
Improvement of Kiteplatin Efficacy by a Benzoato Pt(IV) Prodrug Suitable for Oral Administration


 ,
,  ,
,  ,
,  ,
,  , ,
, , 
Abstract
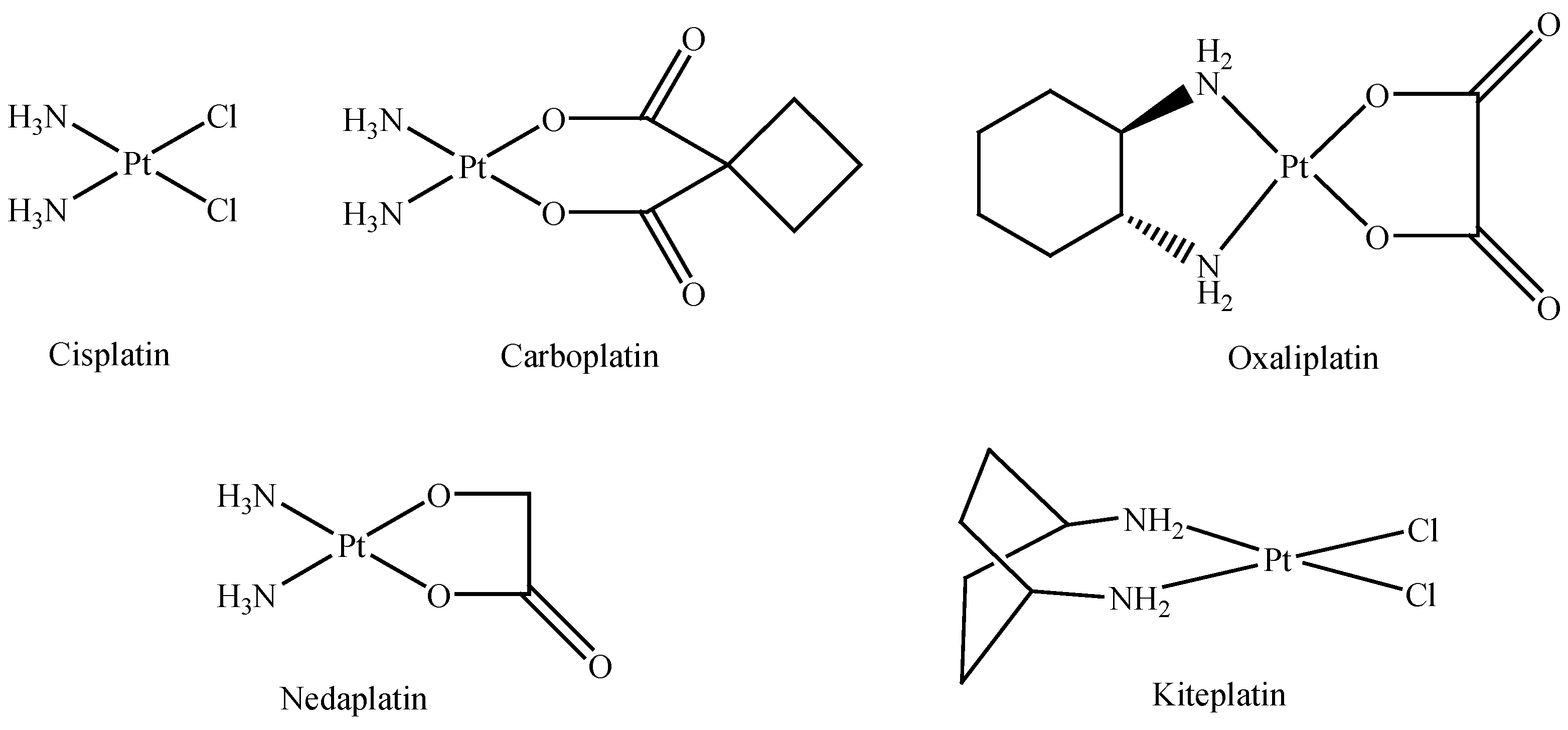
:1. Introduction
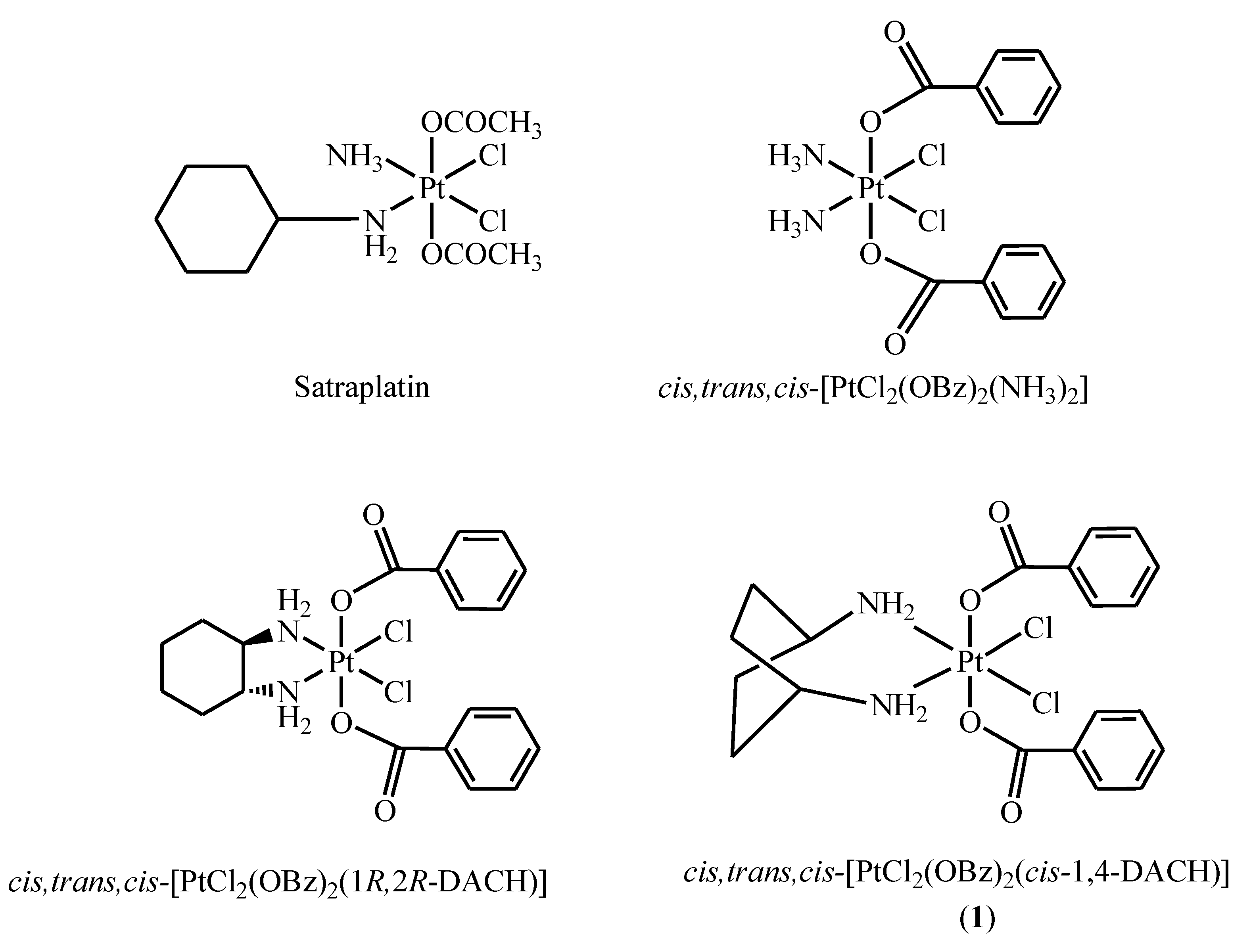
2. Results and Discussion
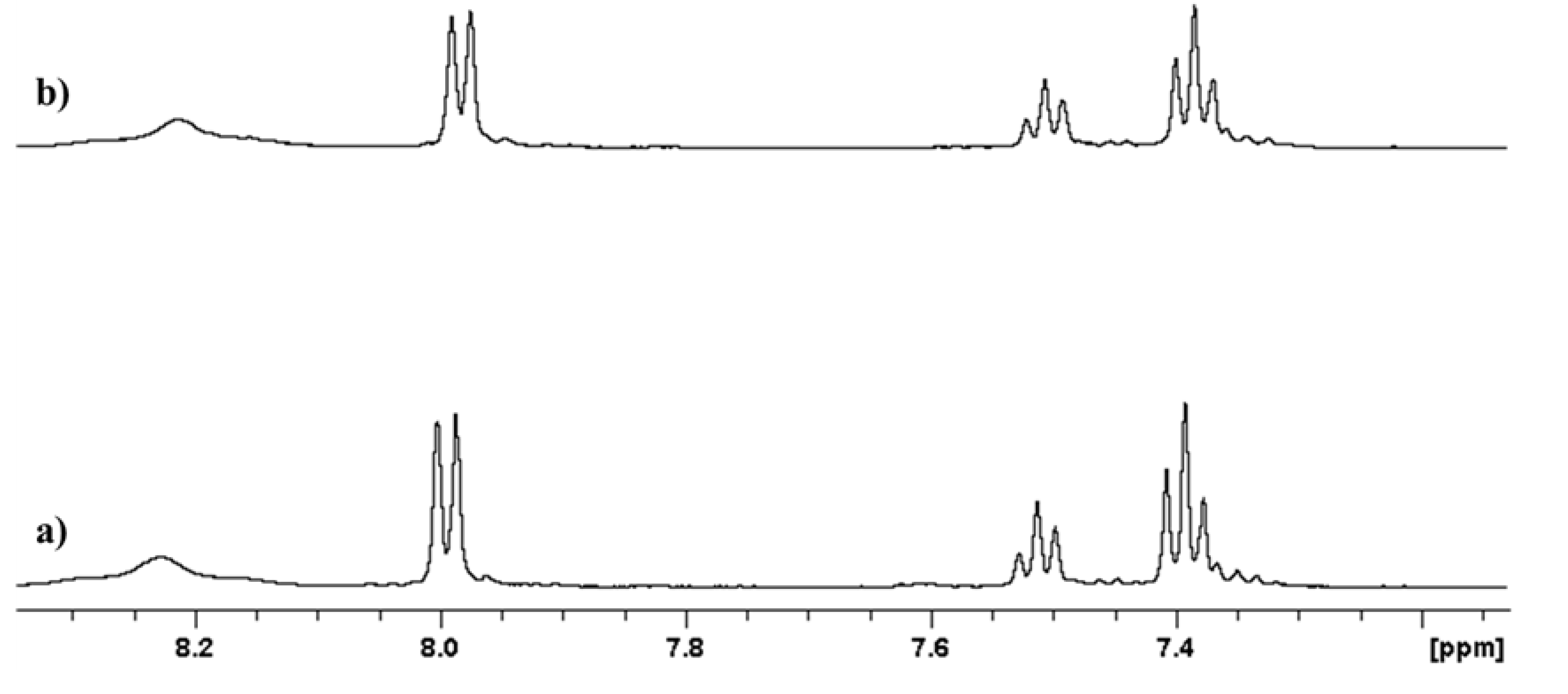
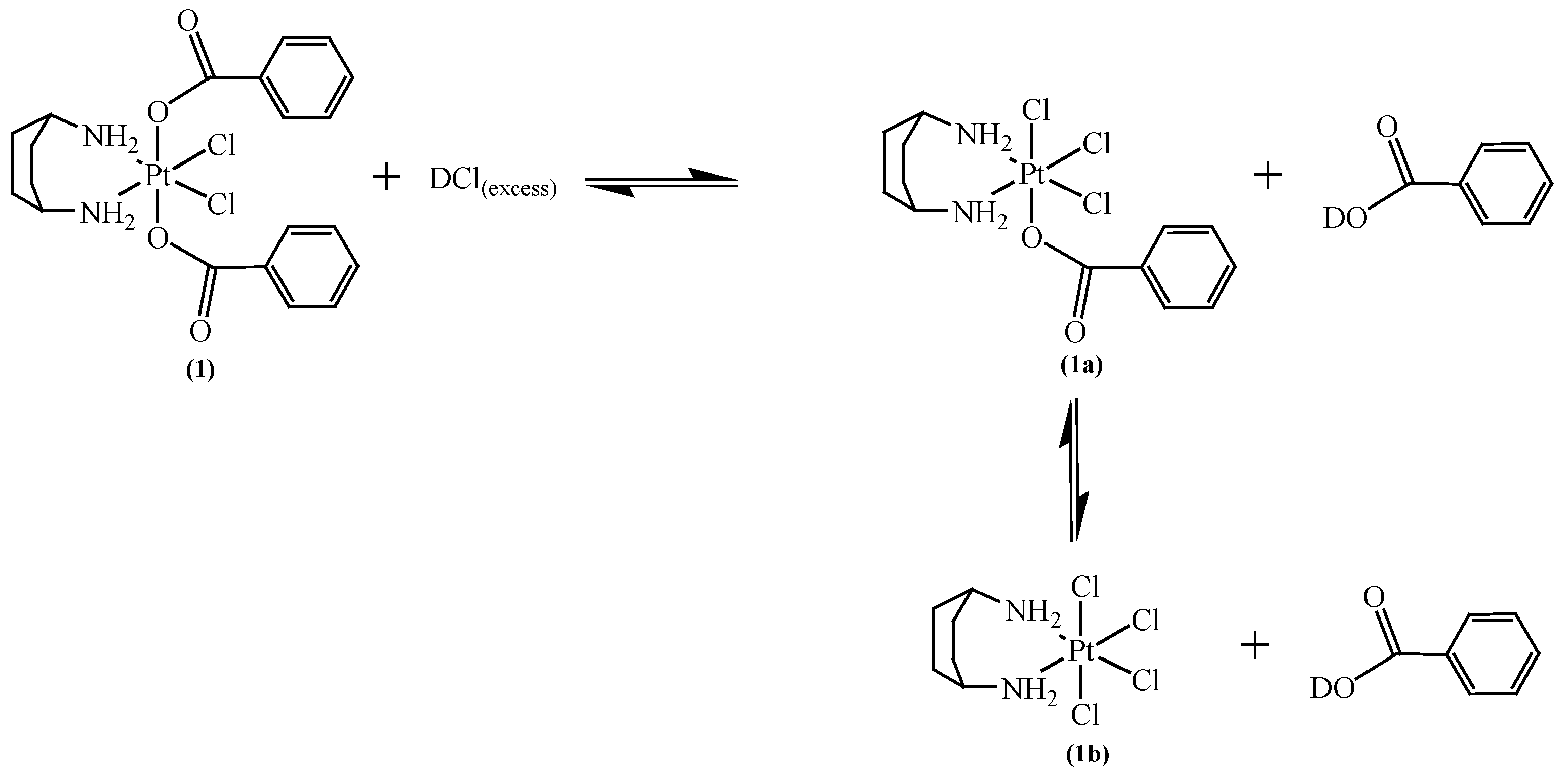
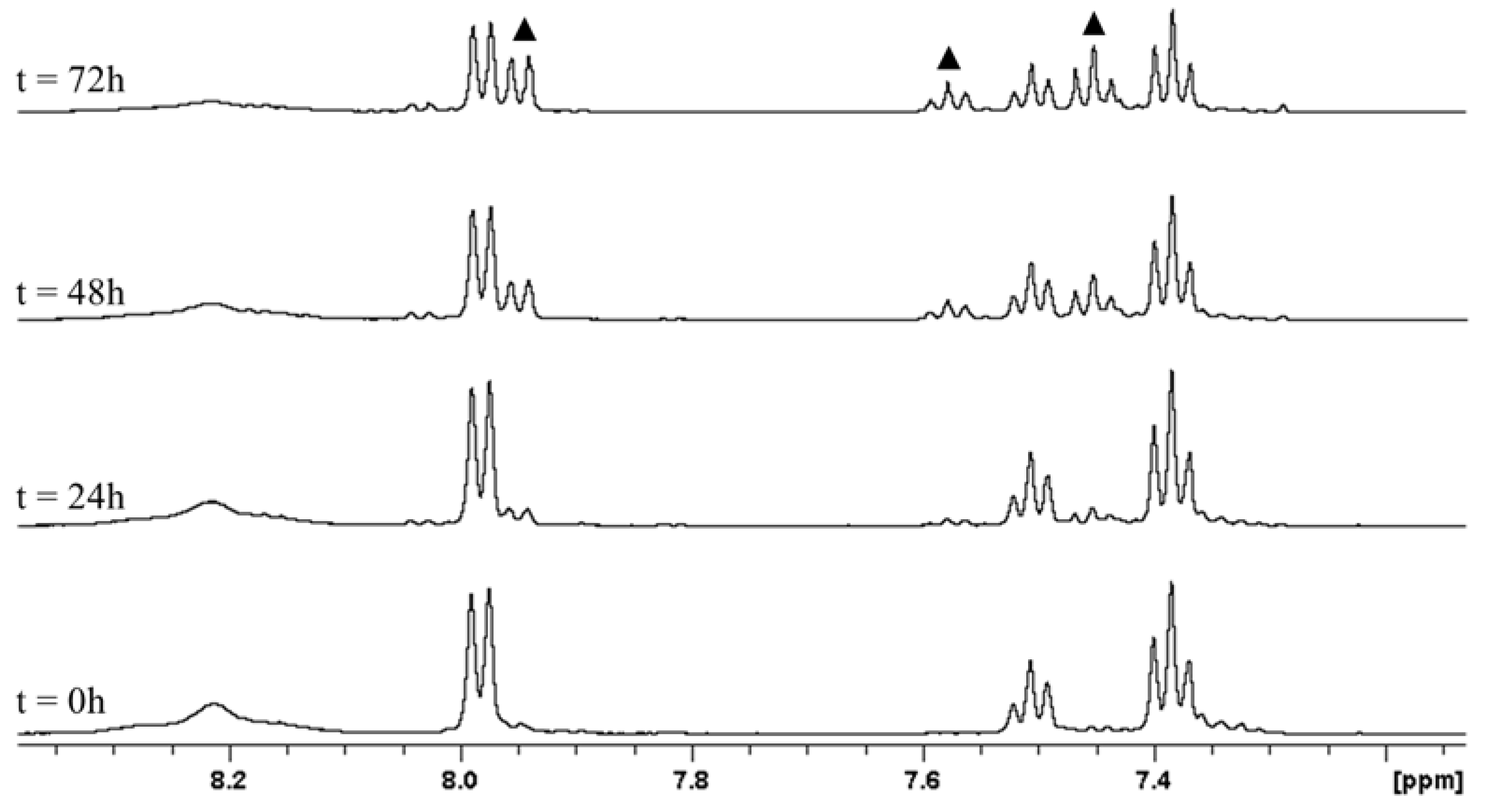
2.1. Stability at Acidic pH
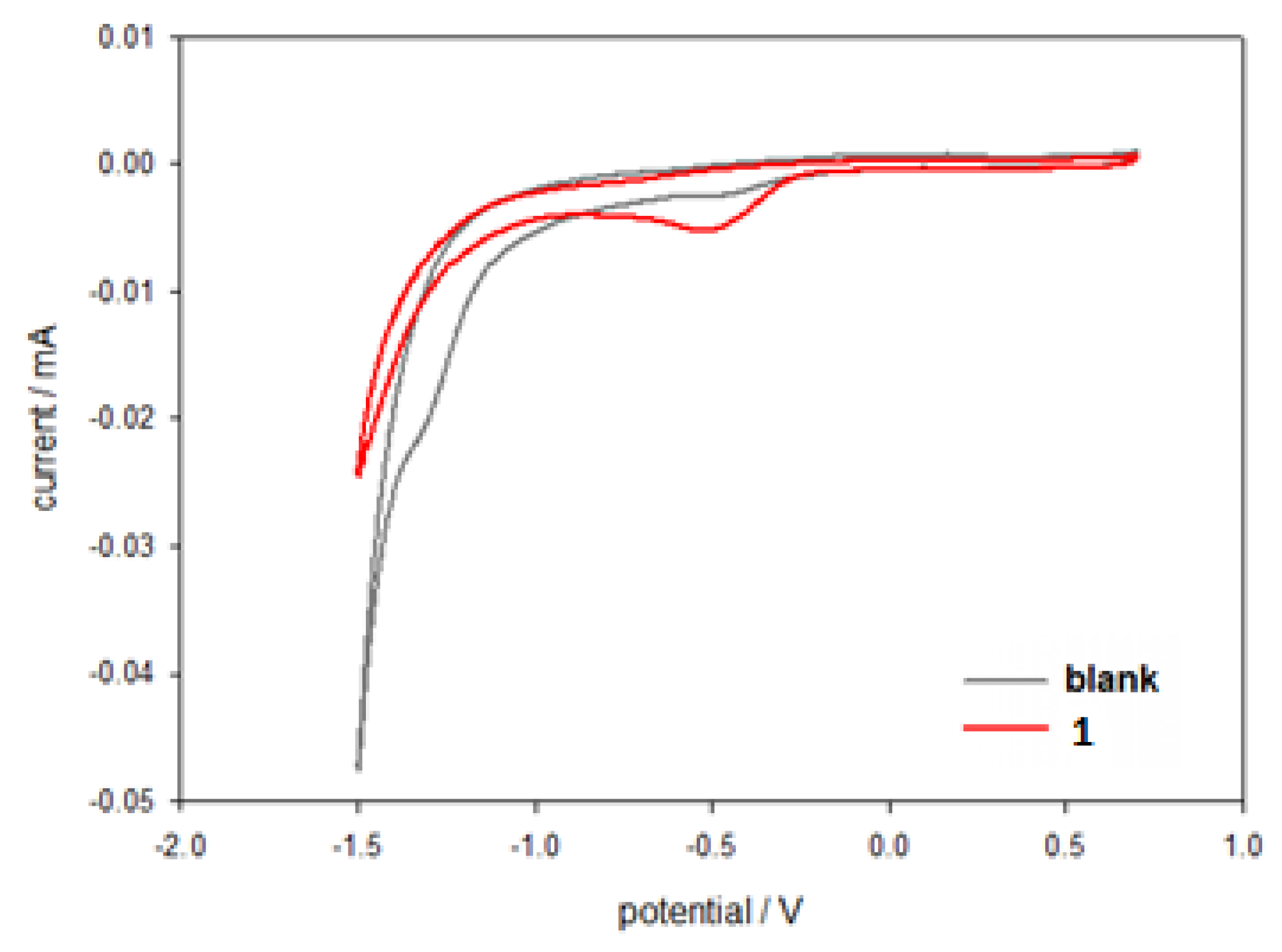
2.2. Electrochemical Characterization
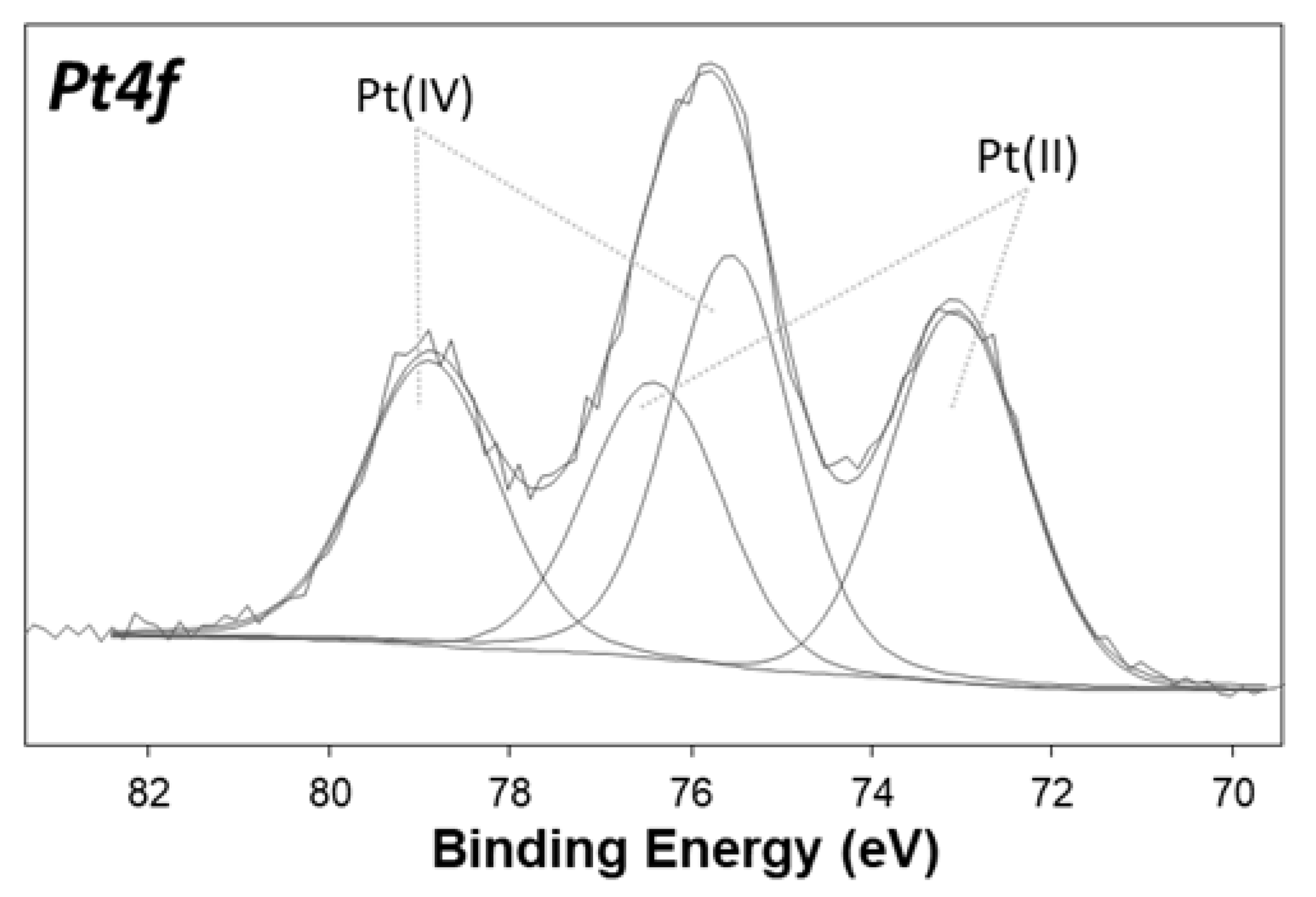
2.3. Characterization of the Surface Reduction
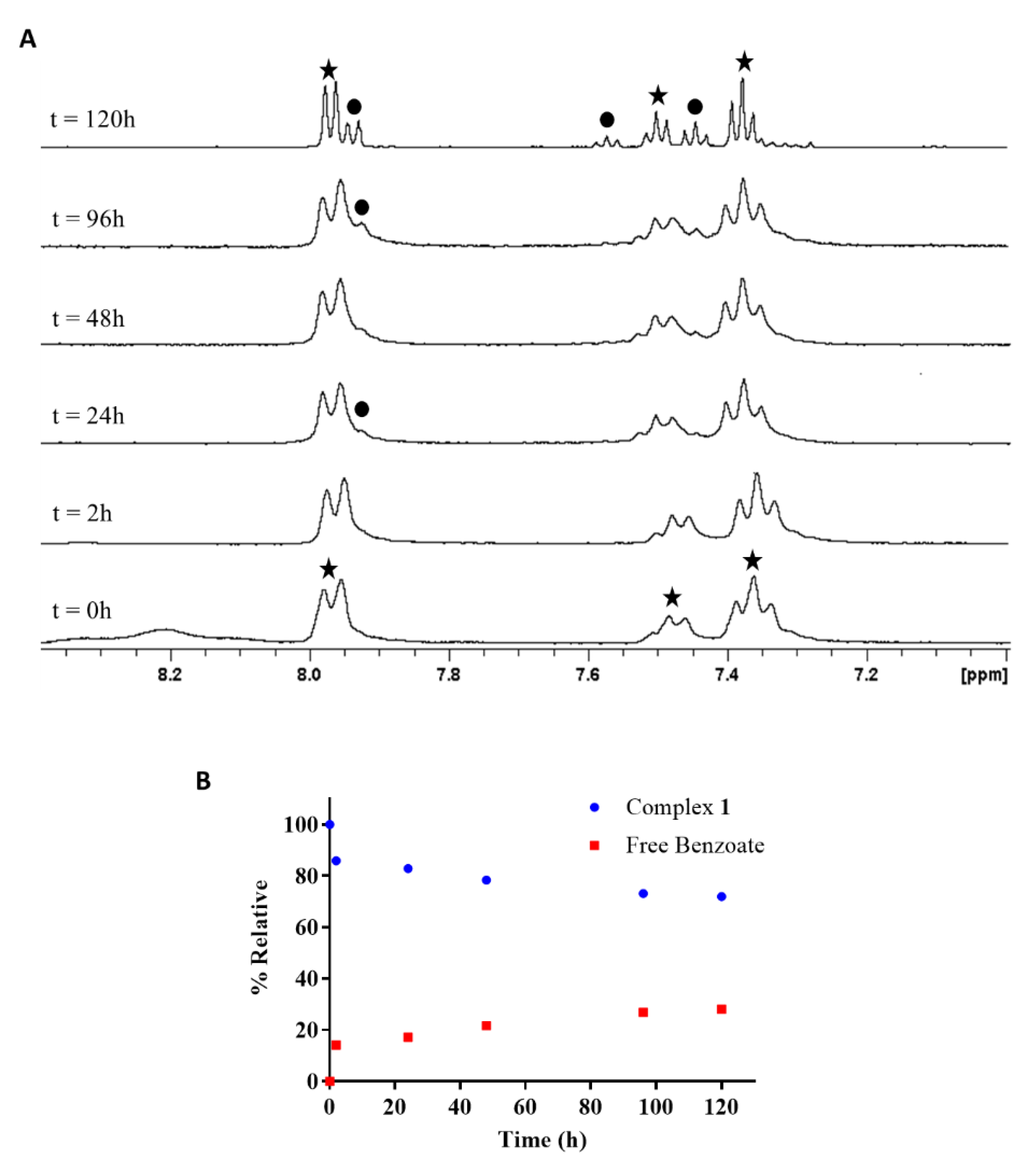
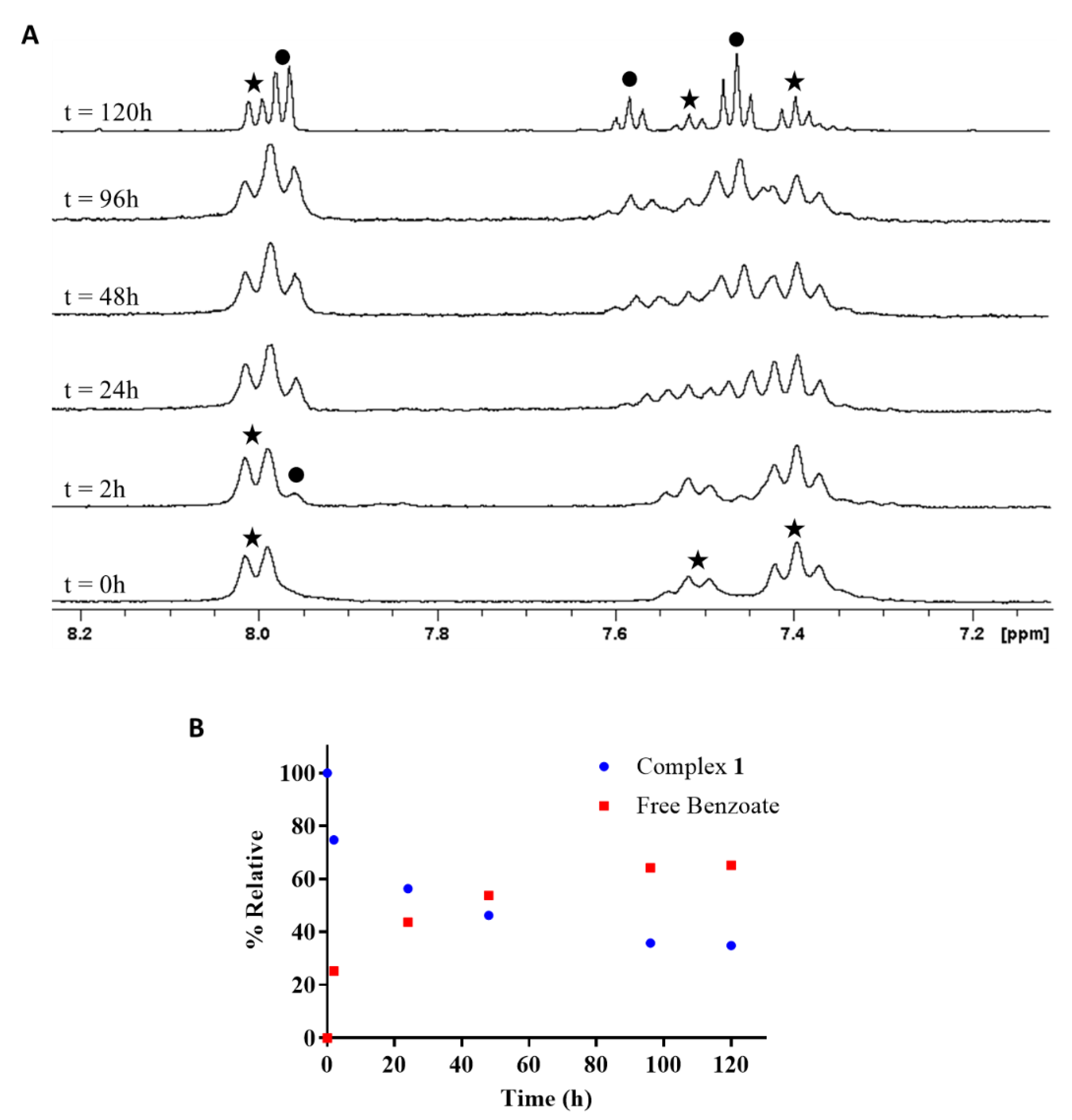
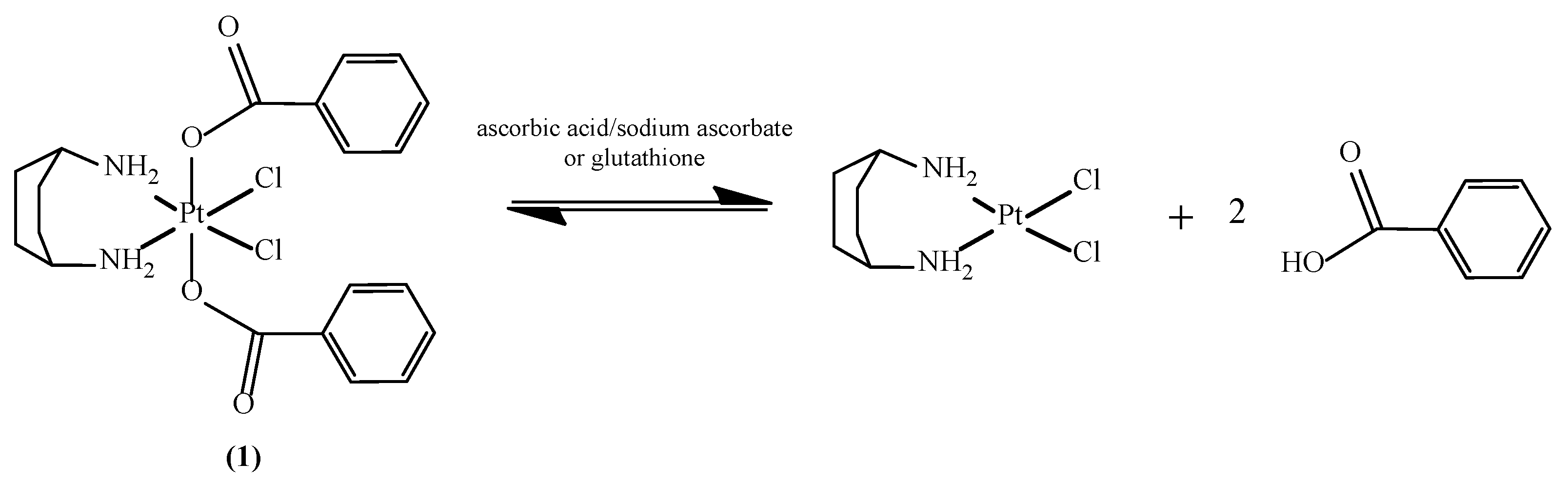
2.4. Reduction of Complex 1 by Bioreductants
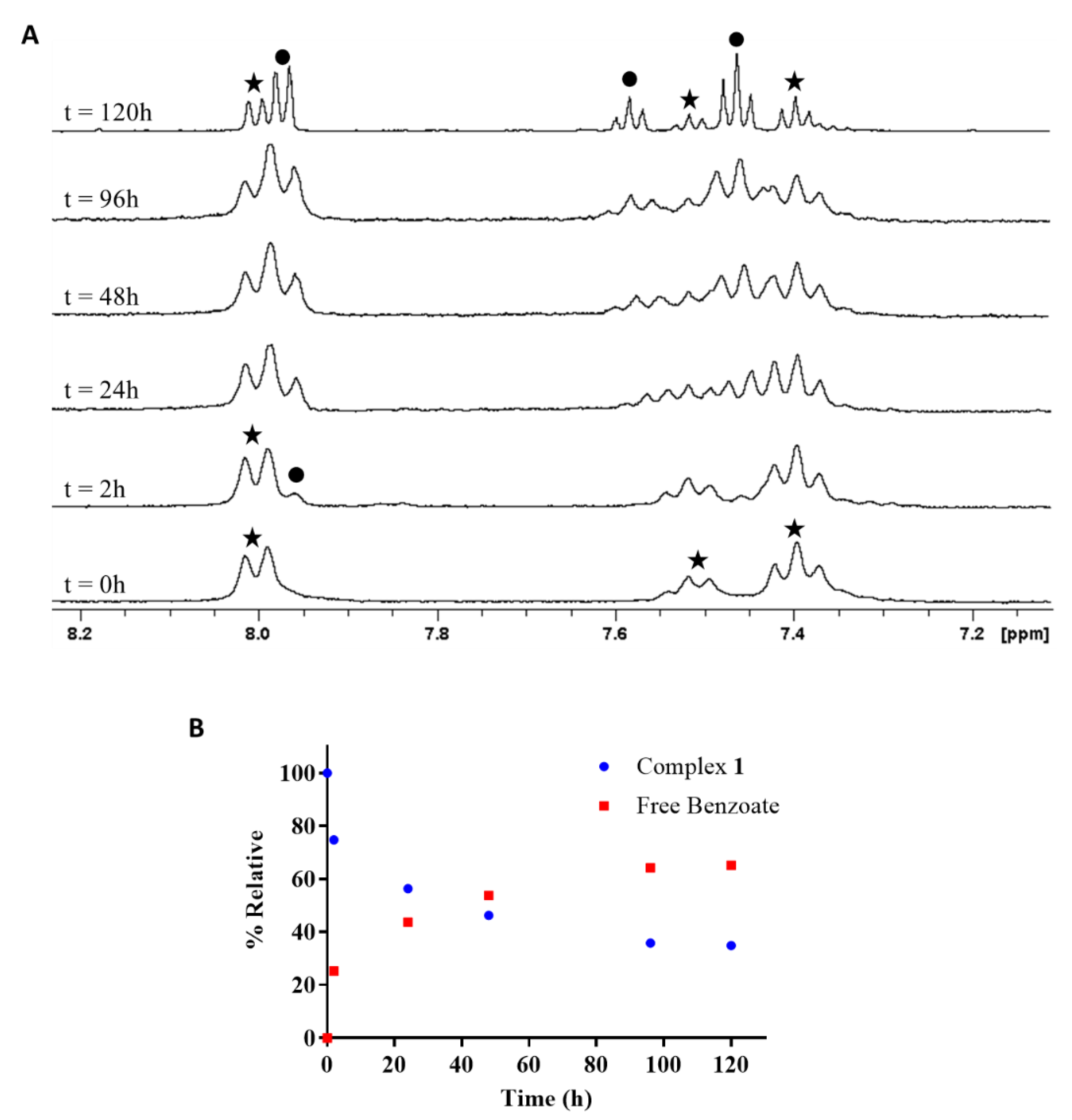
2.4.1. Reduction of cis,trans,cis-[PtCl2(OBz)2(cis-1,4-DACH)] by Ascorbic Acid/Sodium Ascorbate
2.4.2. Reduction of cis,trans,cis-[PtCl2(OBz)2(cis-1,4-DACH)] by Glutathione
2.5. In Vivo Antitumor Activity
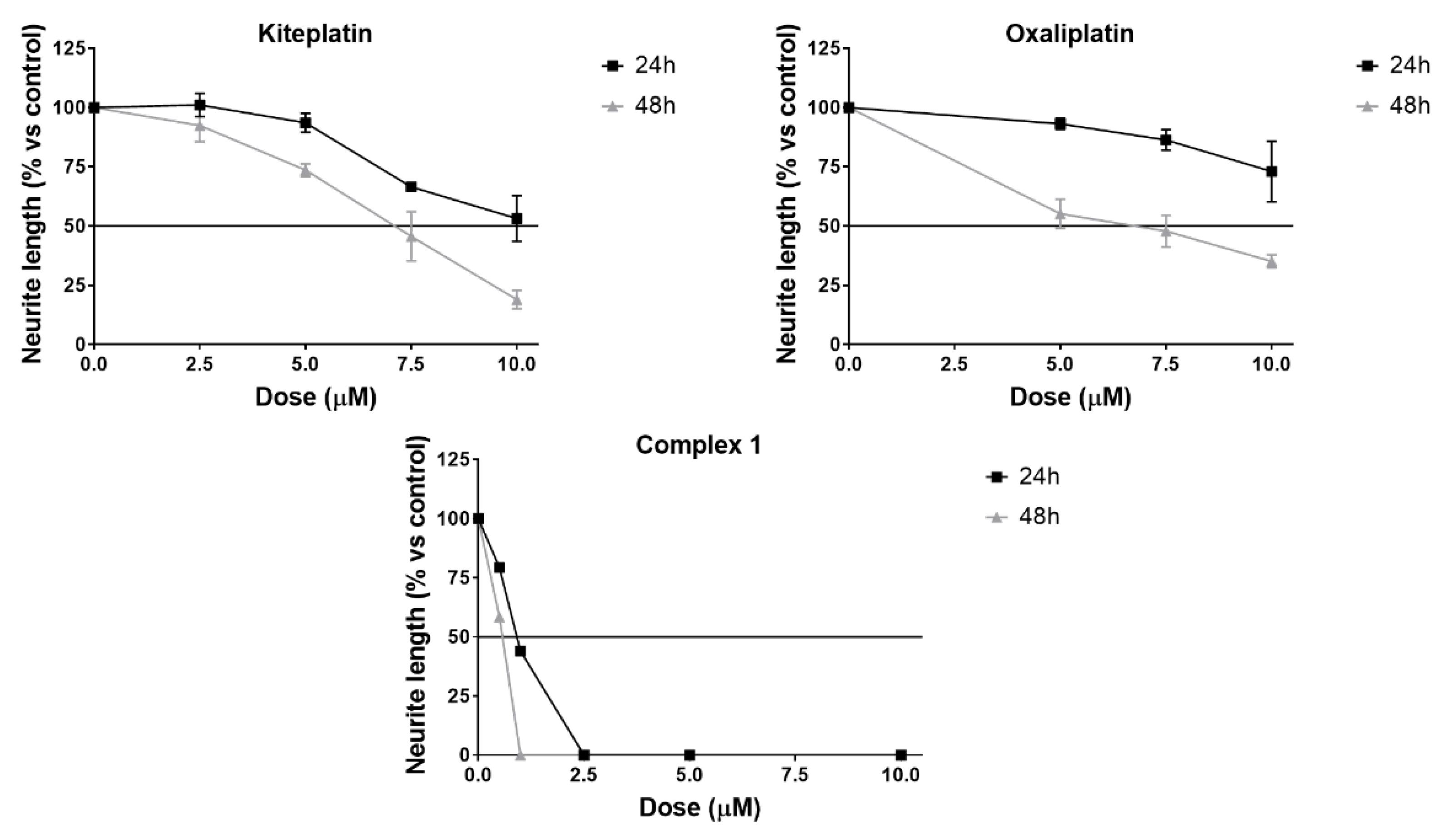
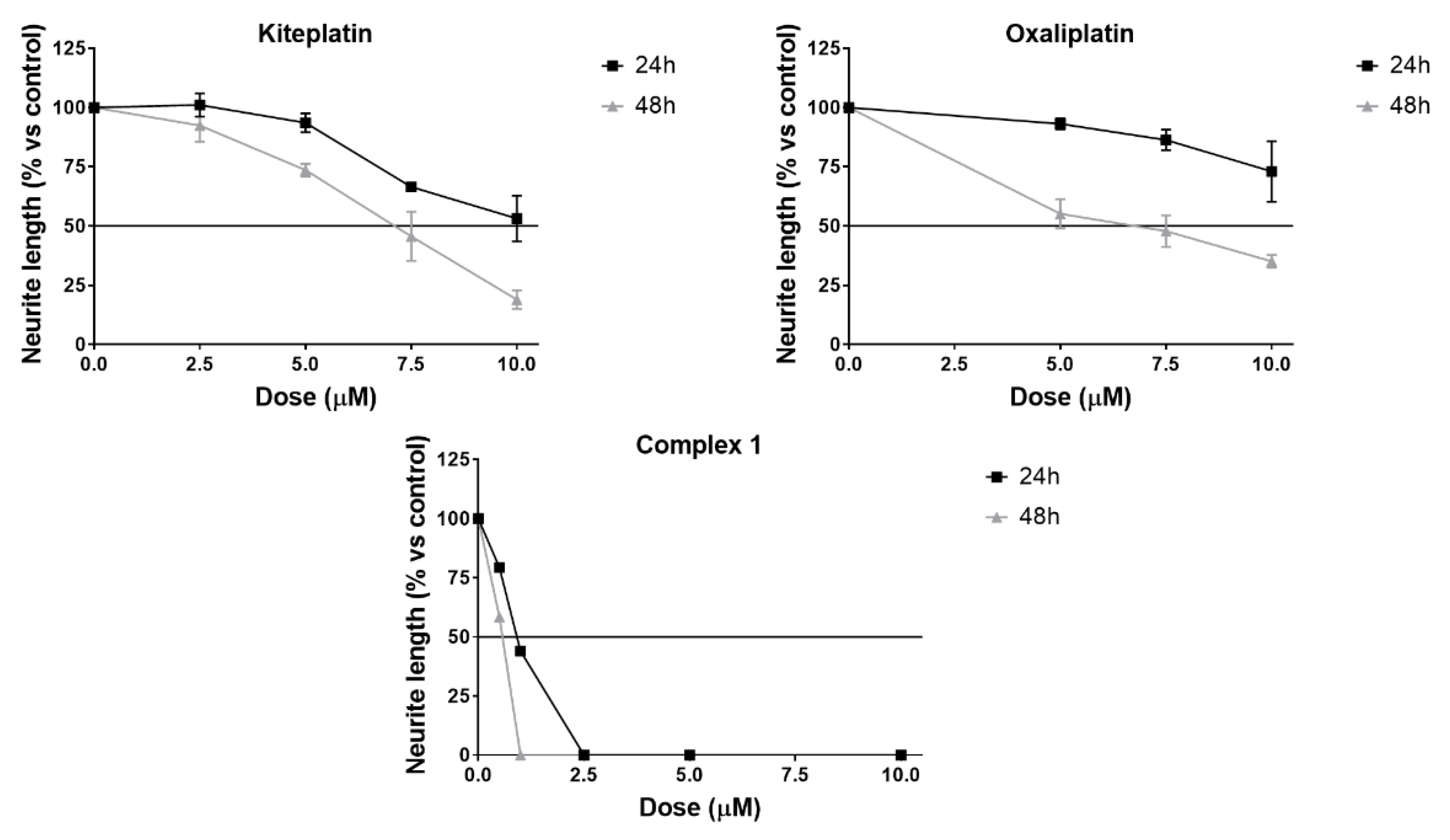
2.6. Neurotoxicity Studies
3. Materials and Methods
3.1. Starting Materials and Instrumental Details
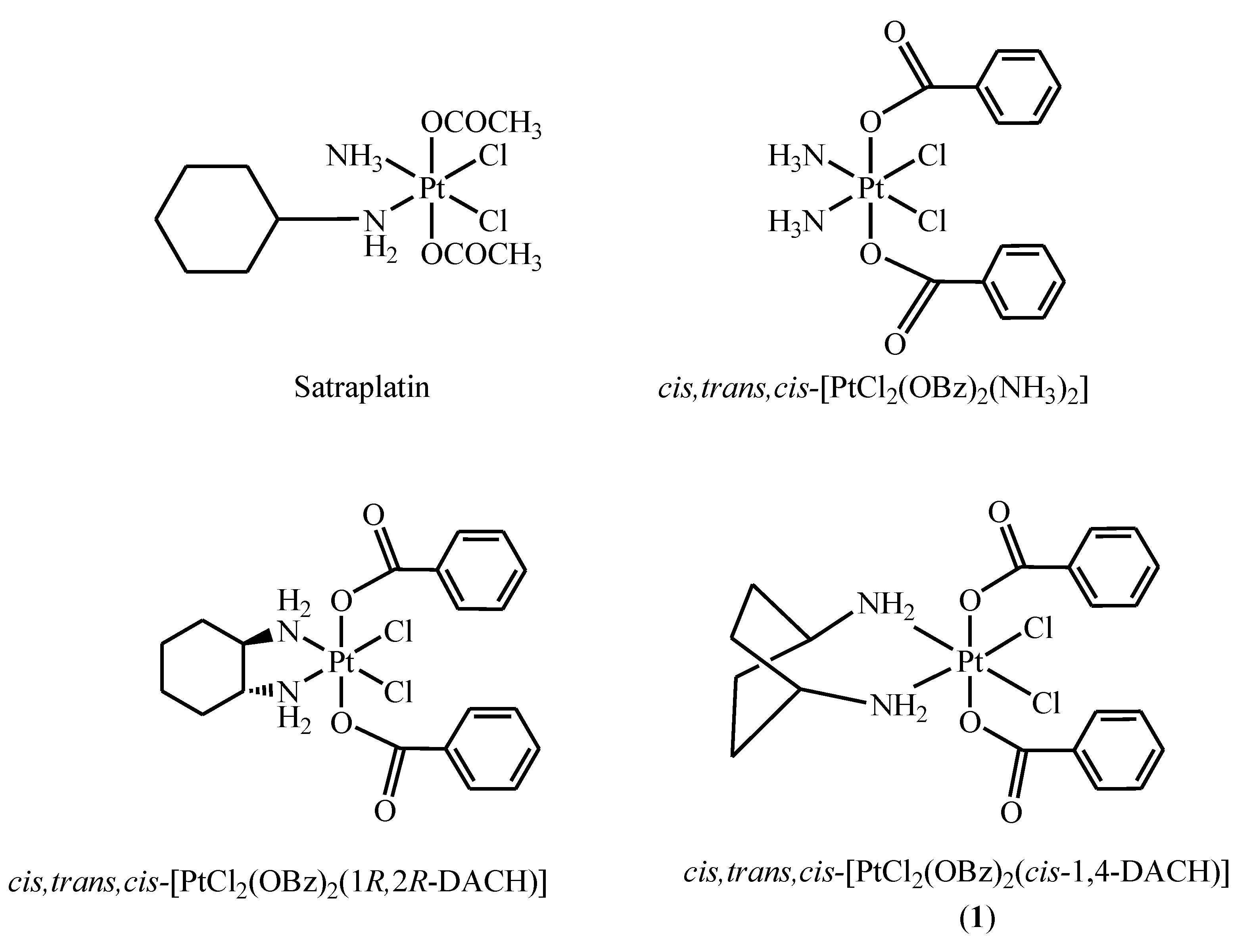
3.2. Synthesis of cis,trans,cis-[PtCl2(OBz)2(cis-1,4-DACH)] (1; OBz = Benzoate = OOCC6H5)
3.3. Stability at Acidic pH
3.4. Reduction by Biological Reducing Agents
3.4.1. Ascorbic Acid/Ascorbate
3.4.2. Glutathione
3.5. In Vivo Antitumor Activity
3.5.1. Experiments with Animals
3.5.2. In Vivo Antitumor Activity in Lewis Lung Carcinoma (LLC)
3.6. Neurotoxicity Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs Are Unique: Opportunities and Challenges of Discovery and Development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef] [PubMed]
- Jakupec, M.A.; Galanski, M.; Arion, V.B.; Hartinger, C.G.; Keppler, B.K. Antitumour Metal Compounds: More than Theme and Variations. Dalton Trans. 2008, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Todd, R.C.; Lippard, S.J. Inhibition of Transcription by Platinum Antitumor Compounds. Metallomics 2009, 1, 280–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, D. Platinum(IV) Anticancer Prodrugs—Hypotheses and Facts. Dalton Trans. 2016, 45, 12983–12991. [Google Scholar] [CrossRef]
