
Molecular Modelling Hurdle in the Next-Generation Sequencing Era


Abstract
:1. Introduction
2. Results
2.1. Genetic Variability
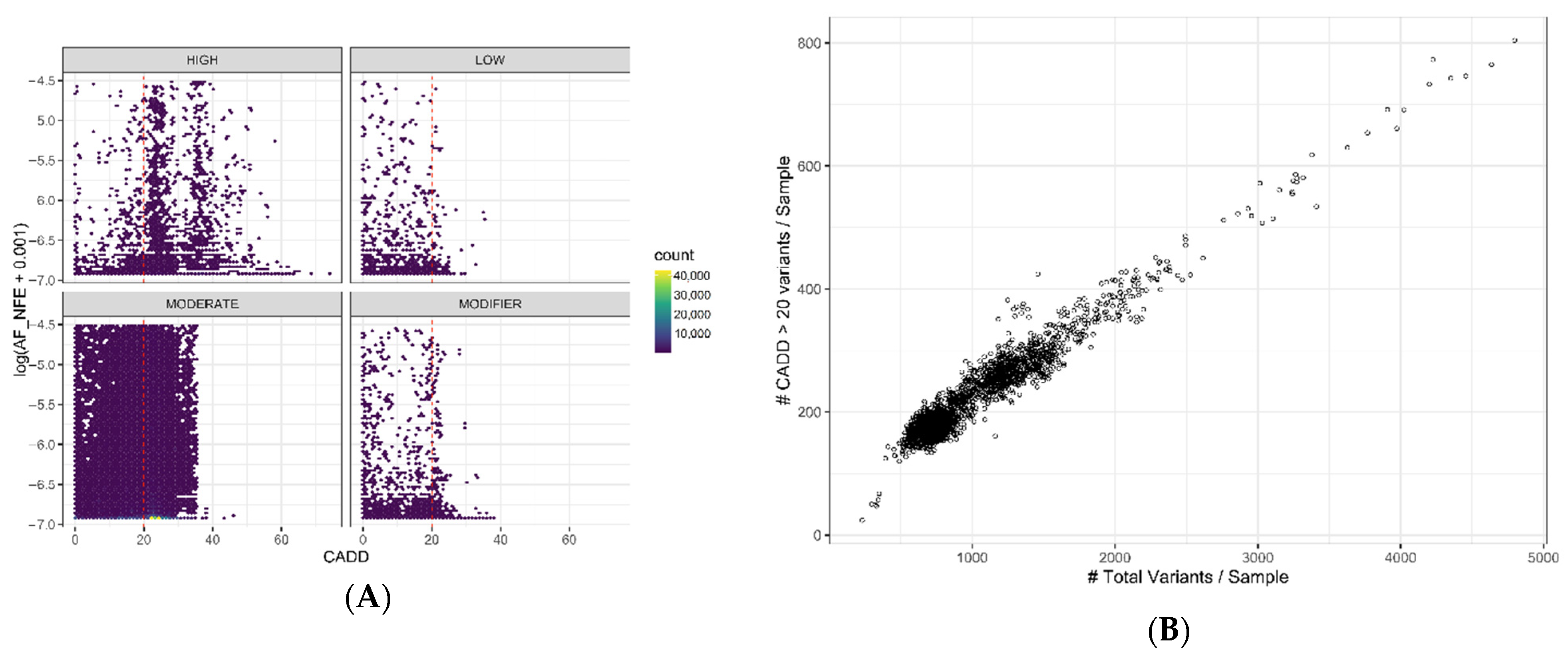
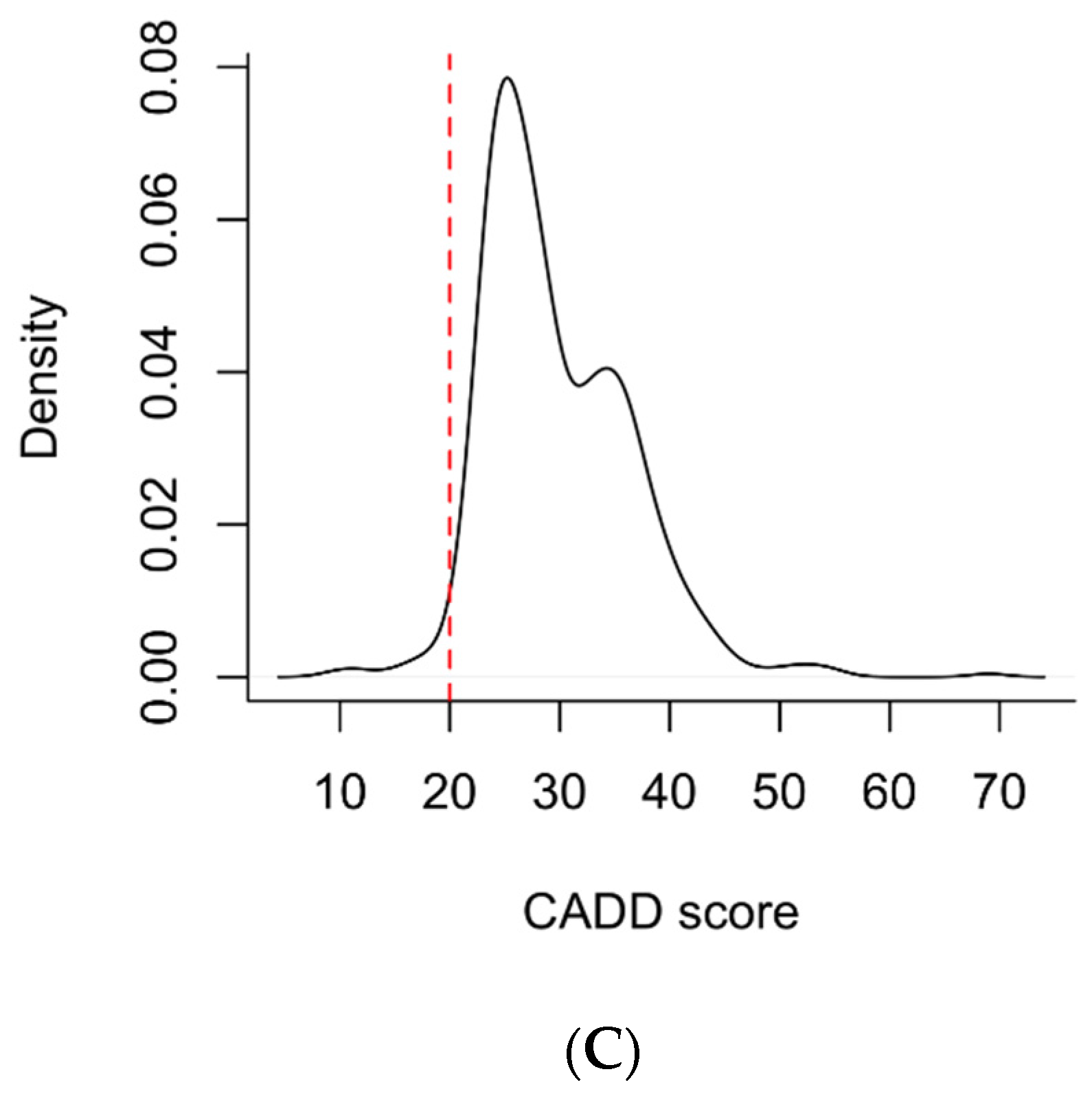
2.2. Variant Conservation Score
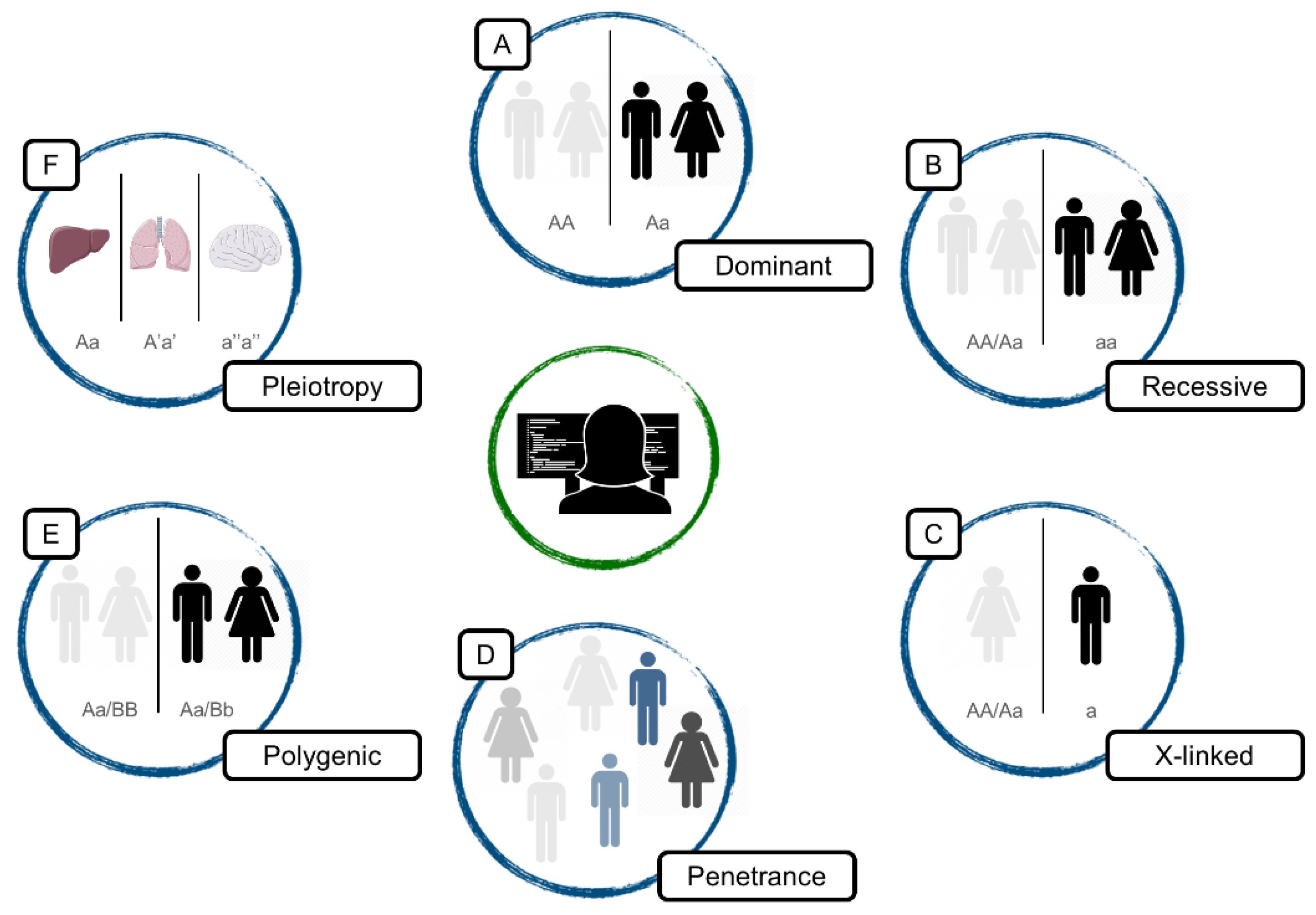
2.3. Variant Classification
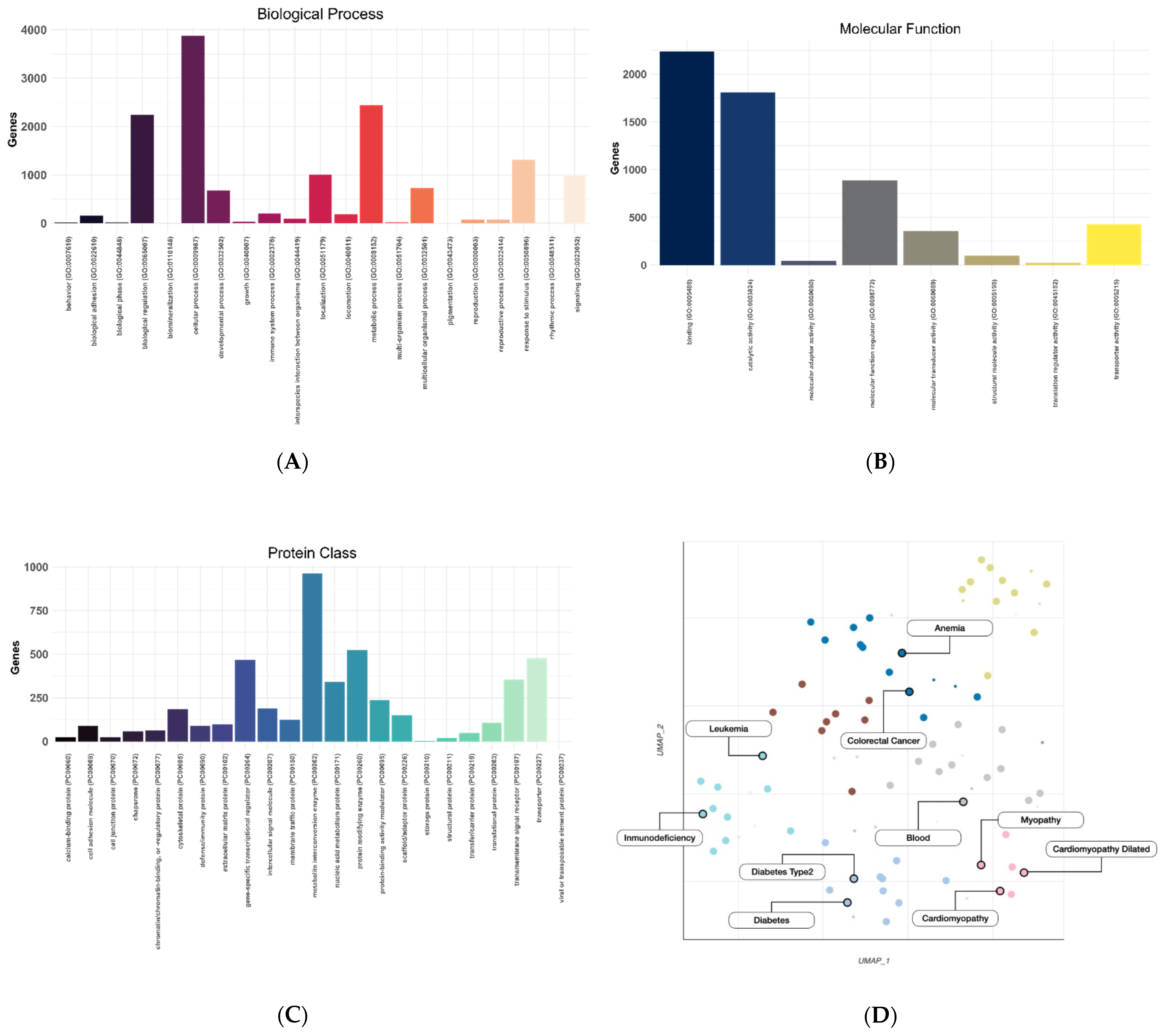
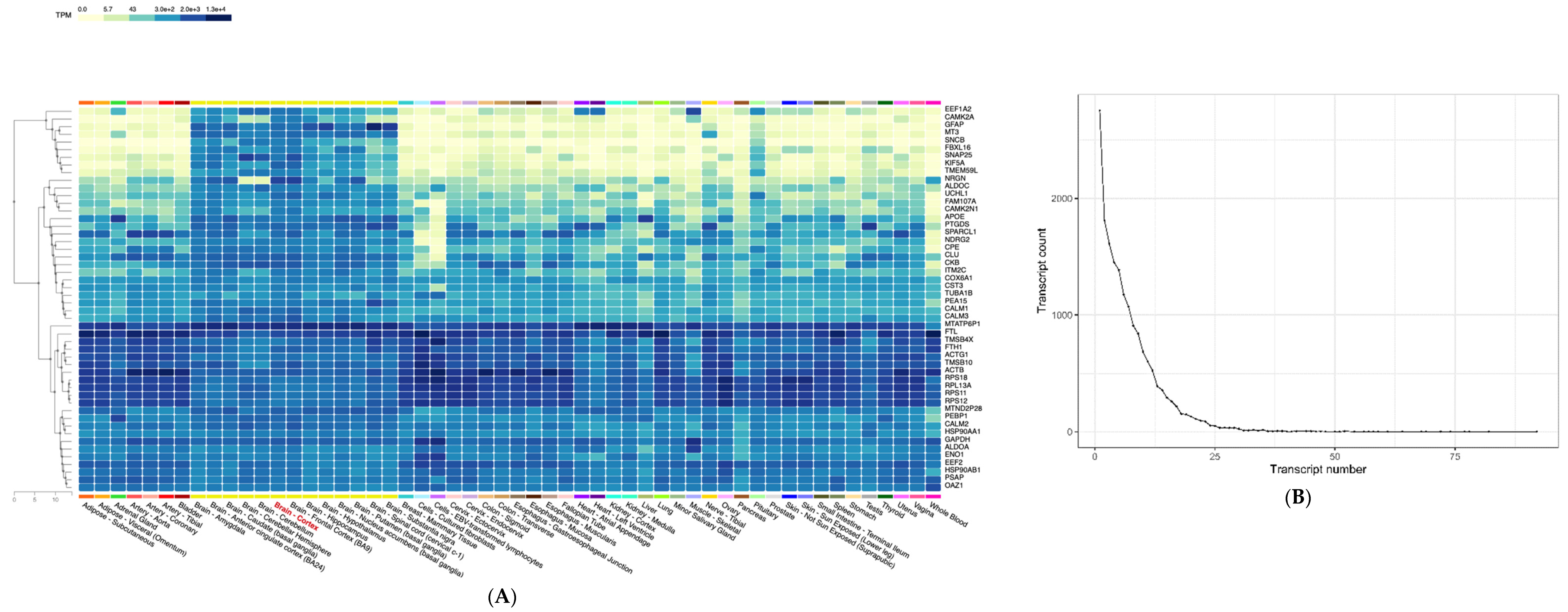
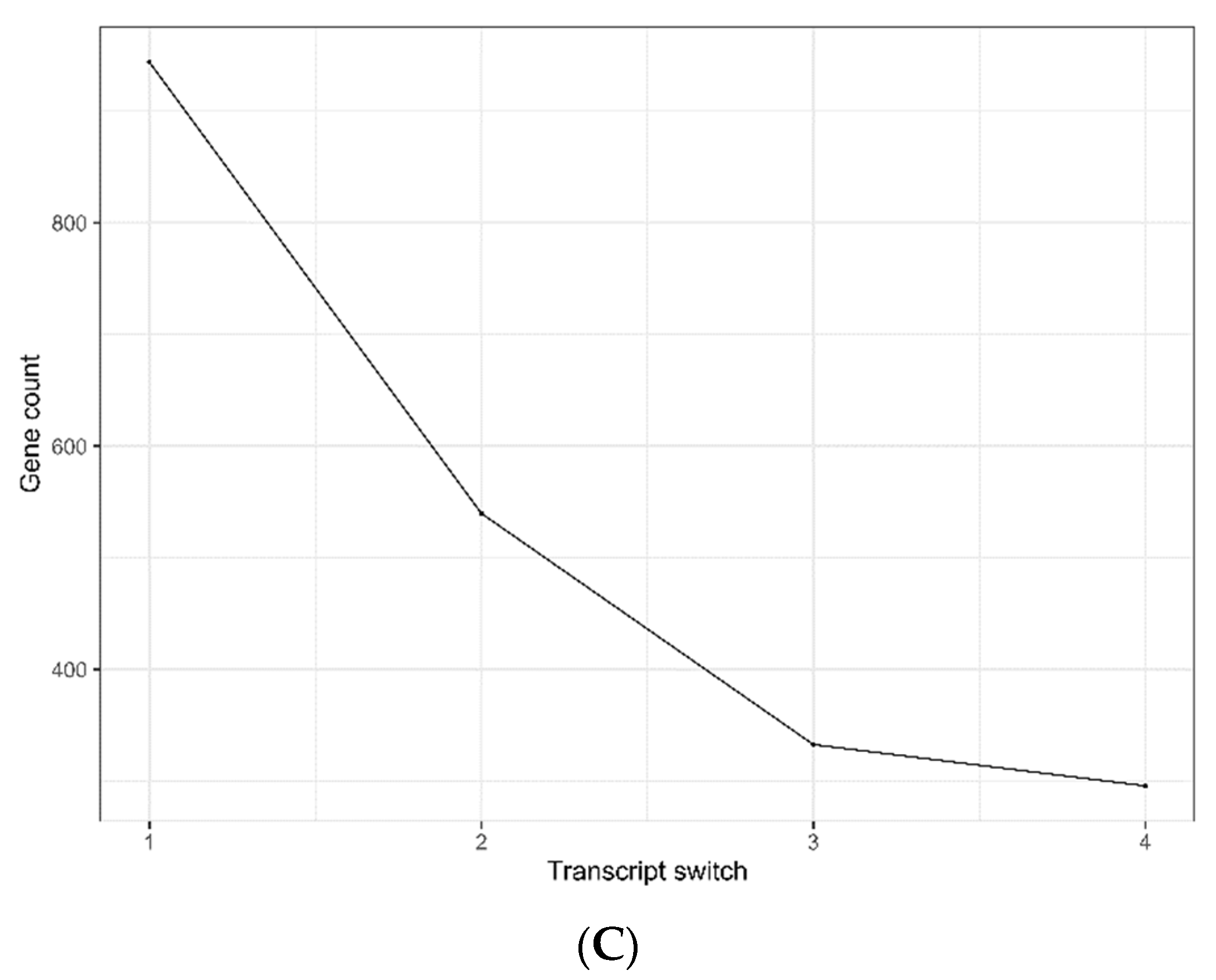
2.4. Expression Variability
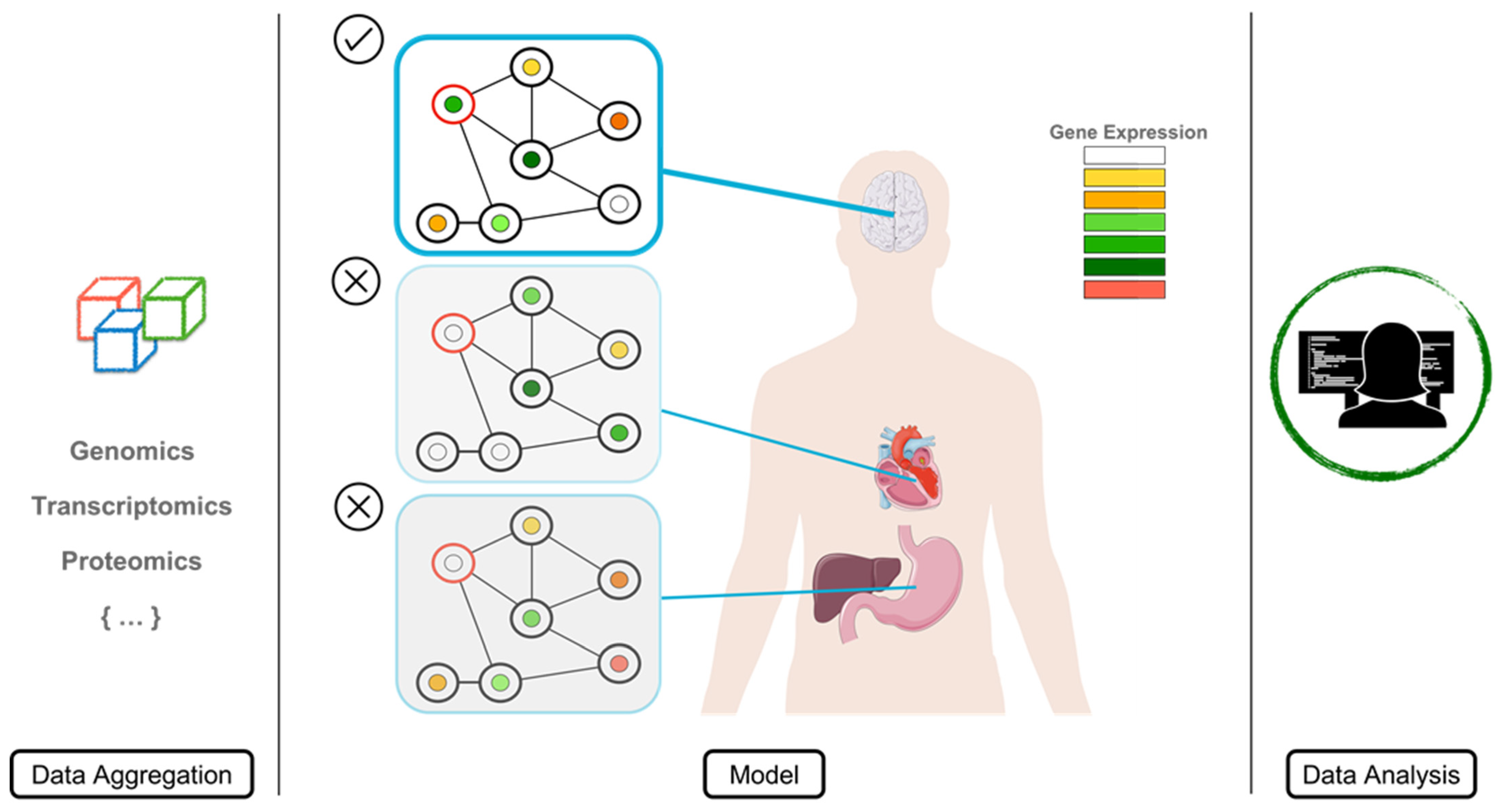
2.5. Protein Variability
2.6. Protein Structure
3. Discussion
4. Materials and Methods
4.1. Samples
4.2. Variant Calling
4.3. Annotation
4.4. Gene/Disease Classification
4.5. Databases
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orphanet. Available online: https://www.orpha.net/consor/cgi-bin/index.php (accessed on 15 January 2022).
- Philippakis, A.A.; Azzariti, D.R.; Beltran, S.; Brookes, A.J.; Brownstein, C.A.; Brudno, M.; Brunner, H.G.; Buske, O.J.; Carey, K.; Doll, C.; et al. The Matchmaker Exchange: A Platform for Rare Disease Gene Discovery. Hum. Mutat. 2015, 36, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Orphanet Database. Available online: www.orphadata.org (accessed on 15 January 2022).
- Ng, S.B.; Turner, E.; Robertson, P.D.; Flygare, S.D.; Bigham, A.W.; Lee, C.; Shaffer, T.; Wong, M.; Bhattacharjee, A.; Eichler, E.E.; et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009, 461, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Bamshad, M.J.; Nickerson, D.A.; Chong, J.X. Mendelian Gene Discovery: Fast and Furious with No End in Sight. Am. J. Hum. Genet. 2019, 105, 448–455. [Google Scholar] [CrossRef] [Green Version]
- Durmaz, A.A.; Karaca, E.; Demkow, U.; Toruner, G.; Schoumans, J.; Cogulu, O. Evolution of Genetic Techniques: Past, Present, and Beyond. BioMed Res. Int. 2015, 2015, 461524. [Google Scholar] [CrossRef]
- Yubero, D.; Brandi, N.; Ormazabal, A.; García-Cazorla, A.; Pérez-Dueñas, B.; Campistol, J.; Ribes, A.; Palau, F.; Artuch, R.; Armstrong, J.; et al. Targeted Next Generation Sequencing in Patients with Inborn Errors of Metabolism. PLoS ONE 2016, 11, e0156359. [Google Scholar] [CrossRef]
- Schlüter, A.; Rodríguez-Palmero, A.; Verdura, E.; Vélez-Santamaría, V.; Ruiz, M.; Fourcade, S.; Planas-Serra, L.; Martínez, J.J.; Guilera, C.; Girós, M.; et al. Diagnosis of Genetic White Matter Disorders by Singleton Whole-Exome and Genome Sequencing Using Interactome-Driven Prioritization. Neurology 2022, 98, e912–e923. [Google Scholar] [CrossRef]
- Boycott, K.M.; Hartley, T.; Biesecker, L.G.; Gibbs, R.A.; Innes, A.M.; Riess, O.; Belmont, J.; Dunwoodie, S.L.; Jojic, N.; Lassmann, T.; et al. A Diagnosis for All Rare Genetic Diseases: The Horizon and the Next Frontiers. Cell 2019, 177, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [Green Version]
- Varsome, The human Genomics Community. Available online: https://varsome.com (accessed on 1 November 2021).
- Tung, K.-F.; Pan, C.-Y.; Chen, C.-H.; Lin, W.-C. Top-ranked expressed gene transcripts of human protein-coding genes investigated with GTEx dataset. Sci. Rep. 2020, 10, 16245. [Google Scholar] [CrossRef]
- Togi, S.; Ura, H.; Niida, Y. Application of Combined Long Amplicon Sequencing (CoLAS) for Genetic Analysis of Neurofibromatosis Type 1: A Pilot Study. Curr. Issues Mol. Biol. 2021, 43, 782–801. [Google Scholar] [CrossRef] [PubMed]
- Bury, A.G.; Robertson, F.M.; Pyle, A.; Payne, B.A.I.; Hudson, G. The Isolation and Deep Sequencing of Mitochondrial DNA. Methods Mol. Biol. 2021, 2277, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, E.; Albion, E.; Modena, C.; Daja, M.; Cecchin, S.; Paolacci, S.; Miertus, J.; Bertelli, M.; Maltese, P.E.; Chiurazzi, P.; et al. PacMAGI: A pipeline including accurate indel detection for the analysis of PacBio sequencing data applied to RPE65. Gene 2022, 832, 146554. [Google Scholar] [CrossRef]
- Noell, G.; Faner, R.; Agustí, A. From systems biology to P4 medicine: Applications in respiratory medicine. Eur. Respir. Rev. 2018, 27, 170110. [Google Scholar] [CrossRef] [Green Version]
- Eraslan, G.; Drokhlyansky, E.; Anand, S.; Fiskin, E.; Subramanian, A.; Slyper, M.; Wang, J.; Van Wittenberghe, N.; Rouhana, J.M.; Waldman, J.; et al. Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Science 2022, 376, eabl4290. [Google Scholar] [CrossRef] [PubMed]
- Kitsak, M.; Sharma, A.; Menche, J.; Guney, E.; Ghiassian, S.D.; Loscalzo, J.; Barabási, A.-L. Tissue Specificity of Human Disease Module. Sci. Rep. 2016, 6, 35241. [Google Scholar] [CrossRef] [PubMed]
- Vidal, S.; Brandi, N.; Pacheco, P.; Maynou, J.; Fernandez, G.; Xiol, C.; Pascual-Alonso, A.; Pineda, M.; Armstrong, J.; del Mar, O.M.; et al. The most recurrent monogenic disorders that overlap with the phenotype of Rett syndrome. Eur. J. Paediatr. Neurol. 2019, 23, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef]
- Martin, A.R.; Williams, E.; Foulger, R.E.; Leigh, S.; Daugherty, L.C.; Niblock, O.; Leong, I.U.S.; Smith, K.R.; Gerasimenko, O.; Haraldsdottir, E.; et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Antonio Nat. Genet. 2019, 51, 1560–1565. [Google Scholar] [CrossRef]
- Martinez-Monseny, A.; Cuadras, D.; Bolasell, M.; Muchart, J.; Arjona, C.; Borregan, M.; Algrabli, A.; Montero, R.; Artuch, R.; Velázquez-Fragua, R.; et al. From gestalt to gene: Early predictive dysmorphic features of PMM2-CDG. J. Med Genet. 2018, 56, 236–245. [Google Scholar] [CrossRef]
- Bossi, A.; Lehner, B. Tissue specificity and the human protein interaction network. Mol. Syst. Biol. 2009, 5, 260. [Google Scholar] [CrossRef] [PubMed]
- Lopes, T.J.S.; Schaefer, M.; Shoemaker, J.; Matsuoka, Y.; Fontaine, J.; Neumann, G.; Andrade-Navarro, M.A.; Kawaoka, Y.; Kitano, H. Tissue-specific subnetworks and characteristics of publicly available human protein interaction databases. Bioinformatics 2011, 27, 2414–2421. [Google Scholar] [CrossRef] [Green Version]
- Bajpai, A.K.; Davuluri, S.; Tiwary, K.; Narayanan, S.; Oguru, S.; Basavaraju, K.; Dayalan, D.; Thirumurugan, K.; Acharya, K.K. Systematic comparison of the protein-protein interaction databases from a user’s perspective. J. Biomed. Inform. 2020, 103, 103380. [Google Scholar] [CrossRef] [PubMed]
- Regev, A.; Teichmann, S.A.; Lander, E.S.; Amit, I.; Benoist, C.; Birney, E.; Bodenmiller, B.; Campbell, P.; Carninci, P.; Clatworthy, M.; et al. Science forum: The Human Cell Atlas. eLife 2017, 6, e27041. [Google Scholar] [CrossRef] [PubMed]
- Glass, K.; Huttenhower, C.; Quackenbush, J.; Yuan, G.-C. Passing Messages between Biological Networks to Refine Predicted Interactions. PLoS ONE 2013, 8, e64832. [Google Scholar] [CrossRef]
- Van Dam, S.; Võsa, U.; Van Der Graaf, A.; Franke, L.; De Magalhães, J.P. Gene co-expression analysis for functional classification and gene–disease predictions. Brief. Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef]
- Matched Annotation from NCBI and EMBL-EBI (MANE). Available online: https://www.ncbi.nlm.nih.gov/refseq/MANE/ (accessed on 15 January 2022).
- Karlebach, G.; Carmody, L.; Sundaramurthi, J.C.; Casiraghi, E.; Hansen, P.; Reese, J.; Mungall, C.J.; Valentini, G.; Robinson, P.N. An algorithmic framework for isoform-specific functional analysis. bioRxiv 2022. [Google Scholar] [CrossRef]
- Weighill, D.; Ben Guebila, M.; Glass, K.; Quackenbush, J.; Platig, J. Predicting genotype-specific gene regulatory networks. Genome Res. 2022, 32, 524–533. [Google Scholar] [CrossRef]
- Menyhárt, O.; Győrffy, B. Multi-omics approaches in cancer research with applications in tumor subtyping, prognosis, and diagnosis. Comput. Struct. Biotechnol. J. 2021, 19, 949–960. [Google Scholar] [CrossRef]
- Ferraro, N.M.; Strober, B.J.; Einson, J.; Abell, N.S.; Aguet, F.; Barbeira, A.N.; Brandt, M.; Bucan, M.; Castel, S.E.; Davis, J.R.; et al. Transcriptomic signatures across human tissues identify functional rare genetic variation. Science 2020, 369, eaaz5900. [Google Scholar] [CrossRef]
- Yépez, V.A.; Mertes, C.; Müller, M.F.; Klaproth-Andrade, D.; Wachutka, L.; Frésard, L.; Gusic, M.; Scheller, I.F.; Goldberg, P.F.; Prokisch, H.; et al. Detection of aberrant gene expression events in RNA sequencing data. Nat. Protoc. 2021, 16, 1276–1296. [Google Scholar] [CrossRef] [PubMed]
- Kopajtich, R.; Smirnov, D.; Stenton, S.L.; Loipfinger, S.; Meng, C.; Scheller, I.F.; Freisinger, P.; Baski, R.; Berutti, R.; Behr, J.; et al. Integration of proteomics with genomics and transcriptomics increases the diagnostic rate of Mendelian disorders. medRxiv 2021, 1–31. [Google Scholar] [CrossRef]
- Du, Y.; Clair, G.C.; Al Alam, D.; Danopoulos, S.; Schnell, D.; Kitzmiller, J.A.; Misra, R.S.; Bhattacharya, S.; Warburton, D.; Mariani, T.J.; et al. Integration of transcriptomic and proteomic data identifies biological functions in cell populations from human infant lung. Am. J. Physiol. Cell. Mol. Physiol. 2019, 317, L347–L360. [Google Scholar] [CrossRef] [PubMed]
- Kustatscher, G.; Collins, T.; Gingras, A.-C.; Guo, T.; Hermjakob, H.; Ideker, T.; Lilley, K.S.; Lundberg, E.; Marcotte, E.M.; Ralser, M.; et al. Understudied proteins: Opportunities and challenges for functional proteomics. Nat. Methods 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Faure, A.J.; Domingo, J.; Schmiedel, J.M.; Hidalgo-Carcedo, C.; Diss, G.; Lehner, B. Global mapping of the energetic and allosteric landscapes of protein binding domains. bioRxiv 2021. [Google Scholar] [CrossRef]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; Del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2013, 42, D358–D363. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2020, 49, D605–D612. [Google Scholar] [CrossRef]
- Fahey, M.E.; Bennett, M.J.; Mahon, C.; Jäger, S.; Pache, L.; Kumar, D.; Shapiro, A.; Rao, K.; Chanda, S.K.; Craik, C.S.; et al. GPS-Prot: A web-based visualization platform for integrating host-pathogen interaction data. BMC Bioinform. 2011, 12, 298. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Gui, J. Prediction of Protein-Protein Interactions from Protein Sequence Using Local Descriptors. Protein Pept. Lett. 2010, 17, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yu, L.; Wen, