder cancer patients (MIBC) correlate with complete response to cisplatin-based neoadjuvant chemotherapy in patients from the clinical trials NCT01031420 and NCT01611662 [81]. In addition, we have previously shown that DNA polymerase beta (Polβ) depletion exquisitely sensitises ovarian cancer cells to cisplatin [82]. We also showed that ovarian tumours with high Polβ expression have poor survival compared to low Polβ expressing tumours. Moreover, we [83] and others [84,85,86] have demonstrated that ERCC1 is a key predictor of cisplatin resistance in ovarian, testicular and lung cancers. Depletion of ERCC1 substantially increased platinum sensitivity in the cell line model [87]. These observations imply that targeting defective DNA repair pathways is a promising approach to modulating cisplatin response.
The Fanconi Anemia pathway (FA) proteins also play a major role in interstrand crosslinks (ICL) repair. ICLs that occur during the S-phase block the replication fork. These ICLs are sensed by the Fanconi Anemia proteins that generate DSBs, which are processed by the homologous recombination pathway. In contrast, the replication-independent ICLs are sensed and processed primarily by the NER pathway. Thus, FA mediated-repair is thought to be directly involved in repairing cisplatin lesions and targeting FA proteins could be a viable approach to modulating cisplatin resistance. Up-regulation of the FA-associated genes, FANCL and RAD18, have been observed in cisplatin-resistant NSCLC cells. Down-regulation of these genes restored platinum sensitisation [88]. A similar approach showed re-sensitisation of the cisplatin-resistant A549/DDP using RNAi for FANCL, FANCD2 or FANCF and inhibition of the FA pathway. A recent study reported that indirect targeting of the FA pathway in squamous cell carcinoma sensitises these tumour cells to cisplatin. Small molecule inhibitor of the deubiquitylase USP28, which is recruited to the DNA damage site in cisplatin-treated cells, indirectly down-regulates FA activation and increases sensitivity to cisplatin [89]. Although the in vitro studies support the inhibition of FA proteins to reverse cisplatin resistance, the clinical development of specific inhibitors has been slow. Early work described several inhibitors of the proteasome machinery as cathepsin B, lysosome, and CHK1 to overcome cisplatin-induced FANCD2 foci formation and FA pathway activation [90]. However, developing more specific inhibitors for key FA proteins will be essential to conducting comprehensive in vivo studies.
6. Cisplatin and Apoptosis
The tumour suppressor gene Tp53 bridges cisplatin-induced DNA adducts to apoptosis signalling. Tp53 regulates the signalling of a plethora of cell cycle progression and apoptosis effector genes and plays a prominent role in the cellular response to DNA damage [4]. Upon cisplatin treatment, the kinases ATM and ATR phosphorylate Tp53 on serine 20, leading to its stabilisation. In breast cell lines, cisplatin mediates Tp53 signalling of the BCL-2 pro-apoptotic activator. Additionally, it mediates the expression of the BH-3 only protein Noxa through the end products of lipid peroxidation [91]. Thus, it appears that platinum cellular response can activate different defence mechanisms that interplay to decide the cell fate [92].
7. Mechanisms of Cisplatin Resistance
Despite the irreversible DNA damage induced by cisplatin, the development of resistance is usually inevitable. Most patients relapse after the initial response to platinum cycles and this is attributed to the development of one or more resistance mechanisms, which is often a multifactorial process involving intrinsic pathways [51,52]. One important mechanism in cisplatin resistance is the reduction in cellular accumulation of the drug, hence, lowering the levels of platinated DNA adducts [93]. One possible explanation for this could be increased cellular efflux to the drug. Some studies revealed the involvement of the copper transporters ATP7A and ATP7B, responsible for copper detoxification, play a role in cisplatin efflux from the cells [94,95]. Previous work has linked the high expression of ATP7A to cisplatin resistance in the lung [96], oesophagus [97], and ovarian cancer patients. Moreover, preclinical studies showed that overexpression of ATP7A in cervical cancer cells leads to platinum resistance [96]. Another key transporter in platinum detoxification is the multidrug resistance-associated protein MRP1, which functions to efflux different anti-tumour drugs mediating cellular resistance. Currently, several members of the ATP-binding cassette (ABC) are identified as crucial transporters in chemoprotection [52].
8. Cisplatin and Immune Response
Cisplatin cell killing mechanisms are not solely attributed to its DNA crosslinking ability but also the ability to interfere with the immune response activation [98]. One suggested mechanism is through induction of the major histocompatibility class complex, MHC class I, which is essential for priming cytotoxic T cells for tumour recognition [99,100]. Early studies reported that cisplatin and vinorelbine doublet upregulate MHC I in lung cancer cell lines [101]. Lung cell lines treated with cisplatin upregulated tumour necrosis factor-α, IL8, CXCL5, and B cell lymphoma-2–like genes (BCL-2) [102]. Similarly, chemotherapeutic drugs, including cisplatin, stimulated MHC I expression through increased interferon-beta signalling in breast cancer cells [103]. More clinical evidence was demonstrated in a study that assessed samples from NSCLC patients treated with cisplatin following radical surgery, showing that 30% of patients had high expression of MHC class I chain-related molecules A and B [99]. Cisplatin-induced MHC class I upregulation was associated with progression-free survival and better prognosis for patients [99].
More promising findings on cisplatin immune response induction are derived from cisplatin combination therapy. The Epitopes-HPV01 and HPV02 trials which investigated the benefit of adding docetaxel to cisplatin plus 5-fluorouracil (DCF) in anal squamous cell carcinoma, found an increase in circulating TH1 T-cells in patients who received the DCF regimen [103,104]. Low levels of myeloid-derived suppressive cells (MDSC) in patients exposed to the DCF treatment were associated with induction of adaptive immune response to hTERT tumour antigen as well as good prognosis [105,106,107]. Combination therapy of low cisplatin dose plus paclitaxel was more effective in immune response induction compared to the maximum tolerated dose of cisplatin in ovarian cancer patients. The low cisplatin dose plus paclitaxel increased IL-2 and IFN-γ associated with cytotoxic CD8(+) T-cell activity [108]. The priming effect of cisplatin has been investigated in combination with a programmed cell death inhibitor (PDL-1). In the ovarian cancer syngeneic mice model, cisplatin plus PDL-1 inhibitor increased CD8+ T-cells and led to tumour regression [109].
Platinum analogues combinations with checkpoint inhibitors PD-1/PD-L-1 have also proved to be promising regimens for suppressing tumour growth, as reported by several investigators [100,110,111]. Wu et al. analysed cisplatin combination with anti-PD-1 antibody (Tislelizumab or Sintilimab) as first-line therapy from the clinical trials (NCT03469557, NCT03748134) [112]. A sublethal dose of cisplatin in oesophageal squamous cell carcinoma (ESCC) induced PD-L1 expression and synergised with an anti-PD-1 antibody [112]. However, this treatment regimen may be limited to certain tumour types. In the Lewis lung carcinoma model, oxaliplatin combination with anti-PD-L1 induced ICD through activation of CD80+ CD86+ dendritic cells and enhanced cytotoxic T cells (CD8+), resulting in tumour regression [113].
Another well-described mechanism is the ability of platinum derivatives to induce immunogenic cell death (ICD), characterised by the relapse of pre-apoptotic calreticulin and the post-apoptotic high-mobility group box 1 protein (HMGB1) [98,114]. Many chemotherapeutic drugs are known to act by damaging the DNA, followed by secondary processes that involve plasma membrane rupture and release of intracellular content during cell death. It is believed that this process can lead to protein expression at the cell surface, as well as cytokine secretion that could trigger an immune response against tumour cells [115,116]. Oxaliplatin appears to have superior efficiency in the induction of ICD compared to other platinum derivatives [98]. In immunocompetent mice bearing CT26 colorectal cancer cells, both oxaliplatin and cisplatin-induced immune response, but not when CRT was inhibited or depleted or when the toll-like receptor 4 (TLR4) was knocked out [117].
Patients from the LARC study (NCT00278694) with locally advanced rectal cancer and who received a full dose of oxaliplatin induction, followed by an adapted chemoradiation regimen dose, showed a significant increase in HMGB1 during the induction course [118]. HMGB1 was used as a biomarker of ICD and positively correlated with metastasis-free disease. Emerging in vivo studies supports the notion that oxaliplatin is a promotor of CD8+ and CD4+ T-cell response [113,117,119,120]. An increase in CD4+ T-cells activates dendritic cells and other antigen-presenting cells leading to immune response activation. Circulating cytotoxic T-cells implies tumour antigen presentation, activation of an immune cascade of pro-inflammatory cytokines, and type I interferon activation. Haung and colleagues have also found activation of the immune cascade following oxaliplatin and PD-L1 antibody treatment in CT26 colorectal tumours in mice [119].