- Wang, D.; Lippard, S.J. Cellular Processing of Platinum Anticancer Drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef]
- Brabec, V.; Hrabina, O.; Kasparkova, J. Cytotoxic Platinum Coordination Compounds. DNA Binding Agents. Coord. Chem. Rev. 2017, 351, 2–31. [Google Scholar] [CrossRef]
- Uchino, H.; Matsumura, Y.; Negishi, T.; Koizumi, F.; Hayashi, T.; Honda, T.; Nishiyama, N.; Kataoka, K.; Naito, S.; Kakizoe, T. Cisplatin-Incorporating Polymeric Micelles (NC-6004) Can Reduce Nephrotoxicity and Neurotoxicity of Cisplatin in Rats. Br. J. Cancer 2005, 93, 678–687. [Google Scholar] [CrossRef] [Green Version]
- Carozzi, V.A.; Marmiroli, P.; Cavaletti, G. The Role of Oxidative Stress and Anti-Oxidant Treatment in Platinum-Induced Peripheral Neurotoxicity. Curr. Cancer Drug Targets 2010, 10, 670–682. [Google Scholar] [CrossRef]
- Brabec, V.; Kasparkova, J. Modifications of DNA by Platinum Complexes. Relation to Resistance of Tumors to Platinum Antitumor Drugs. Drug Resist. Updates 2005, 8, 131–146. [Google Scholar] [CrossRef]
- Torigoe, T.; Izumi, H.; Ishiguchi, H.; Yoshida, Y.; Tanabe, M.; Yoshida, T.; Igarashi, T.; Niina, I.; Wakasugi, T.; Imaizumi, T.; et al. Cisplatin Resistance and Transcription Factors. Curr. Med. Chem. Anticancer Agents 2005, 5, 15–27. [Google Scholar] [CrossRef]
- Wang, X.; Guo, Z. Targeting and Delivery of Platinum-Based Anticancer Drugs. Chem. Soc. Rev. 2013, 42, 202–224. [Google Scholar] [CrossRef]
- Galanski, M.; Jakupec, M.; Keppler, B. Update of the Preclinical Situation of Anticancer Platinum Complexes: Novel Design Strategies and Innovative Analytical Approaches. Curr. Med. Chem. 2005, 12, 2075–2094. [Google Scholar] [CrossRef]
- Wang, X.; Wang, X.; Jin, S.; Muhammad, N.; Guo, Z. Stimuli-Responsive Therapeutic Metallodrugs. Chem. Rev. 2019, 119, 1138–1192. [Google Scholar] [CrossRef]
- Gibson, D. The Mechanism of Action of Platinum Anticancer Agents—What Do We Really Know about It? Dalt. Trans. 2009, 10681–10689. [Google Scholar] [CrossRef]
- Kenny, R.G.; Marmion, C.J. Toward Multi-Targeted Platinum and Ruthenium Drugs—A New Paradigm in Cancer Drug Treatment Regimens? Chem. Rev. 2019, 119, 1058–1137. [Google Scholar] [CrossRef]
- Kasparkova, J.; Suchankova, T.; Halamikova, A.; Zerzankova, L.; Vrana, O.; Margiotta, N.; Natile, G.; Brabec, V. Cytotoxicity, Cellular Uptake, Glutathione and DNA Interactions of an Antitumor Large-Ring PtII Chelate Complex Incorporating the Cis-1,4-Diaminocyclohexane Carrier Ligand. Biochem. Pharmacol. 2010, 79, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Margiotta, N.; Marzano, C.; Gandin, V.; Osella, D.; Ravera, M.; Gabano, E.; Platts, J.A.; Petruzzella, E.; Hoeschele, J.D.; Natile, G. Revisiting [PtCl₂(Cis-1,4-DACH)]: An Underestimated Antitumor Drug with Potential Application to the Treatment of Oxaliplatin-Refractory Colorectal Cancer. J. Med. Chem. 2012, 55, 7182–7192. [Google Scholar] [CrossRef]
- Brabec, V.; Malina, J.; Margiotta, N.; Natile, G.; Kasparkova, J. Thermodynamic and Mechanistic Insights into Translesion DNA Synthesis Catalyzed by Y-Family DNA Polymerase across a Bulky Double-Base Lesion of an Antitumor Platinum Drug. Chemistry 2012, 18, 15439–15448. [Google Scholar] [CrossRef]
- Barbanente, A.; Galliani, A.; Iacobazzi, R.M.; Lasorsa, A.; Nardella, M.I.; Pennetta, A.; Margiotta, N.; Arnesano, F. Interaction of Copper Trafficking Proteins with the Platinum Anticancer Drug Kiteplatin. ChemMedChem 2022, 17, e202100593. [Google Scholar] [CrossRef]
- Kenny, R.G.; Chuah, S.W.; Crawford, A.; Marmion, C.J. Platinum(IV) Prodrugs—A Step Closer to Ehrlich’s Vision? Eur. J. Inorg. Chem. 2017, 2017, 1596–1612. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.D.; Hambley, T.W. Platinum(IV) Antitumour Compounds: Their Bioinorganic Chemistry. Coord. Chem. Rev. 2002, 232, 49–67. [Google Scholar] [CrossRef]
