
Genes Possessing the Most Frequent DNA DSBs Are Highly Associated with Development and Cancers, and Essentially Overlap with the rDNA-Contacting Genes

 ,
,
Abstract
1. Introduction
2. Results
2.1. Nature of the Genes That Are the Main Targets of DSBs in HEK293T Cells
2.2. Associations of the Genes That Are Targeted by DSBs with Diseases
2.3. Frequent DSBs and rDNA Contacts in Both Silent and Active Genes
2.4. Expression Patterns of Genes Frequently Targeted by DSBs in HEK293T Cells
2.5. Genetic Annotations of DSBs Mapped in HEK293T Cells
2.6. Epigenetic Profiles at the Targets of DSBs in HEK293T Cells
2.7. DSBs at Bi- and Unidirectional Promoters in HEK293T Cells
3. Discussion
4. Methods
4.1. DSB Library Preparation
4.2. C-rDNA Procedure
4.3. DSBs Mapping and Processing
4.4. 4C Mapping and Processing
4.5. Genome-Wide Profiles
4.6. RNA-Seq Analysis
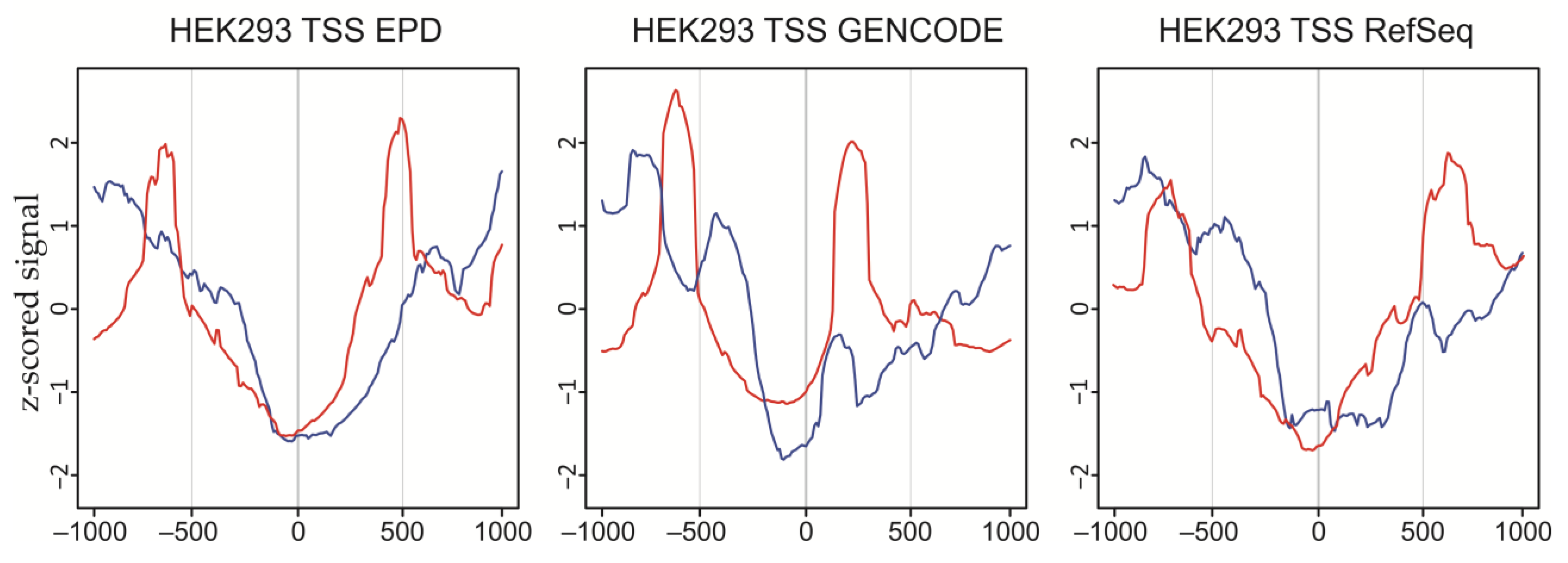
4.7. Transcription Start-Site Analysis
4.8. Analysis of the Distribution of Genes and Their Expression Levels by Violin Plots
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Teves, S.S.; Henikoff, S. DNA torsion as a feedback mediator of transcription and chromatin dynamics. Nucleus 2014, 5, 211–218. [Google Scholar] [CrossRef]
- Shimizu, A.; Fujimori, H.; Minakawa, Y.; Matsuno, Y.; Hyodo, M.; Murakami, Y.; Yoshioka, K.-I. Onset of deaminase APOBEC3B induction in response to DNA double-strand breaks. Biochem. Biophys. Rep. 2018, 16, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.; Pizzul, P.; Longhese, M.P.; Bonetti, D. Sensing R-Loop-Associated DNA Damage to Safeguard Genome Stability. Front. Cell Dev. Biol. 2021, 8, 618157. [Google Scholar] [CrossRef] [PubMed]
- Vilenchik, M.M.; Knudson, A.G. Radiation dose-rate effects, endogenous DNA damage, and signaling resonance. Proc. Natl. Acad. Sci. USA 2006, 103, 17874–17879. [Google Scholar] [CrossRef] [PubMed]
- Tchurikov, N.A.; Kretova, O.V.; Sosin, D.V.; Zykov, I.A.; Zhimulev, I.F.; Kravatsky, Y.V. Genome-wide profiling of forum domains in Drosophila melanogaster. Nucleic Acids Res. 2011, 39, 3667–3685. [Google Scholar] [CrossRef]
- Tchurikov, N.A.; Kretova, O.V.; Fedoseeva, D.M.; Sosin, D.V.; Grachev, S.A.; Serebraykova, M.V.; Romanenko, S.A.; Vorobieva, N.V.; Kravatsky, Y.V. DNA Double-Strand Breaks Coupled with PARP1 and HNRNPA2B1 Binding Sites Flank Coordinately Expressed Domains in Human Chromosomes. PLoS Genet. 2013, 9, e1003429. [Google Scholar] [CrossRef]
- Tchurikov, N.A.; Fedoseeva, D.M.; Sosin, D.V.; Snezhkina, A.V.; Melnikova, N.V.; Kudryavtseva, A.V.; Kravatsky, Y.V.; Kretova, O.V. Hot spots of DNA double-strand breaks and genomic contacts of human rDNA units are involved in epigenetic regulation. J. Mol. Cell Biol. 2015, 7, 366–382. [Google Scholar] [CrossRef]