Elevated levels of pro-inflammatory cytokines such as CCL2, CXCL12, and CXCL13, as well as CXCL9 and CXCL10, which favour T-cell infiltration into the tumour, were found post combination treatment. In addition, their results illustrated activation of Th1-type cytokines IFN-γ and TNF-α, besides IL-4 and IL-10, which are surrogate markers of Th2 response that could promote tumour tolerance [119]. Thus, immune response activation by oxaliplatin checkpoint inhibitor combination is evident. However, harnessing this response through optimising therapeutic doses and regimens is necessary to achieve benefits.
9. Cisplatin Uptake Transporters
Several membrane transporters have been proposed to carry cisplatin into the cells through passive diffusion, including Na+, K+-ATPase, and the solute carriers SLC family of transporters. SLC22A2 (OCT2) and CTR1 are the most described members of the SLC involved in platinum uptake, and OCT2 plays a role in cisplatin transport into renal cells [121]. Mice deleted for OCT1 and OCT2 showed no ototoxicity and mild nephrotoxicity upon cisplatin treatment compared to wild-type mice [122]. Additionally, administration of the OCT2 inhibitor cimetidine with cisplatin in wild-type mice was effective in nephroprotection and significantly reduced cisplatin renal uptake [122]. Another study illustrated that cimetidine reduces acute kidney injury without affecting cisplatin anti-tumour activities [123], indicating the significance of cisplatin transport mechanisms in overcoming toxicities.
The role of copper transporters in the mechanisms of cisplatin resistance is well described, and CTR1 is the primary platinum uptake transporter [97,124,125]. Cisplatin interacts with CTR1 resulting in conformational changes in its methionine residues, allowing the formation of a smaller CTR1 intermediate to promote the drug uptake. However, cisplatin can trigger the degradation of CTR1 and reduce its cellular uptake, thereby promoting resistance to the drug [93].
Importantly, in serous epithelial ovarian cancer patients who received post-operative platinum therapy, high CTR1 mRNA levels were observed in resistant tumours, although the CTR1 protein level was not determined [124,126]. In contrast, NSCLC patients with low CTR1 expression had less intracellular platinum accumulation and poor response [127]. In another study of 54 patients of stage III NSCLC, high CTR1 expression correlated with longer progression-free survival and overall survival [128].
Another copper transporter CTR2 has been identified for the mechanisms of platinum transport, but it functions as an efflux transporter [93]. Knockdown of CTR2 is associated with increased platinum cellular accumulation and efficacy. CTR2 is expressed mainly in lysosomes and late endosomal formations; therefore, regulation of endocytosis is suggested to be involved in cisplatin efflux by CTR2 [125]. There are other transporters, such as OCT1, OCT2 and OCT3, and small GTPases that are believed to serve as regulators of cisplatin trafficking [4].
10. Increased Cisplatin Detoxification
Glutathione conjugation to cisplatin mediated by glutathione transferase has been linked to nephrotoxicity and ototoxicity. Cisplatin has a high affinity to glutathione because of its nucleophilic nature. Platinum-glutathione conjugates are subjected to extracellular transport by MRP proteins. Cisplatin sequesters with glutathione S-transferase (GST P1-1) resulted in cisplatin inactivation and inhibition of apoptosis signalling by c-JUN terminal kinase [129]. Similarly, metallothionein (MT) can promote cisplatin diffusion and detoxification. MT are cysteine-rich proteins responsible for metal homeostasis. MT2 overexpression was previously described in bladder carcinomas resistant to platinum [130]. Comparably, in non-small cell lung cancer, MT2 upregulation was evident in patients following platinum therapy as well as in vivo murine models [131]. More recently, MT3 upregulation in neuroblastoma was correlated with refractory mechanisms to cisplatin [132]. Thus, inhibitors of the specific metallothionein may serve to diminish the cisplatin dose while maintaining its cytotoxic and genotoxic effects.
11. Epigenetics Changes
As cisplatin primarily targets the DNA, it is predicted that cellular resistance mechanisms can extend to epigenetic regulations. DNA methylation is a key mechanism for acquired cisplatin resistance. Studies using the DNA hypomethylating agent 2-deoxy-5-aza-cytidine revealed over hundred genes that are hypermethylated in platinum-resistant cell lines and could be reactivated via azacytidine [53]. Methylation of the folate-binding gene (FBP) was shown as a mechanism of cisplatin resistance in hepatocellular carcinoma [133]. Epigenetic profiling aided in understanding the molecular landscape of platinum resistance. A study analysing CpG promoter islands methylation in germ cell tumours revealed hypermethylation of key genes, including the stem cell marker NANOG and POU5F1, to be drivers of platinum resistance [134].
Histone modifications are also described to be a mechanism for cisplatin resistance [51], and post-translational modifications of histones can regulate many of the processes involved in the resistance, including DNA repair effectors, transcription and signalling. It was found that in head and neck squamous cell carcinomas, NFkβ activation can drive chemoresistance. Active NFkβ signalling promotes histone deacetylation and reduces nuclear BRCA1 levels and increases genomic instability [135]. The combination of belinostat, an inhibitor of histone deacetylase, with decitabine increased the expression of epigenetically silenced MLH1 and MAGE-A1 and increased cisplatin sensitivity in ovarian cancer xenografts [136]. In fact, Histone deacetylase inhibitors seem promising clinically to circumvent cisplatin resistance. Cisplatin plus belinostat are in phase I clinical trials in advanced solid tumours [137], as well as panobinostat which is another FDA histone deacetylase inhibitor [138].
12. Upregulation of DNA Repair Capacity
The prominent role of DNA repair pathways in overcoming cisplatin toxicity is well established. The majority of cisplatin lesions are recognised by NER and MMR pathways. Therefore, increased expression of NER and MMR genes is a key mechanism for repairing platinum DNA adducts and controlling chemoresistance [58]. ERCC1 expression is associated with platinum resistance and poor survival in ovarian, bladder, oesophageal, head and neck cancers, as well as in NSCLC [87]. Additionally, MMR-related proteins that participate in the recognition of GpG inter-strand adducts, including MSH2 and MLH1, are mutated in some cisplatin refractory tumours [56]. In fact, microsatellite instability or mismatch repair deficiency is a predictor of patient prognosis and chemotherapy response in endometrial and colorectal cancers [139].
Interestingly, a study compared the microsatellite instability status in ovarian cancers between the primary resected tumours and the secondary resected tumours of the same patients. The amplification of 10 microsatellite loci and immunohistochemical detection of hMSH2 and hMLH1 expression in 24 cases of ovarian cancers revealed that all the secondary resected tumours showed microsatellite instability (MSI). Of the 24 primary tumours, 15 had MSS status; however, their residual tumours after 5 or 6 courses of platinum exhibited MSI status through loss of the expression of MLH1 [140].
On the contrary, a similar analysis for MMR markers, including MLH1, was performed on cervical cancer patients pre and post-neoadjuvant chemotherapy and showed no change in microsatellite stability post-chemo exposure [140]. Thus, the role of mismatch repair deficiency in platinum resistance appears to be dependent on the tumour-specific microenvironment.
Another crucial mechanism for repairing cisplatin-induced DNA adducts is translesion synthesis by a group of DNA polymerases belonging to the Y family (Polymerase η (Polη), Polι, Polκ and Rev1) and B families (such as Polζ) of DNA polymerases. Translesion DNA synthesis (TLS) is a tolerance mechanism by which the cells circumvent deleterious double-strand breaks caused by replication stalling [141].
When the replication fork encounters DNA lesions on the leading strand, replication is stalled. While the lagging strand can still go through the replication mechanism, normal base pairing cannot progress, and eventually, replication will be blocked. The cells develop mechanisms to prevent this hazardous form of DNA damage and continue replication. The cells switch to translesion polymerases to insert bases and fill the gap. Yet this is done with low fidelity and thus induces mutations. Polη can bypass several bulky DNA adducts, including cisplatin-GG adducts. Polη deficient cells display more sensitivity to cisplatin treatment compared to proficient cell lines [141,142].
Another TLS polymerase, Polζ, composed of two subunits, Rev3L and Rev7, is implied in bypassing cisplatin-induced intra-strand adducts. Similarly, it was shown in murine studies that Polζ deficient tumours are more sensitive to cisplatin [143]. Knockdown of the catalytic subunit Rev3L desensitised cells to platinum [144]. Importantly, in a cohort of head and neck squamous cell carcinomas with high polη expression showed an association with poor platinum response and worse outcomes for patients [145]. These findings imply that TLS polymerases play a crucial role in bypassing cisplatin lesions. However, the exact signalling by which the cells activate the polymerase of choice is not clear.
13. Future Prospective
Cisplatin is one of the most effective chemotherapeutic anticancer agents. Since it acquired FDA approval five decades ago for the treatment of testicular cancer, it remains very effective in combination with bleomycin and etoposides [146,147]. Other tumours remain responsive to cisplatin like head and neck, ovarian and non-small cell lung cancers due to its actions on various cellular components and the activation of multiple pathways for cell killing [128]. Cisplatin also elects complex mechanisms of resistance. Response to cisplatin involves several DNA repair pathways and epigenetic changes, which drive the cells to develop defence mechanisms against the toxic effects of the drug [51]. As a result, tumours recur with profound molecular and genetic changes that favour cell survival, DNA methylation, gene silencing, or activation that inhibits apoptosis [4]. Thus, current new therapeutic regimes combine platinum with other molecular targeted therapies aiming to inhibit resistance mechanisms. The combination of cisplatin with bevacizumab, a vascular endothelial growth factor inhibitor, remains attractive in non-small cell lung cancers as well as cervical cancers [148,149]. Another interesting approach is the combination of cisplatin with olaparib, the first FDA-approved DNA-targeted therapy. A promising clinical study investigated the approach of combining olaparib with cisplatin and irinotecan for pancreatic ductal carcinoma and found a durable clinical response [150]. Additional clinical studies can explore how to maximise the benefit of this strategy. Approaches for combining cisplatin with checkpoint inhibitors also seem attractive.
Cisplatin nephrotoxicity is a challenging obstacle. A recent report showed that co-administration of cilastatin with cisplatin significantly reduced nephrotoxicity in a manner that permits escalating cisplatin dose. Cilastatin acts as a blocker of megalin, which is an endocytic receptor at the apical membrane of the tubular epithelial cells [151]. Several studies have shown that the transporters CTR1 and OCT2 are responsible for cisplatin uptake into cells. The abundant expression of these transporters on renal tubular cells has a major role in kidney toxicity [121]. Thus, developing small molecule inhibitors that inhibit these transporters or compete with cisplatin for the transporter binding site could have clinical significance by reducing nephrotoxicity. The current advent in proteomic and transcriptomic studies is expected to yield new targets that will aid in stratifying patients and when combined with DNA repair defects, should enhance the antitumour effect of cisplatin. At the moment, the challenge remains to find ways to bypass the molecular processes causing cisplatin resistance and with the many targets highlighted herein, it seems that downregulating multiple processes would be required to maximise the anticancer benefits of cisplatin.
Author Contributions
R.A., M.A., S.M. and D.R. wrote the manuscript. R.A., A.A.S. and M.A. prepared the figures and R.A. and A.A.S. prepared the Tables. R.A., S.M., M.A. and D.R. reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by funding from Qatar Foundation to the College of Health and Life Sciences.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
College of Health and Life sciences, Hamad Bin Khalifa University, Education city, Doha, Qatar.
Acknowledgments
This work was supported with funding from the Qatar foundation to the college. Open Access funding was provided by the Qatar National Library, Doha, Qatar.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Omez-Ruiz, S.G.; Maksimović-Ivanić, D.; Mijatović, S.; Kaluđerović, G.N. On the Discovery, Biological Effects, and Use of Cisplatin and Metallocenes in Anticancer Chemotherapy. Bioinorg. Chem. Appl. 2012, 2012, 140284. [Google Scholar] [CrossRef]
- Alderden, R.A.; Hall, M.D.; Hambley, T.W.; Kauffman, G.B. Chemistry for Everyone the Discovery and Development of Cisplatin Products of Chemistry edited by. J. Chem. Educ. 2006, 83, 728. [Google Scholar] [CrossRef]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Krishnamurthy, S. Cellular responses to cisplatin-induced DNA damage. J. Nucleic Acids 2010, 2010, 201367. [Google Scholar] [CrossRef]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin Nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef]
- Townsend, D.M.; Hanigan, M.H. Inhibition of γ-glutamyl transpeptidase or cysteine sconjugate β-lyase activity blocks the nephrotoxicity of cisplatin in mice. J. Pharmacol. Exp. Ther. 2002, 300, 142–148. [Google Scholar] [CrossRef]
- Daubeuf, S.; Balin, D.; Leroy, P.; Visvikis, A. Different mechanisms for γ-glutamyltransferase-dependent resistance to carboplatin and cisplatin. Biochem. Pharmacol. 2003, 66, 595–604. [Google Scholar] [CrossRef]
- Ha, S.S.; Rubaina, K.; Lee, C.-S.; John, V.; Seetharamu, N. Amifostine is a Nephro-Protectant in Patients Receiving Treatment with Cisplatin- Myth, Mystery or Matter-of-Fact? J. Nephrol. Sci. 2021, 3, 4–8. [Google Scholar]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Vasconcellos, V.F.; Marta, G.N.; da Silva, E.M.K.; Gois, A.F.T.; de Castria, T.B.; Riera, R. Cisplatin versus carboplatin in combination with third-generation drugs for advanced non-small cell lung cancer. Cochrane Database Syst. Rev. 2020, 1, CD009256. [Google Scholar] [CrossRef]
- Zhou, J.; Kang, Y.; Chen, L.; Wang, H.; Liu, J.; Zeng, S.; Yu, L. The Drug-Resistance Mechanisms of Five Platinum-Based Antitumor Agents. Front. Pharmacol. 2020, 11, 343. [Google Scholar] [CrossRef] [PubMed]
- Ho, G.Y.; Woodward, N.; Coward, J.I.G. Cisplatin versus carboplatin: Comparative review of therapeutic management in solid malignancies. Crit. Rev. Oncol. Hematol. 2016, 102, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Holsinger, F.C.; Colevas, A.D.; Hara, W.; Le, Q.T.; Beadle, B.M. Cisplatin or Carboplatin-Based Chemoradiation for Locoregionally Advanced Squamous Cell Carcinoma of the Head and Neck: A Population-Based Comparison. Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, e371. [Google Scholar] [CrossRef]
- de Man, F.M.; van Eerden, R.A.G.; Oomen-De Hoop, E.; Veraart, J.N.; van Doorn, N.; van Doorn, L.; van der Gaast, A.; Mathijssen, R.H.J. Efficacy and Toxicity of Weekly Carboplatin and Paclitaxel as Induction or Palliative Treatment in Advanced Esophageal Cancer Patients. Cancers 2019, 11, 826. [Google Scholar] [CrossRef] [PubMed]
- Marr, B.P.; Dunkel, I.J.; Linker, A.; Abramson, D.H. Periocular carboplatin for retinoblastoma: Long-term report (12 years) on efficacy and toxicity. Br. J. Ophthalmol. 2012, 96, 881–883. [Google Scholar] [CrossRef]
- Murray, L.J.; Bridgewater, C.H.; Levy, D. Carboplatin Chemotherapy in Patients with Recurrent High-grade Glioma. Clin. Oncol. 2011, 23, 55–61. [Google Scholar] [CrossRef]
- Moens, S.; Zhao, P.; Baietti, M.F.; Marinelli, O.; Van Haver, D.; Impens, F.; Floris, G.; Marangoni, E.; Neven, P.; Annibali, D.; et al. The mitotic checkpoint is a targetable vulnerability of carboplatin-resistant triple negative breast cancers. Sci. Rep. 2021, 11, 3176. [Google Scholar] [CrossRef]
- Lopez-Beltran, A.; Cimadamore, A.; Blanca, A.; Massari, F.; Vau, N.; Scarpelli, M.; Cheng, L.; Montironi, R. Immune Checkpoint Inhibitors for the Treatment of Bladder Cancer. Cancers 2021, 13, 131. [Google Scholar] [CrossRef]
- Oh, D.-Y.; Lee, K.-H.; Lee, D.-W.; Yoon, J.; Kim, T.-Y.; Bang, J.-H.; Nam, A.-R.; Oh, K.-S.; Kim, J.-M.; Lee, Y.; et al. Gemcitabine and cisplatin plus durvalumab with or without tremelimumab in chemotherapy-naive patients with advanced biliary tract cancer: An open-label, single-centre, phase 2 study. Lancet Gastroenterol. Hepatol. 2022, 7, 522–532. [Google Scholar] [CrossRef]
- Hussain, S.A.; Lester, J.F.; Jackson, R.; Gornall, M.; Qureshi, M.; Elliott, A.; Crabb, S.J.; Huddart, R.A.; Vasudev, N.; Birtle, A.J.; et al. Addition of nintedanib or placebo to neoadjuvant gemcitabine and cisplatin in locally advanced muscle-invasive bladder cancer (NEOBLADE): A double-blind, randomised, phase 2 trial. Lancet Oncol. 2022, 23, 650–658. [Google Scholar] [CrossRef]
- Bourhis, J.; Burtness, B.; Licitra, L.F.; Nutting, C.; Schoenfeld, J.D.; Omar, M.; Bouisset, F.; Nauwelaerts, H.; Urfer, Y.; Zanna, C.; et al. Xevinapant or placebo plus chemoradiotherapy in locally advanced squamous cell carcinoma of the head and neck: TrilynX phase III study design. Future Oncol. 2022, 18, 1669–1678. [Google Scholar] [CrossRef]
- Frankart, A.J.; Sadraei, N.H.; Huth, B.; Redmond, K.P.; Barrett, W.L.; Kurtzweil, N.; Riaz, M.K.; Wise-Draper, T.; Rodriguez, C.P.; Adelstein, D.J.; et al. A phase I/II trial of concurrent immunotherapy with chemoradiation in locally advanced larynx cancer. Laryngoscope Investig. Otolaryngol. 2022, 7, 437–443. [Google Scholar] [CrossRef]
- Kubicek, G.J.; Khrizman, P.; Squillante, C.; Callahan, K.; Xu, Q.; Abouzgheib, W.; Boujaoude, Z.; Patel, A.; Hageboutros, A. Stereotactic Body Radiotherapy and Systemic Dose Chemotherapy for Locally Advanced Lung Cancer: Single Arm Phase 2 Study. Am. J. Clin. Oncol. 2022, 45, 129–133. [Google Scholar] [CrossRef]
- Biswas, T.; Dowlati, A.; Kunos, C.A.; Pink, J.J.; Oleinick, N.L.; Malik, S.; Fu, P.; Cao, S.; Bruno, D.S.; Bajor, D.L.; et al. Adding Base-Excision Repair Inhibitor TRC102 to Standard Pemetrexed-Platinum-Radiation in Patients with Advanced Nonsquamous Non-Small Cell Lung Cancer: Results of a Phase I Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 646–652. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Huang, H.Q.; Filiaci, V.L.; Randall, M.; DiSilvestro, P.A.; Moxley, K.M.; Fowler, J.M.; Powell, M.A.; Spirtos, N.M.; Tewari, K.S.; et al. Patient reported outcomes for cisplatin and radiation followed by carboplatin/paclitaxel versus carboplatin/paclitaxel for locally advanced endometrial carcinoma: An NRG oncology study. Gynecol. Oncol. 2022, 164, 428–436. [Google Scholar] [CrossRef]
- Xu, B.; Zeng, M.; Zeng, J.; Feng, J.; Yu, L. Meta-analysis of clinical trials comparing the efficacy and safety of liposomal cisplatin versus conventional nonliposomal cisplatin in nonsmall cell lung cancer (NSCLC) and squamous cell carcinoma of the head and neck (SCCHN). Medicine 2018, 97, e13169. [Google Scholar] [CrossRef]
- Liu, Y.; Xiao, Q.; He, J.; Hu, H.; Du, J.; Zhu, Y.; Chen, J.; Liu, Z.; Wang, J.; Sun, L.; et al. Phase II study of anlotinib in combination with oxaliplatin and capecitabine for patients with RAS/BRAF wild-type metastatic colorectal adenocarcinoma as the first-line therapy. BMC Med. 2022, 20, 155. [Google Scholar] [CrossRef]
- Alcindor, T.; Beauger, N. Oxaliplatin: A review in the era of molecularly targeted therapy. Curr. Oncol. 2011, 18, 18–25. [Google Scholar] [CrossRef]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A subset of platinum-containing chemotherapeutic agents kill cells by inducing ribosome biogenesis stress rather than by engaging a DNA damage response. Nat. Med. 2017, 23, 461. [Google Scholar] [CrossRef]
- Sutton, E.C.; DeRose, V.J. Early nucleolar responses differentiate mechanisms of cell death induced by oxaliplatin and cisplatin. J. Biol. Chem. 2021, 296, 100633. [Google Scholar] [CrossRef]
- Bordonaro, R.; Calvo, A.; Auriemma, A.; Hollebecque, A.; Rubovszky, G.; Saunders, M.P.; Pápai, Z.; Prager, G.; Stein, A.; André, T.; et al. Trifluridine/tipiracil in combination with oxaliplatin and either bevacizumab or nivolumab in metastatic colorectal cancer: A dose-expansion, phase I study. ESMO Open 2021, 6, 100270. [Google Scholar] [CrossRef] [PubMed]
- Shimada, M.; Itamochi, H.; Kigawa, J. Nedaplatin: A cisplatin derivative in cancer chemotherapy. Cancer Manag. Res. 2013, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkova, D.; Ivanova, S. Application of Approved Cisplatin Derivatives in Combination Therapy against Different Cancer Diseases. Molecules 2022, 27, 2466. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.Z.; Xu, H.Y.; Zhao, Z.M.; Zhang, G.M.; Lin, F.W. Comparison of efficacy and toxicity between nedaplatin and cisplatin in treating malignant pleural effusion. OncoTargets Ther. 2018, 11, 5509–5512. [Google Scholar] [CrossRef]
- El-Shafie, S.; Fahmy, S.A.; Ziko, L.; Elzahed, N.; Shoeib, T.; Kakarougkas, A. Encapsulation of Nedaplatin in Novel PEGylated Liposomes Increases Its Cytotoxicity and Genotoxicity against A549 and U2OS Human Cancer Cells. Pharmaceutics 2020, 12, 863. [Google Scholar] [CrossRef]
- Hamilton, G.; Olszewski, U. Picoplatin pharmacokinetics and chemotherapy of non-small cell lung cancer. Expert Opin. Drug Metab. Toxicol. 2013, 9, 1381–1390. [Google Scholar] [CrossRef]
- Eckardt, J.R.; Oncology, J.; Lipatov, O.N. Phase II Study of Picoplatin As Second-Line Therapy for Patients With Small-Cell Lung Cancer. Artic. J. Clin. Oncol. 2009, 27, 2046–2051. [Google Scholar] [CrossRef]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef]
- Ciuleanu, T.; Samarzjia, M.; Demidchik, Y.; Beliakouski, V.; Rancic, M.; Bentsion, D.L.; Orlov, S.V.; Schaeffler, B.A.; De Jager, R.L.; Breitz, H.B. Randomized phase III study (SPEAR) of picoplatin plus best supportive care (BSC) or BSC alone in patients (pts) with SCLC refractory or progressive within 6 months after first-line platinum-based chemotherapy. J. Clin. Oncol. 2010, 28, 7002. [Google Scholar] [CrossRef]
- Malik-Gajewska, M.; Trynda, J.; Zierkiewicz, W.; Helios, K.; Latajka, R.; Wietrzyk, J.; Michalska, D. Picoplatin-based complexes with the bioactive orotate and 5-fluoroorotate ligands: Synthesis, DFT calculations, structure, spectroscopic characterization and in vitro cytotoxicity. J. Mol. Struct. 2018, 1171, 155–167. [Google Scholar] [CrossRef]
- Boulikas, T. Clinical overview on LipoplatinTM: A successful liposomal formulation of cisplatin. Drugs 2009, 18, 1197–1218. [Google Scholar] [CrossRef]
- Farooq, M.A.; Aquib, M.; Farooq, A.; Haleem Khan, D.; Joelle Maviah, M.B.; Sied Filli, M.; Kesse, S.; Boakye-Yiadom, K.O.; Mavlyanova, R.; Parveen, A.; et al. Recent progress in nanotechnology-based novel drug delivery systems in designing of cisplatin for cancer therapy: An overview. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1674–1692. [Google Scholar] [CrossRef]