- Gabano, E.; Ravera, M.; Osella, D. Pros and Cons of Bifunctional Platinum(IV) Antitumor Prodrugs: Two Are (Not Always) Better than One. Dalton Trans. 2014, 43, 9813–9820. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.F.; Tian, Q.; Setyawati, M.I.; Fang, W.; Tan, E.S.Q.; Leong, D.T.; Ang, W.H. Tuning the Activity of Platinum(IV) Anticancer Complexes through Asymmetric Acylation. J. Med. Chem. 2012, 55, 7571–7582. [Google Scholar] [CrossRef]
- Kelland, L. The Resurgence of Platinum-Based Cancer Chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Hall, M.D.; Mellor, H.R.; Callaghan, R.; Hambley, T.W. Basis for Design and Development of Platinum(IV) Anticancer Complexes. J. Med. Chem. 2007, 50, 3403–3411. [Google Scholar] [CrossRef]
- Tabrizi, L.; Thompson, K.; Mnich, K.; Chintha, C.; Gorman, A.M.; Morrison, L.; Luessing, J.; Lowndes, N.F.; Dockery, P.; Samali, A.; et al. Novel Pt (IV) Prodrugs Displaying Antimitochondrial Effects. Mol. Pharm. 2020, 17, 3009–3023. [Google Scholar] [CrossRef]
- Petruzzella, E.; Margiotta, N.; Ravera, M.; Natile, G. NMR Investigation of the Spontaneous Thermal- and/or Photoinduced Reduction of Trans Dihydroxido Pt(IV) Derivatives. Inorg. Chem. 2013, 52, 2393–2403. [Google Scholar] [CrossRef]
- Barbanente, A.; Gandin, V.; Ditaranto, N.; Marzano, C.; Hoeschele, J.D.; Suranna, G.P.; Papadia, P.; Natile, G.; Margiotta, N. A Pt (IV) Prodrug of Kiteplatin with the Bone-Targeting Pyrophosphate Ligand. Inorg. Chim. Acta 2019, 494, 98–104. [Google Scholar] [CrossRef]
- Curci, A.; Denora, N.; Iacobazzi, R.M.; Ditaranto, N.; Hoeschele, J.D.; Margiotta, N.; Natile, G. Synthesis, Characterization, and in Vitro Cytotoxicity of a Kiteplatin-Ibuprofen Pt (IV) Prodrug. Inorg. Chim. Acta 2018, 472, 221–228. [Google Scholar] [CrossRef]
- Savino, S.; Gandin, V.; Hoeschele, J.D.; Marzano, C.; Natile, G.; Margiotta, N. Dual-Acting Antitumor Pt (IV) Prodrugs of Kiteplatin with Dichloroacetate Axial Ligands. Dalton Trans. 2018, 47, 7144–7158. [Google Scholar] [CrossRef] [PubMed]