- Tchurikov, N.A.; Ponomarenko, N.A. Detection of DNA domains in Drosophila, human, and plant chromosomes possessing mainly 50- to 150-kilobase stretches of DNA. Proc. Natl. Acad. Sci. USA 1992, 89, 6751–6755. [Google Scholar] [CrossRef]
- Trenner, A.; Sartori, A.A. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front. Oncol. 2019, 9, 1388. [Google Scholar] [CrossRef]
- Ui, A.; Chiba, N.; Yasui, A. Relationship among DNA double-strand break (DSB), DSB repair, and transcription prevents genome instability and cancer. Cancer Sci. 2020, 111, 1443–1451. [Google Scholar] [CrossRef]
- Zhu, L.S.; Wang, D.Q.; Cui, K.; Liu, D.; Zhu, L.Q. Emerging Perspectives on DNA Double-strand Breaks in Neurodegenerative Diseases. Curr. Neuropharmacol. 2019, 17, 1146–1157. [Google Scholar] [CrossRef] [PubMed]
- Tchurikov, N.A.; Fedoseeva, D.M.; Klushevskaya, E.S.; Slovohotov, I.Y.; Chechetkin, V.R.; Kravatsky, Y.V.; Kretova, O.V. rDNA Clusters Make Contact with Genes that Are Involved in Differentiation and Cancer and Change Contacts after Heat Shock Treatment. Cells 2019, 8, 1393. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Williamson, A.K.; Zhu, Z.; Yuan, Z.M. Epigenetic mechanisms behind cellular sensitivity to DNA damage. Cell Stress 2018, 2, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Takata, H.; Hanafusa, T.; Mori, T.; Shimura, M.; Iida, Y.; Ishikawa, K.; Yoshikawa, K.; Yoshikawa, Y.; Maeshima, K. Chromatin Compaction Protects Genomic DNA from Radiation Damage. PLoS ONE 2013, 8, e75622. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Ingelsson, M.; Fukumoto, H.; Ramasamy, K.; Kowa, H.; Frosch, M.P.; Irizarry, M.C.; Hyman, B.T. Expression of APP pathway mRNAs and proteins in Alzheimer’s disease. Brain Res. 2007, 1161, 116–123. [Google Scholar] [CrossRef]
- Lee, D.; Zhang, J.; Liu, J.; Gerstein, M. Epigenome-based splicing prediction using a recurrent neural network. PLOS Comput. Biol. 2020, 16, e1008006. [Google Scholar] [CrossRef]
- Dreos, R.; Ambrosini, G.; Périer, R.C.; Bucher, P. The Eukaryotic Promoter Database: Expansion of EPDnew and new promoter analysis tools. Nucleic Acids Res. 2015, 43, D92–D96. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Scruggs, B.S.; Gilchrist, D.A.; Nechaev, S.; Muse, G.W.; Burkholder, A.; Fargo, D.C.; Adelman, K. Bidirectional Transcription Arises from Two Distinct Hubs of Transcription Factor Binding and Active Chromatin. Mol. Cell 2015, 58, 1101–1112. [Google Scholar] [CrossRef]
- Kravatsky, Y.V.; Chechetkin, V.R.; Tchurikov, N.A.; Kravatskaya, G.I. Genome-wide study of correlations between genomic features and their relationship with the regulation of gene expression. DNA Res. 2015, 22, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Vítor, A.C.; Huertas, P.; Legube, G.; De Almeida, S.F. Studying DNA Double-Strand Break Repair: An Ever-Growing Toolbox. Front. Mol. Biosci. 2020, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Rouet, P.; Smih, F.; Jasin, M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol. 1994, 14, 8096–8106. [Google Scholar] [CrossRef] [PubMed]