- Song, H.; Quan, F.; Yu, Z.; Zheng, M.; Ma, Y.; Xiao, H.; Ding, F. Carboplatin prodrug conjugated Fe3O4 nanoparticles for magnetically targeted drug delivery in ovarian cancer cells. J. Mater. Chem. B 2019, 7, 433–442. [Google Scholar] [CrossRef]
- Esim, O.; Gedik, M.E.; Dogan, A.L.; Gunaydin, G.; Hascicek, C. Development of carboplatin loaded bovine serum albumin nanoparticles and evaluation of its effect on an ovarian cancer cell line. J. Drug Deliv. Sci. Technol. 2021, 64, 102655. [Google Scholar] [CrossRef]
- Roudsari, M.H.; Saeidi, N.; Kabiri, N.; Ahmadi, A.; Tabrizi, M.M.; Shahmabadi, H.E.; Khiyavi, A.A.; Reghbati, B. Investigation of Characteristics and Behavior of Loaded Carboplatin on the, Liposomes Nanoparticles, on the Lung and Ovarian Cancer: An In-Vitro Evaluation. Asian Pac. J. Cancer Biol. 2016, 1, 9. [Google Scholar] [CrossRef]
- Sadasivam, M.; Avci, P.; Gupta, G.K.; Lakshmanan, S.; Chandran, R.; Huang, Y.Y.; Kumar, R.; Hamblin, M.R. Self-assembled liposomal nanoparticles in photodynamic therapy. Eur. J. Nanomed. 2013, 5, 115–129. [Google Scholar] [CrossRef]
- Liu, D.; Poon, C.; Lu, K.; He, C.; Lin, W. Self-assembled nanoscale coordination polymers with trigger release properties for effective anticancer therapy. Nat. Commun. 2014, 5, 4182. [Google Scholar] [CrossRef]
- Mi, P.; Miyata, K.; Kataoka, K.; Cabral, H. Clinical translation of self-assembled cancer nanomedicines. Adv. Ther. 2021, 4, 2000159. [Google Scholar] [CrossRef]
- Yang, G.G.; Pan, Z.Y.; Zhang, D.Y.; Cao, Q.; Ji, L.N.; Mao, Z.W. Precisely assembled nanoparticles against cisplatin resistance via cancer-specific targeting of mitochondria and imaging-guided chemo-photothermal therapy. ACS Appl. Mater. Interfaces 2020, 12, 43444–43455. [Google Scholar] [CrossRef] [PubMed]
- Du, X.F.; Li, Y.; Long, J.; Zhang, W.; Wang, D.; Li, C.R.; Zhao, M.X.; Lai, Y. Fabrication of cisplatin-loaded polydopamine nanoparticles via supramolecular self-assembly for photoacoustic imaging guided chemo-photothermal cancer therapy. Appl. Mater. Today 2021, 23, 101019. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2011, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA repair pathways and cisplatin resistance: An intimate relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-Y.; Guan, Y.-D.; Chen, X.-S.; Yang, J.-M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2021, 11, 629266. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J.S.; Wu, X.; Follen, M.; Gershenson, D. Nucleotide excision repair pathway review I: Implications in ovarian cancer and platinum sensitivity. Gynecol. Oncol. 2007, 107, S56–S71. [Google Scholar] [CrossRef] [PubMed]
- Kusakabe, M.; Onishi, Y.; Tada, H.; Kurihara, F.; Kusao, K.; Furukawa, M.; Iwai, S.; Yokoi, M.; Sakai, W.; Sugasawa, K. Mechanism and regulation of DNA damage recognition in nucleotide excision repair. Genes Environ. 2019, 41, 2. [Google Scholar] [CrossRef] [PubMed]
- Sawant, A.; Kothandapani, A.; Zhitkovich, A.; Sobol, R.W.; Patrick, S.M. Role of mismatch repair proteins in the processing of cisplatin interstrand cross-links. DNA Repair 2015, 35, 126. [Google Scholar] [CrossRef]
- Takahashi, M.; Koi, M.; Balaguer, F.; Boland, C.R.; Goel, A. MSH3 Mediates Sensitization of Colorectal Cancer Cells to Cisplatin, Oxaliplatin, and a Poly(ADP-ribose) Polymerase Inhibitor*. J. Biol. Chem. 2011, 286, 12157–12165. [Google Scholar] [CrossRef]
- Martin, L.P.; Hamilton, T.C.; Schilder, R.J. Platinum Resistance: The Role of DNA Repair Pathways. Clin. Cancer Res. 2008, 14, 1291–1295. [Google Scholar] [CrossRef]
- Tentori, L.; Muzi, A.; Dorio, A.S.; Dolci, S.; Campolo, F.; Vernole, P.; Lacal, P.M.; Praz, F.; Graziani, G. MSH3 expression does not influence the sensitivity of colon cancer HCT116 cell line to oxaliplatin and poly(ADP-ribose) polymerase (PARP) inhibitor as monotherapy or in combination. Cancer Chemother. Pharmacol. 2013, 72, 117–125. [Google Scholar] [CrossRef]
- Kunitomi, H.; Banno, K.; Yanokura, M.; Takeda, T.; Iijima, M.; Nakamura, K.; Iida, M.; Adachi, M.; Watanabe, K.; Matoba, Y.; et al. New use of microsatellite instability analysis in endometrial cancer. Oncol. Lett. 2017, 14, 3297. [Google Scholar] [CrossRef]
- Karamurzin, Y.; Rutgers, J.K.L. DNA mismatch repair deficiency in endometrial carcinoma. Int. J. Gynecol. Pathol. 2009, 28, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Alldredge, J.K.; Eskander, R.N. EZH2 inhibition in ARID1A mutated clear cell and endometrioid ovarian and endometrioid endometrial cancers. Gynecol. Oncol. Res. Pract. 2017, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.W.; Tang, J.J.; Sun, W.; Wang, H. PGK1 facilities cisplatin chemoresistance by triggering HSP90/ERK pathway mediated DNA repair and methylation in endometrial endometrioid adenocarcinoma. Mol. Med. 2019, 25, 11. [Google Scholar] [CrossRef] [PubMed]
- Fazal, Z.; Singh, R.; Fang, F.; Bikorimana, E.; Baldwin, H.; Corbet, A.; Tomlin, M.; Yerby, C.; Adra, N.; Albany, C.; et al. Hypermethylation and global remodelling of DNA methylation is associated with acquired cisplatin resistance in testicular germ cell tumours. Epigenetics 2021, 16, 1071–1084. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Constâncio, V.; Leite-Silva, P.; Guimarães, R.; Cantante, M.; Braga, I.; Maurício, J.; Looijenga, L.H.J.; Henrique, R.; Jerónimo, C. Differential methylation EPIC analysis discloses cisplatin-resistance related hypermethylation and tumor-specific heterogeneity within matched primary and metastatic testicular germ cell tumor patient tissue samples. Clin. Epigenet. 2021, 13, 70. [Google Scholar] [CrossRef]
- Diggle, C.P.; Bentley, J.; Knowles, M.A.; Kiltie, A.E. Inhibition of double-strand break non-homologous end-joining by cisplatin adducts in human cell extracts. Nucleic Acids Res. 2005, 33, 2531–2539. [Google Scholar] [CrossRef][Green Version]
- Boeckman, H.J.; Trego, K.S.; Turchi, J.J. Cisplatin sensitizes cancer cells to ionizing radiation via inhibition of non-homologous end joining. Mol. Cancer Res. MCR 2005, 3, 277. [Google Scholar] [CrossRef]
- West, R.B.; Yaneva, M.; Lieber, M.R. Productive and Nonproductive Complexes of Ku and DNA-Dependent Protein Kinase at DNA Termini. Mol. Cell. Biol. 1998, 18, 5908. [Google Scholar] [CrossRef]
- Borrego-Soto, G.; Ortiz-López, R.; Rojas-Martínez, A. Ionizing radiation-induced DNA injury and damage detection in patientswith breast cancer. Genet. Mol. Biol. 2015, 38, 420. [Google Scholar] [CrossRef]
- Sears, C.R.; Turchi, J.J. Complex Cisplatin-Double Strand Break (DSB) Lesions Directly Impair Cellular Non-Homologous End-Joining (NHEJ) Independent of Downstream Damage Response (DDR) Pathways. J. Biol. Chem. 2012, 287, 24263. [Google Scholar] [CrossRef]
- Usanova, S.; Piée-Staffa, A.; Sied, U.; Thomale, J.; Schneider, A.; Kaina, B.; Köberle, B. Cisplatin sensitivity of testis tumour cells is due to deficiency in interstrand-crosslink repair and low ERCC1-XPF expression. Mol. Cancer 2010, 9, 248. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Kothandapani, A.; Tillison, K.; Kalman-Maltese, V.; Patrick, S.M. Downregulation of XPF–ERCC1 enhances cisplatin efficacy in cancer cells. DNA Repair 2010, 9, 745. [Google Scholar] [CrossRef] [PubMed]
- Teraoka, S.; Muguruma, M.; Takano, N.; Miyahara, K.; Kawate, T.; Kaise, H.; Yamada, K.; Miyazawa, K.; Ishikawa, T. Association of BRCA Mutations and BRCAness Status With Anticancer Drug Sensitivities in Triple-Negative Breast Cancer Cell Lines. J. Surg. Res. 2020, 250, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Alli, E.; Sharma, V.B.; Hartman, A.R.; Lin, P.S.; McPherson, L.; Ford, J.M. Enhanced sensitivity to cisplatin and gemcitabine in Brca1-deficient murine mammary epithelial cells. BMC Pharmacol. 2011, 11, 7. [Google Scholar] [CrossRef]
- Panzarino, N.J.; Krais, J.J.; Cong, K.; Peng, M.; Mosqueda, M.; Nayak, S.U.; Bond, S.M.; Calvo, J.A.; Doshi, M.B.; Bere, M.; et al. Replication gaps underlie BRCA deficiency and therapy response. Cancer Res. 2021, 81, 1388–1397. [Google Scholar] [CrossRef]
- Deo, K.M.; Ang, D.L.; McGhie, B.; Rajamanickam, A.; Dhiman, A.; Khoury, A.; Holland, J.; Bjelosevic, A.; Pages, B.; Gordon, C.; et al. Platinum coordination compounds with potent anticancer activity. Coord. Chem. Rev. 2018, 375, 148–163. [Google Scholar] [CrossRef]
- Zhang, S.; Zhong, X.; Yuan, H.; Guo, Y.; Song, D.; Qi, F.; Zhu, Z.; Wang, X.; Guo, Z. Interfering in apoptosis and DNA repair of cancer cells to conquer cisplatin resistance by platinumn (iv) prodrugs. Chem. Sci. 2020, 11, 3829–3835. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116. [Google Scholar] [CrossRef]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Jackson, L.M.; Dhoonmoon, A.; Hale, A.; Dennis, K.A.; Schleicher, E.M.; Nicolae, C.M.; Moldovan, G.L. Loss of MED12 activates the TGFβ pathway to promote chemoresistance and replication fork stability in BRCA-deficient cells. Nucleic Acids Res. 2021, 49, 12855–12869. [Google Scholar] [CrossRef]
- Miron, B.; Hoffman-Censits, J.H.; Anari, F.; O’Neill, J.; Geynisman, D.M.; Zibelman, M.R.; Kutikov, A.; Viterbo, R.; Greenberg, R.E.; Chen, D.; et al. Defects in DNA Repair Genes Confer Improved Long-term Survival after Cisplatin-based Neoadjuvant Chemotherapy for Muscle-invasive Bladder Cancer. Eur. Urol. Oncol. 2020, 3, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Alblihy, A.; Miligy, I.M.; Alabdullah, M.L.; Alsaleem, M.; Toss, M.S.; Algethami, M.; Abdel-Fatah, T.; Moseley, P.; Chan, S.; et al. Molecular disruption of DNA polymerase β for platinum sensitisation and synthetic lethality in epithelial ovarian cancers. Oncogene 2021, 40, 2496–2508. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, K.A.; Alabdullah, M.; Griffin, M.; Toss, M.S.; Fatah, T.M.A.A.; Alblihy, A.; Moseley, P.; Chan, S.Y.T.; Rakha, E.A.; Madhusudan, S. ERCC1-XPF deficiency is a predictor of olaparib induced synthetic lethality and platinum sensitivity in epithelial ovarian cancers. Gynecol. Oncol. 2019, 153, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Zazuli, Z.; Otten, L.S.; Drögemöller, B.I.; Medeiros, M.; Monzon, J.G.; Wright, G.E.B.; Kollmannsberger, C.K.; Bedard, P.L.; Chen, Z.; Gelmon, K.A.; et al. Outcome Definition Influences the Relationship between Genetic Polymorphisms of ERCC1, ERCC2, SLC22A2 and Cisplatin Nephrotoxicity in Adult Testicular Cancer Patients. Genes 2019, 10, 364. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chen, D.; Yi, Y.; Zeng, S.; Liu, S.; Li, P.; Xie, H.; Yu, P.; Jiang, G.; Liu, H. Histone Deacetylase Inhibitor Sensitizes ERCC1-High Non-small-Cell Lung Cancer Cells to Cisplatin via Regulating miR-149. Mol. Ther. Oncolytics 2020, 17, 448–459. [Google Scholar] [CrossRef]
- Pan, C.H.; Chen, S.Y.; Wang, J.Y.; Tsao, S.P.; Huang, H.Y.; Wei-Chen Chiu, P.; Wu, C.H. Sclareol ameliorated ERCC1-mediated cisplatin resistance in A549 human lung adenocarcinoma cells and a murine xenograft tumor model by suppressing AKT-GSK3β-AP1/Snail and JNK-AP1 pathways. Chem. Biol. Interact. 2020, 332, 109304. [Google Scholar] [CrossRef]
- Du, P.; Wang, Y.; Chen, L.; Gan, Y.; Wu, Q. High ERCC1 expression is associated with platinum-resistance, but not survival in patients with epithelial ovarian cancer. Oncol. Lett. 2016, 12, 857–862. [Google Scholar] [CrossRef]
- Chen, P.; Li, J.; Chen, Y.C.; Qian, H.; Chen, Y.J.; Su, J.Y.; Wu, M.; Lan, T. The functional status of DNA repair pathways determines the sensitization effect to cisplatin in non-small cell lung cancer cells. Cell. Oncol. 2016, 39, 511–522. [Google Scholar] [CrossRef]
- Prieto-Garcia, C.; Hartmann, O.; Reissland, M.; Fischer, T.; Maier, C.R.; Rosenfeldt, M.; Schülein-Völk, C.; Klann, K.; Kalb, R.; Dikic, I.; et al. Inhibition of USP28 overcomes Cisplatin-resistance of squamous tumors by suppression of the Fanconi anemia pathway. Cell Death Differ. 2021, 29, 568–584. [Google Scholar] [CrossRef]
- Jacquemont, C.; Simon, J.A.; D’Andrea, A.D.; Taniguchi, T. Non-specific chemical inhibition of the Fanconi anemia pathway sensitizes cancer cells to cisplatin. Mol. Cancer 2012, 11, 26. [Google Scholar] [CrossRef]
- Kutuk, O.; Arisan, E.D.; Tezil, T.; Shoshan, M.C.; Basaga, H. Cisplatin overcomes Bcl-2-mediated resistance to apoptosis via preferential engagement of Bak: Critical role of Noxa-mediated lipid peroxidation. Carcinogenesis 2009, 30, 1517–1527. [Google Scholar] [CrossRef]
- Fraser, M.; Bai, T.; Tsang, B.K. Akt promotes cisplatin resistance in human ovarian cancer cells through inhibition of p53 phosphorylation and nuclear function. Int. J. Cancer 2008, 122, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Kilari, D.; Guancial, E.; Kim, E.S. Role of copper transporters in platinum resistance. World J. Clin. Oncol. 2016, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Kalayda, G.V.; Wagner, C.H.; Buß, I.; Reedijk, J.; Jaehde, U. Altered localisation of the copper efflux transporters ATP7A and ATP7B associated with cisplatin resistance in human ovarian carcinoma cells. BMC Cancer 2008, 8, 175. [Google Scholar] [CrossRef]
- Zhu, S.; Shanbhag, V.; Wang, Y.; Lee, J.; Petris, M. A Role for The ATP7A Copper Transporter in Tumorigenesis and Cisplatin Resistance. J. Cancer 2017, 8, 1952. [Google Scholar] [CrossRef]
- Li, Z.H.; Qiu, M.Z.; Zeng, Z.L.; Luo, H.Y.; Wu, W.J.; Wang, F.; Wang, Z.Q.; Zhang, D.S.; Li, Y.H.; Xu, R.H. Copper-transporting P-type adenosine triphosphatase (ATP7A) is associated with platinum-resistance in non-small cell lung cancer (NSCLC). J. Transl. Med. 2012, 10, 21. [Google Scholar] [CrossRef]
- Li, Z.H.; Zheng, R.; Chen, J.T.; Jia, J.; Qiu, M. The role of copper transporter ATP7A in platinum-resistance of esophageal squamous cell cancer (ESCC). J. Cancer 2016, 7, 2085. [Google Scholar] [CrossRef]
- Rébé, C.; Demontoux, L.; Pilot, T.; Ghiringhelli, F. Platinum Derivatives Effects on Anticancer Immune Response. Biomolecules 2019, 10, 13. [Google Scholar] [CrossRef]