- Savino, S.; Marzano, C.; Gandin, V.; Hoeschele, J.D.J.D.; Natile, G.; Margiotta, N. Multi-Acting Mitochondria-Targeted Platinum(IV) Prodrugs of Kiteplatin with α-Lipoic Acid in the Axial Positions. Int. J. Mol. Sci. 2018, 19, 2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margiotta, N.; Savino, S.; Denora, N.; Marzano, C.; Laquintana, V.; Cutrignelli, A.; Hoeschele, J.D.J.D.; Gandin, V.; Natile, G. Encapsulation of Lipophilic Kiteplatin Pt (IV) Prodrugs in PLGA-PEG Micelles. Dalton Trans. 2016, 45, 13070–13081. [Google Scholar] [CrossRef] [PubMed]
- Papadia, P.; Micoli, K.; Barbanente, A.; Ditaranto, N.; Hoeschele, J.D.; Natile, G.; Marzano, C.; Gandin, V.; Margiotta, N. Platinum(IV) Complexes of Trans-1,2-Diamino-4-Cyclohexene: Prodrugs Affording an Oxaliplatin Analogue That Overcomes Cancer Resistance. Int. J. Mol. Sci. 2020, 21, 2325. [Google Scholar] [CrossRef] [Green Version]
- Barnes, K.R.; Kutikov, A.; Lippard, S.J. Synthesis, Characterization, and Cytotoxicity of a Series of Estrogen-Tethered Platinum(IV) Complexes. Chem. Biol. 2004, 11, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Sternberg, C.N.; Whelan, P.; Hetherington, J.; Paluchowska, B.; Slee, P.H.T.J.; Vekemans, K.; van Erps, P.; Theodore, C.; Koriakine, O.; Oliver, T.; et al. Phase III Trial of Satraplatin, an Oral Platinum plus Prednisone vs. Prednisone Alone in Patients with Hormone-Refractory Prostate Cancer. Oncology 2005, 68, 2–9. [Google Scholar] [CrossRef]
- Hanessian, S.; Zhan, L.; Bovey, R.; Saavedra, O.M.; Juillerat-Jeanneret, L. Functionalized Glycomers as Growth Inhibitors and Inducers of Apoptosis in Human Glioblastoma Cells. J. Med. Chem. 2003, 46, 3600–3611. [Google Scholar] [CrossRef]
- Ang, W.H.; Khalaila, I.; Allardyce, C.S.; Juillerat-Jeanneret, L.; Dyson, P.J. Rational Design of Platinum(IV) Compounds to Overcome Glutathione-S-Transferase Mediated Drug Resistance. J. Am. Chem. Soc. 2005, 127, 1382–1383. [Google Scholar] [CrossRef]