- Hussmann, J.A.; Ling, J.; Ravisankar, P.; Yan, J.; Cirincione, A.; Xu, A.; Simpson, D.; Yang, D.; Bothmer, A.; Cotta-Ramusino, C.; et al. Mapping the genetic landscape of DNA double-strand break repair. Cell 2021, 184, 5653–5669.e25. [Google Scholar] [CrossRef]
- Tchurikov, N.A.; Yudkin, D.V.; Gorbacheva, M.A.; Kulemzina, A.I.; Grischenko, I.V.; Fedoseeva, D.M.; Sosin, D.V.; Kravatsky, Y.V.; Kretova, O.V. Hot spots of DNA double-strand breaks in human rDNA units are produced in vivo. Sci. Rep. 2016, 6, 25866. [Google Scholar] [CrossRef]
- Tchurikov, N.; Krasnov, A.N.; Ponomarenko, N.A.; Golova, Y.B.; Chernov, B.K. Forum domain in Drosophila melanogaster cut locus possesses looped domains inside. Nucleic Acids Res. 1998, 26, 3221–3227. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.R. The Comet Assay for DNA Damage and Repair: Principles, Applications, and Limitations. Mol. Biotechnol. 2004, 26, 249–261. [Google Scholar] [CrossRef]
- Tchurikov, N.A.; Kravatsky, Y.V.; Kretova, O.V. Link Between Double-Strand DNA Break Hotspots and Transcription Regulation: Forum Domains—50–250 kb Chromosome Regions Containing Coordinately Expressed Genes. Biochemistry 2018, 83, 437–449. [Google Scholar] [CrossRef]
- Mocciaro, E.; Runfola, V.; Ghezzi, P.; Pannese, M.; Gabellini, D. DUX4 Role in Normal Physiology and in FSHD Muscular Dystrophy. Cells 2021, 10, 3322. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Murga, M.; Jaco, I.; Fan, Y.; Soria, R.; Martinez-Pastor, B.; Cuadrado, M.; Yang, S.-M.; Blasco, M.A.; Skoultchi, A.I.; Fernandez-Capetillo, O. Global chromatin compaction limits the strength of the DNA damage response. J. Cell Biol. 2007, 178, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Tchurikov, N.A.; Kravatsky, Y.V. The Role of rDNA Clusters in Global Epigenetic Gene Regulation. Front. Genet. 2021, 12, 730633. [Google Scholar] [CrossRef]
- Monroe, J.G.; Srikant, T.; Carbonell-Bejerano, P.; Becker, C.; Lensink, M.; Exposito-Alonso, M.; Klein, M.; Hildebrandt, J.; Neumann, M.; Kliebenstein, D.; et al. Mutation bias reflects natural selection in Arabidopsis thaliana. Nature 2022, 602, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Important genomic regions mutate less often than do other regions. Nature 2022, 602, 38–39. [Google Scholar] [CrossRef] [PubMed]
- Tchurikov, N.A.; Kretova, O.V.; Fedoseeva, D.M.; Chechetkin, V.R.; Gorbacheva, M.A.; Karnaukhov, A.A.; Kravatskaya, G.I.; Kravatsky, Y.V. Mapping of genomic double-strand breaks by ligation of biotinylated oligonucleotides to forum domains: Analysis of the data obtained for human rDNA units. Genom. Data 2014, 3, 15–18. [Google Scholar] [CrossRef]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing Chromosome Conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef]