- Okita, R.; Yukawa, T.; Nojima, Y.; Maeda, A.; Saisho, S.; Shimizu, K.; Nakata, M. MHC class I chain-related molecule A and B expression is upregulated by cisplatin and associated with good prognosis in patients with non-small cell lung cancer. Cancer Immunol. Immunother. 2016, 65, 499–509. [Google Scholar] [CrossRef]
- De Biasi, A.R.; Villena-Vargas, J.; Adusumilli, P.S. Cisplatin-Induced Antitumor Immunomodulation: A Review of Preclinical and Clinical Evidence. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 5384. [Google Scholar] [CrossRef]
- Gameiro, S.R.; Caballero, J.A.; Hodge, J.W. Defining the molecular signature of chemotherapy-mediated lung tumor phenotype modulation and increased susceptibility to T-cell killing. Cancer Biother. Radiopharm. 2012, 27, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Pestka, S.; Jubin, R.G.; Lyu, Y.L.; Tsai, Y.C.; Liu, L.F. Chemotherapeutics and radiation stimulate MHC class I expression through elevated interferon-beta signaling in breast cancer cells. PLoS ONE 2012, 7, e32542. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Meurisse, A.; Spehner, L.; Stouvenot, M.; François, E.; Buecher, B.; André, T.; Samalin, E.; Jary, M.; Nguyen, T.; et al. Pooled analysis of 115 patients from updated data of Epitopes-HPV01 and Epitopes-HPV02 studies in first-line advanced anal squamous cell carcinoma. Ther. Adv. Med Oncol. 2020, 12, 1758835920975356. [Google Scholar] [CrossRef] [PubMed]
- Spehner, L.; Boustani, J.; Cabel, L.; Doyen, J.; Vienot, A.; Borg, C.; Kim, S. Present and Future Research on Anal Squamous Cell Carcinoma. Cancers 2021, 13, 3895. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Meurisse, A.; Stouvenot, M.; Jary, M.; Hon, T.N.T.; Francois, E.; Buecher, B.; Andre, T.; Samalin, E.; Boulbair, F.; et al. Updated data of epitopes-HPV02 trial and external validation of efficacy of DCF in prospective epitopes-HPV01 study in advanced anal squamous cell carcinoma. Pooled analysis of 115 patients. Ann. Oncol. 2019, 30, v203. [Google Scholar] [CrossRef]
- Spehner, L.; Kim, S.; Vienot, A.; François, E.; Buecher, B.; Adotevi, O.; Vernerey, D.; Abdeljaoued, S.; Meurisse, A.; Borg, C. Anti-Telomerase CD4+ Th1 Immunity and Monocytic-Myeloid-Derived-Suppressor Cells Are Associated with Long-Term Efficacy Achieved by Docetaxel, Cisplatin, and 5-Fluorouracil (DCF) in Advanced Anal Squamous Cell Carcinoma: Translational Study of Epitopes-HPV01 and 02 Trials. Int. J. Mol. Sci. 2020, 21, 6838. [Google Scholar] [CrossRef]
- Kim, S.; François, E.; André, T.; Samalin, E.; Jary, M.; El Hajbi, F.; Baba-Hamed, N.; Pernot, S.; Kaminsky, M.C.; Bouché, O.; et al. Docetaxel, cisplatin, and fluorouracil chemotherapy for metastatic or unresectable locally recurrent anal squamous cell carcinoma (Epitopes-HPV02): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 1094–1106. [Google Scholar] [CrossRef]
- Chang, C.L.; Hsu, Y.T.; Wu, C.C.; Lai, Y.Z.; Wang, C.; Yang, Y.C.; Wu, T.C.; Hung, C.F. Dose-dense chemotherapy improves mechanisms of antitumor immune response. Cancer Res. 2013, 73, 119. [Google Scholar] [CrossRef]
- Grabosch, S.; Bulatovic, M.; Zeng, F.; Ma, T.; Zhang, L.; Ross, M.; Brozick, J.; Fang, Y.S.; Tseng, G.; Kim, E.; et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene 2019, 38, 2380. [Google Scholar] [CrossRef]
- Liu, X.; He, S.; Wu, H.; Xie, H.; Zhang, T.; Deng, Z. Blocking the PD-1/PD-L1 axis enhanced cisplatin chemotherapy in osteosarcoma in vitro and in vivo. Environ. Health Prev. Med. 2019, 24, 79. [Google Scholar] [CrossRef]
- Wei, H.; Zhao, L.; Li, W.; Fan, K.; Qian, W.; Hou, S.; Wang, H.; Dai, M.; Hellstrom, I.; Hellstrom, K.E.; et al. Combinatorial PD-1 Blockade and CD137 Activation Has Therapeutic Efficacy in Murine Cancer Models and Synergizes with Cisplatin. PLoS ONE 2013, 8, 84927. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Qian, J.; Wang, H.; Ren, L.; Yang, Y.; Chen, C.; Chen, X.; Huang, Y.; Liu, J.; Xu, N.; et al. Cisplatin plus anti-PD-1 antibody enhanced treatment efficacy in advanced esophageal squamous cell carcinoma. Am. J. Cancer Res. 2022, 12, 451. [Google Scholar]
- Sun, F.; Cui, L.; Li, T.; Chen, S.; Song, J.; Li, D. Oxaliplatin induces immunogenic cells death and enhances therapeutic efficacy of checkpoint inhibitor in a model of murine lung carcinoma. J. Recept. Signal Transduct. 2019, 39, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Martins, I.; Kepp, O.; Schlemmer, F.; Adjemian, S.; Tailler, M.; Shen, S.; Michaud, M.; Menger, L.; Gdoura, A.; Tajeddine, N.; et al. Restoration of the immunogenicity of cisplatin-induced cancer cell death by endoplasmic reticulum stress. Oncogene 2010, 30, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Tesniere, A.; Schlemmer, F.; Boige, V.; Kepp, O.; Martins, I.; Ghiringhelli, F.; Aymeric, L.; Michaud, M.; Apetoh, L.; Barault, L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2009, 29, 482–491. [Google Scholar] [CrossRef]
- Bains, S.J.; Abrahamsson, H.; Flatmark, K.; Dueland, S.; Hole, K.H.; Seierstad, T.; Redalen, K.R.; Meltzer, S.; Ree, A.H. Immunogenic cell death by neoadjuvant oxaliplatin and radiation protects against metastatic failure in high-risk rectal cancer. Cancer Immunol. Immunother. 2020, 69, 355. [Google Scholar] [CrossRef]
- Song, W.; Shen, L.; Wang, Y.; Liu, Q.; Goodwin, T.J.; Li, J.; Dorosheva, O.; Liu, T.; Liu, R.; Huang, L. Indeed, self-assembled nanomedicine for cisplatin and lipoplatin as well as other chemotherapeutic drugs is an accelerating field of research that is believed to overcome the major toxicity problem limiting their use. Molecular self- assembly is a process. Nat. Commun. 2018, 9, 2237. [Google Scholar] [CrossRef]
- Zhu, H.; Shan, Y.; Ge, K.; Lu, J.; Kong, W.; Jia, C. Oxaliplatin induces immunogenic cell death in hepatocellular carcinoma cells and synergizes with immune checkpoint blockade therapy. Cell. Oncol. 2020, 43, 1203–1214. [Google Scholar] [CrossRef]
- Sprowl, J.A.; Lancaster, C.S.; Pabla, N.; Hermann, E.; Kosloske, A.M.; Gibson, A.A.; Li, L.; Zeeh, D.; Schlatter, E.; Janke, L.J.; et al. Cisplatin-induced renal injury is independently mediated by OCT2 and p53. Clin. Cancer Res. 2014, 20, 4026–4035. [Google Scholar] [CrossRef] [PubMed]
- Ciarimboli, G.; Deuster, D.; Knief, A.; Sperling, M.; Holtkamp, M.; Edemir, B.; Pavenstädt, H.; Lanvers-Kaminsky, C.; Zehnhoff-Dinnesen, A.A.; Schinkel, A.H.; et al. Organic Cation Transporter 2 Mediates Cisplatin-Induced Oto- and Nephrotoxicity and Is a Target for Protective Interventions. Am. J. Pathol. 2010, 176, 1169. [Google Scholar] [CrossRef] [PubMed]
- Katsuda, H.; Yamashita, M.; Katsura, H.; Yu, J.; Waki, Y.; Nagata, N.; Sai, Y.; Miyamoto, K.I. Protecting cisplatin-induced nephrotoxicity with cimetidine does not affect antitumor activity. Biol. Pharm. Bull. 2010, 33, 1867–1871. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef]
- Blair, B.G.; Larson, C.; Safaei, R.; Howell, S.B. Copper transporter 2 regulates the cellular accumulation and cytotoxicity of cisplatin and carboplatin. Clin. Cancer Res. 2009, 15, 4312–4321. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; McCormick, F.; Smith-McCune, K.; Hanahan, D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell 2010, 17, 574. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.S.; Tang, X.M.; Peterson, D.R.; Kilari, D.; Chow, C.W.; Fujimoto, J.; Kalhor, N.; Swisher, S.G.; Stewart, D.J.; Wistuba, I.I.; et al. Copper transporter CTR1 expression and tissue platinum concentration in non-small cell lung cancer. Lung Cancer 2014, 85, 88–93. [Google Scholar] [CrossRef]
- Rose, M.C.; Kostyanovskaya, E.; Huang, R.S. Pharmacogenomics of Cisplatin Sensitivity in Non-small Cell Lung Cancer. Genom. Proteom. Bioinform. 2014, 12, 198–209. [Google Scholar] [CrossRef]
- Sawers, L.; Ferguson, M.J.; Ihrig, B.R.; Young, H.C.; Chakravarty, P.; Wolf, C.R.; Smith, G. Glutathione S-transferase P1 (GSTP1) directly influences platinum drug chemosensitivity in ovarian tumour cell lines. Br. J. Cancer 2014, 111, 1150–1158. [Google Scholar] [CrossRef]
- Wülfing, C.; van Ahlen, H.; Eltze, E.; Piechota, H.; Hertle, L.; Schmid, K.W. Metallothionein in bladder cancer: Correlation of overexpression with poor outcome after chemotherapy. World J. Urol. 2007, 25, 199–205. [Google Scholar] [CrossRef]
- Werynska, B.; Pula, B.; Muszczynska-Bernhard, B.; Gomulkiewicz, A.; Piotrowska, A.; Prus, R.; Podhorska-Okolow, M.; Jankowska, R.; Dziegiel, P. Metallothionein 1F and 2A overexpression predicts poor outcome of non-small cell lung cancer patients. Exp. Mol. Pathol. 2013, 94, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, M.A.M.; Michalkova, H.; Strmiska, V.; Casar, B.; Crespo, P.; de los Rios, V.; Ignacio Casal, J.; Haddad, Y.; Guran, R.; Eckschlager, T.; et al. Metallothionein-3 promotes cisplatin chemoresistance remodelling in neuroblastoma. Sci. Rep. 2021, 11, 5496. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tang, Y.; Yang, D.; Zheng, L. MicroRNA-591 Functions as a Tumor Suppressor in Hepatocellular Carcinoma by Lowering Drug Resistance through Inhibition of Far-Upstream Element-Binding Protein 2-Mediated Phosphoinositide 3-Kinase/Akt/Mammalian Target of Rapamycin Axis. Pharmacology 2019, 104, 173–186. [Google Scholar] [CrossRef]
- Williams, L.A.; Mills, L.; Hooten, A.J.; Langer, E.; Roesler, M.; Frazier, A.L.; Krailo, M.; Nelson, H.H.; Bestrashniy, J.; Amatruda, J.F.; et al. Differences in DNA methylation profiles by histologic subtype of paediatric germ cell tumours: A report from the Children’s Oncology Group. Br. J. Cancer 2018, 119, 864–872. [Google Scholar] [CrossRef]
- Vander Broek, R.; Snow, G.E.; Chen, Z.; Van Waes, C. Chemoprevention of Head and Neck Squamous Cell Carcinoma through Inhibition of NF-κB Signaling. Oral Oncol. 2014, 50, 930. [Google Scholar] [CrossRef]
- Steele, N.; Finn, P.; Brown, R.; Plumb, J.A. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br. J. Cancer 2009, 100, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Redon, C.E.; Peer, C.J.; Bryla, C.; Lee, M.J.; Trepel, J.B.; Tomita, Y.; Rajan, A.; Giaccone, G.; Bonner, W.M.; et al. Phase I trial of belinostat with cisplatin and etoposide in advanced solid tumors, with a focus on neuroendocrine and small cell cancers of the lung. Anti-Cancer Drugs 2018, 29, 457. [Google Scholar] [CrossRef]
- Lobo, J.; Guimarães-Teixeira, C.; Barros-Silva, D.; Miranda-Gonçalves, V.; Camilo, V.; Guimarães, R.; Cantante, M.; Braga, I.; Maurício, J.; Oing, C.; et al. Efficacy of HDAC Inhibitors Belinostat and Panobinostat against Cisplatin-Sensitive and Cisplatin-Resistant Testicular Germ Cell Tumors. Cancers 2020, 12, 2903. [Google Scholar] [CrossRef]
- Kim, T.M.; Laird, P.W.; Park, P.J. XThe landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell 2013, 155, 858. [Google Scholar] [CrossRef]
- Watanabe, Y.; Koi, M.; Hemmi, H.; Hoshai, H.; Noda, K. A change in microsatellite instability caused by cisplatin-based chemotherapy of ovarian cancer. Br. J. Cancer 2001, 85, 1064–1069. [Google Scholar] [CrossRef]
- Nicolay, N.H.; Helleday, T.; Sharma, R.A. Biological Relevance of DNA Polymerase Beta and Translesion Synthesis Polymerases to Cancer and its Treatment. Curr. Mol. Pharmacol. 2011, 5, 54–67. [Google Scholar] [CrossRef]
- Shilkin, E.S.; Boldinova, E.O.; Stolyarenko, A.D.; Goncharova, R.I.; Chuprov-Netochin, R.N.; Smal, M.P.; Makarova, A.V. Translesion DNA Synthesis and Reinitiation of DNA Synthesis in Chemotherapy Resistance. Biochemistry 2020, 85, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Ling, H. Structural Basis for Human DNA Polymerase Kappa to Bypass Cisplatin Intrastrand Cross-Link (Pt-GG) Lesion as an Efficient and Accurate Extender. J. Mol. Biol. 2018, 430, 1577–1589. [Google Scholar] [CrossRef] [PubMed]
- Doles, J.; Oliver, T.G.; Cameron, E.R.; Hsu, G.; Jacks, T.; Walker, G.C.; Hemann, M.T. Suppression of Rev3, the catalytic subunit of Pol?, sensitizes drug-resistant lung tumors to chemotherapy. Proc. Natl. Acad. Sci. USA 2010, 107, 20786–20791. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chen, Y.W.; Liu, X.; Chu, P.; Loria, S.; Wang, Y.; Yen, Y.; Chou, K.M. Expression of DNA Translesion Synthesis Polymerase η in Head and Neck Squamous Cell Cancer Predicts Resistance to Gemcitabine and Cisplatin-Based Chemotherapy. PLoS ONE 2013, 8, e83978. [Google Scholar] [CrossRef]
- Einhorn, L.H. Treatment of testicular cancer: A new and improved model. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1990, 8, 1777–1781. [Google Scholar] [CrossRef]
- de Vries, G.; Rosas-Plaza, X.; van Vugt, M.A.T.M.; Gietema, J.A.; de Jong, S. Testicular cancer: Determinants of cisplatin sensitivity and novel therapeutic opportunities. Cancer Treat. Rev. 2020, 88, 102054. [Google Scholar] [CrossRef]
- Mok, T.S.K.; Hsia, T.C.; Tsai, C.M.; Tsang, K.; Chang, G.C.; Chang, J.W.C.; Thitiya, S.; Sriuranpong, V.; Thongprasert, S.; Chua, D.T.; et al. Efficacy of bevacizumab with cisplatin and gemcitabine in Asian patients with advanced or recurrent non-squamous non-small cell lung cancer who have not received prior chemotherapy: A substudy of the Avastin in Lung trial. Asia Pac. J. Clin. Oncol. 2011, 7, 4–12. [Google Scholar] [CrossRef]
- Chu, G.; Liu, X.; Yu, W.; Chen, M.; Dong, L. Cisplatin plus paclitaxel chemotherapy with or without bevacizumab in postmenopausal women with previously untreated advanced cervical cancer: A retrospective study. BMC Cancer 2021, 21, 133. [Google Scholar] [CrossRef]
- Yarchoan, M.; Myzak, M.C.; Johnson, B.A.; De Jesus-Acosta, A.D.; Le, D.T.; Jaffee, E.M.; Azad, N.S.; Donehower, R.C.; Zheng, L.; Oberstein, P.E.; et al. Olaparib in combination with irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic cancer. Oncotarget 2017, 8, 44073–44081. [Google Scholar] [CrossRef]
- Arita, M.; Watanabe, S.; Aoki, N.; Kuwahara, S.; Suzuki, R.; Goto, S.; Abe, Y.; Takahashi, M.; Sato, M.; Hokari, S.; et al. Combination therapy of cisplatin with cilastatin enables an increased dose of cisplatin, enhancing its antitumor effect by suppression of nephrotoxicity. Sci. Rep. 2021, 11, 750. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).