- Ang, W.H.; Pilet, S.; Scopelliti, R.; Bussy, F.; Juillerat-Jeanneret, L.; Dyson, P.J. Synthesis and Characterization of Platinum(IV) Anticancer Drugs with Functionalized Aromatic Carboxylate Ligands: Influence of the Ligands on Drug Efficacies and Uptake. J. Med. Chem. 2005, 48, 8060–8069. [Google Scholar] [CrossRef]
- Gandin, V.; Marzano, C.; Pelosi, G.; Ravera, M.; Gabano, E.; Osella, D. Trans, Cis, Cis-Bis(Benzoato)Dichlorido(Cyclohexane-1 R,2 R-Diamine)Platinum(IV): A Prodrug Candidate for the Treatment of Oxaliplatin-Refractory Colorectal Cancer. ChemMedChem 2014, 9, 1299–1305. [Google Scholar] [CrossRef]
- Margiotta, N.; Savino, S.; Marzano, C.; Pacifico, C.; Hoeschele, J.D.; Gandin, V.; Natile, G. Cytotoxicity-Boosting of Kiteplatin by Pt(IV) Prodrugs with Axial Benzoate Ligands. J. Inorg. Biochem. 2016, 160, 85–93. [Google Scholar] [CrossRef]
- Wexselblatt, E.; Raveendran, R.; Salameh, S.; Friedman-Ezra, A.; Yavin, E.; Gibson, D. On the Stability of Pt IV Pro-Drugs with Haloacetato Ligands in the Axial Positions. Chem. Eur. J. 2015, 21, 3108–3114. [Google Scholar] [CrossRef]
- Choi, S.; Filotto, C.; Bisanzo, M.; Delaney, S.; Lagasee, D.; Whitworth, J.L.; Jusko, A.; Li, C.; Wood, N.A.; Willingham, J.; et al. Reduction and Anticancer Activity of Platinum(IV) Complexes. Inorg. Chem. 1998, 37, 2500–2504. [Google Scholar] [CrossRef]
- Fakih, S.; Munk, V.P.; Shipman, M.A.; del Murdoch, P.S.; Parkinson, J.A.; Sadler, P.J. Novel Adducts of the Anticancer Drug Oxaliplatin with Glutathione and Redox Reactions with Glutathione Disulfide. Eur. J. Inorg. Chem. 2003, 2003, 1206–1214. [Google Scholar] [CrossRef]
- Papadia, P.; Margiotta, N.; Bergamo, A.; Sava, G.; Natile, G. Platinum(II) Complexes with Antitumoral/Antiviral Aromatic Heterocycles: Effect of Glutathione upon in Vitro Cell Growth Inhibition. J. Med. Chem. 2005, 48, 3364–3371. [Google Scholar] [CrossRef]
- Margiotta, N.; Petruzzella, E.; Platts, J.A.; Mutter, S.T.; Deeth, R.J.; Ranaldo, R.; Papadia, P.; Marzilli, P.A.; Marzilli, L.G.; Hoeschele, J.D.; et al. DNA Fragment Conformations in Adducts with Kiteplatin. Dalton Trans. 2015, 44, 3544–3556. [Google Scholar] [CrossRef] [Green Version]
- Lemma, K.; Berglund, J.; Farrell, N.; Elding, L.I. Kinetics and Mechanism for Reduction of Anticancer-Active Tetrachloroam(m)Ine Platinum(IV) Compounds by Glutathione. JBIC J. Biol. Inorg. Chem. 2000, 5, 300–306. [Google Scholar] [CrossRef]
- Yuan, S.; Zhu, Y.; Dai, Y.; Wang, Y.; Jin, D.; Liu, M.; Tang, L.; Arnesano, F.; Natile, G.; Liu, Y. 19 F NMR Allows the Investigation of the Fate of Platinum(IV) Prodrugs in Physiological Conditions. Angew. Chem. Int. Ed. 2022, 61, e202114250. [Google Scholar] [CrossRef]
- Hoeschele, J.D.; Margiotta, N.; Gandin, V.; Petruzzella, E.; Marzano, C. Method of Treating Colorectal Cancer. U.S. Patent US9220705B2, 29 December 2015. [Google Scholar]
- Saltz, L.B.; Meropol, N.J.; Loehrer, P.J.; Needle, M.N.; Kopit, J.; Mayer, R.J. Phase II Trial of Cetuximab in Patients with Refractory Colorectal Cancer That Expresses the Epidermal Growth Factor Receptor. J. Clin. Oncol. 2004, 22, 1201–1208. [Google Scholar] [CrossRef]
- Meyerhardt, J.A.; Mayer, R.J. Systemic Therapy for Colorectal Cancer. N. Engl. J. Med. 2005, 352, 476–487. [Google Scholar] [CrossRef]
- Staff, N.P.; Podratz, J.L.; Grassner, L.; Bader, M.; Paz, J.; Knight, A.M.; Loprinzi, C.L.; Trushina, E.; Windebank, A.J. Bortezomib Alters Microtubule Polymerization and Axonal Transport in Rat Dorsal Root Ganglion Neurons. Neurotoxicology 2013, 39, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Maggioni, D.; Nicolini, G.; Chiorazzi, A.; Meregalli, C.; Cavaletti, G.; Tredici, G. Different Effects of Erythropoietin in Cisplatin- and Docetaxel-Induced Neurotoxicity: An in Vitro Study. J. Neurosci. Res. 2010, 88, 3171–3179. [Google Scholar] [CrossRef]
- Scuteri, A.; Nicolini, G.; Miloso, M.; Bossi, M.; Cavaletti, G.; Windebank, A.J.; Tredici, G. Paclitaxel Toxicity in Post-Mitotic Dorsal Root Ganglion (DRG) Cells. Anticancer Res. 2006, 26, 1065–1070. [Google Scholar]
- Krüger, K.; Thomale, J.; Stojanović, N.; Osmak, M.; Henninger, C.; Bormann, S.; Fritz, G. Platinum-Induced Kidney Damage: Unraveling the DNA Damage Response (DDR) of Renal Tubular Epithelial and Glomerular Endothelial Cells Following Platinum Injury. Biochim. Biophys. Acta 2015, 1853, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Feltham, R.D.; Hayter, R.G. The Electrolyte Type of Ionized Complexes. J. Chem. Soc. 1964, 4587, 4587–4591. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Daily Dose (mg/Kg) | Average Tumor Weight (Mean ± S.D., g) | Inhibition of Tumor Growth (%) |
|---|---|---|---|
| Control a | - | 0.40 ± 0.07 | - |
| 1 | 5 | 0.11 ± 0.08 | 72.5 |
| cisplatin | 1.5 | 0.09 ± 0.02 | 77.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbanente, A.; Gandin, V.; Ceresa, C.; Marzano, C.; Ditaranto, N.; Hoeschele, J.D.; Natile, G.; Arnesano, F.; Pacifico, C.; Intini, F.P.; et al. Improvement of Kiteplatin Efficacy by a Benzoato Pt(IV) Prodrug Suitable for Oral Administration. Int. J. Mol. Sci. 2022, 23, 7081. https://doi.org/10.3390/ijms23137081
Barbanente A, Gandin V, Ceresa C, Marzano C, Ditaranto N, Hoeschele JD, Natile G, Arnesano F, Pacifico C, Intini FP, et al. Improvement of Kiteplatin Efficacy by a Benzoato Pt(IV) Prodrug Suitable for Oral Administration. International Journal of Molecular Sciences. 2022; 23(13):7081. https://doi.org/10.3390/ijms23137081
Chicago/Turabian StyleBarbanente, Alessandra, Valentina Gandin, Cecilia Ceresa, Cristina Marzano, Nicoletta Ditaranto, James D. Hoeschele, Giovanni Natile, Fabio Arnesano, Concetta Pacifico, Francesco P. Intini, and et al. 2022. "Improvement of Kiteplatin Efficacy by a Benzoato Pt(IV) Prodrug Suitable for Oral Administration" International Journal of Molecular Sciences 23, no. 13: 7081. https://doi.org/10.3390/ijms23137081
APA StyleBarbanente, A., Gandin, V., Ceresa, C., Marzano, C., Ditaranto, N., Hoeschele, J. D., Natile, G., Arnesano, F., Pacifico, C., Intini, F. P., & Margiotta, N. (2022). Improvement of Kiteplatin Efficacy by a Benzoato Pt(IV) Prodrug Suitable for Oral Administration. International Journal of Molecular Sciences, 23(13), 7081. https://doi.org/10.3390/ijms23137081