- Osborne, C.S.; Chakalova, L.; Brown, K.E.; Carter, D.; Horton, A.; Debrand, E.; Goyenechea, B.; Mitchell, J.A.; Lopes, S.; Reik, W.; et al. Active genes dynamically colocalize to shared sites of ongoing transcription. Nat. Genet. 2004, 36, 1065–1071. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Boyle, A.; Guinney, J.; Crawford, G.E.; Furey, T.S. F-Seq: A feature density estimator for high-throughput sequence tags. Bioinformatics 2008, 24, 2537–2538. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Zweig, A.S.; Barber, G.; Hinrichs, A.; Karolchik, D. BigWig and BigBed: Enabling browsing of large distributed datasets. Bioinformatics 2010, 26, 2204–2207. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Storer, J.; Hubley, R.; Rosen, J.; Wheeler, T.J.; Smit, A.F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob. DNA 2021, 12, 2. [Google Scholar] [CrossRef]
- Neph, S.; Kuehn, M.S.; Reynolds, A.P.; Haugen, E.; Thurman, R.E.; Johnson, A.K.; Rynes, E.; Maurano, M.T.; Vierstra, J.; Thomas, S.; et al. BEDOPS: High-performance genomic feature operations. Bioinformatics 2012, 28, 1919–1920. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Johnson, N.; Juettemann, T.; Wilder, S.P.; Flicek, P. WiggleTools: Parallel processing of large collections of genome-wide datasets for visualization and statistical analysis. Bioinformatics 2014, 30, 1008–1009. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed]
- Stempor, P.; Ahringer, J. SeqPlots—Interactive software for exploratory data analyses, pattern discovery and visualization in genomics. Wellcome Open Res. 2016, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- FANTOM Consortium aand the RIKEN PMI and CLST (DGT). A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef]
- Liu, H.-M.; Yang, D.; Liu, Z.-F.; Hu, S.-Z.; Yan, S.-H.; He, X.-W. Density distribution of gene expression profiles and evaluation of using maximal information coefficient to identify differentially expressed genes. PLoS ONE 2019, 14, e0219551. [Google Scholar] [CrossRef]
- Tchurikov, N.A.; Klushevskaya, E.S.; Alembekov, I.R.; Bukreeva, A.S.; Kretova, A.N.; Chechetkin, V.R.; Kravatskaya, G.I.; Kravatsky, Y.V. Fragments of rDNA Genes Scattered over the Human Genome Are Targets of Small RNAs. Int. J. Mol. Sci. 2022, 23, 3014. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Index | Name | Adjusted p-Value |
|---|---|---|
| 1 | Kidney cancer | 3.358 × 10−56 |
| 2 | Liver cancer | 1.896 × 10−26 |
| 3 | Skin cancer | 8.424 × 10−16 |
| 4 | Melanoma | 2.769 × 10−15 |
| 5 | Breast cancer | 3.143 × 10−12 |
| 6 | Pancreatic cancer | 3.595 × 10−11 |
| 7 | Endometrial cancer | 8.648 × 10−11 |
| 8 | Acquired metabolic disease | 2.167 × 10−11 |
| 9 | Carcinoma | 3.577 × 10−8 |
| 10 | Ovarian cancer | 0.00003196 |
| 11 | Attention deficit hyperactivity disorder | 0.00003272 |
| 12 | Type 2 diabetes mellitus | 0.00005111 |
| 13 | Bipolar disorder | 0.0001426 |
| 14 | Schizophrenia | 0.0002438 |
| 15 | Anorexia nervosa | 0.0006728 |
| 16 | Retinal disease | 0.0008184 |
| 17 | Refractive error | 0.001096 |
| 18 | Immune system cancer | 0.001685 |
| 19 | Cognitive disorder | 0.001689 |
| 20 | Obesity | 0.003255 |
| 21 | Alzheimer’s disease | 0.003277 |
| 22 | Large intestine cancer | 0.003464 |
| 23 | Anemia | 0.003785 |
| 24 | Osteoporosis | 0.004880 |
| 25 | Globe disease | 0.008163 |
| 26 | DOID:2627 | 0.008163 |
| 27 | Pericholangitis | 0.01312 |
| 28 | Lymphoid leukemia | 0.02868 |
| 29 | Parkinson’s disease | 0.04334 |
| 30 | Chronic obstructive pulmonary disease | 0.04528 |
| 31 | Restless legs syndrome | 0.04648 |
| 32 | Lung cancer | 0.04799 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tchurikov, N.A.; Alembekov, I.R.; Klushevskaya, E.S.; Kretova, A.N.; Keremet, A.M.; Sidorova, A.E.; Meilakh, P.B.; Chechetkin, V.R.; Kravatskaya, G.I.; Kravatsky, Y.V. Genes Possessing the Most Frequent DNA DSBs Are Highly Associated with Development and Cancers, and Essentially Overlap with the rDNA-Contacting Genes. Int. J. Mol. Sci. 2022, 23, 7201. https://doi.org/10.3390/ijms23137201
Tchurikov NA, Alembekov IR, Klushevskaya ES, Kretova AN, Keremet AM, Sidorova AE, Meilakh PB, Chechetkin VR, Kravatskaya GI, Kravatsky YV. Genes Possessing the Most Frequent DNA DSBs Are Highly Associated with Development and Cancers, and Essentially Overlap with the rDNA-Contacting Genes. International Journal of Molecular Sciences. 2022; 23(13):7201. https://doi.org/10.3390/ijms23137201
Chicago/Turabian StyleTchurikov, Nickolai A., Ildar R. Alembekov, Elena S. Klushevskaya, Antonina N. Kretova, Ann M. Keremet, Anastasia E. Sidorova, Polina B. Meilakh, Vladimir R. Chechetkin, Galina I. Kravatskaya, and Yuri V. Kravatsky. 2022. "Genes Possessing the Most Frequent DNA DSBs Are Highly Associated with Development and Cancers, and Essentially Overlap with the rDNA-Contacting Genes" International Journal of Molecular Sciences 23, no. 13: 7201. https://doi.org/10.3390/ijms23137201
APA StyleTchurikov, N. A., Alembekov, I. R., Klushevskaya, E. S., Kretova, A. N., Keremet, A. M., Sidorova, A. E., Meilakh, P. B., Chechetkin, V. R., Kravatskaya, G. I., & Kravatsky, Y. V. (2022). Genes Possessing the Most Frequent DNA DSBs Are Highly Associated with Development and Cancers, and Essentially Overlap with the rDNA-Contacting Genes. International Journal of Molecular Sciences, 23(13), 7201. https://doi.org/10.3390/ijms23137201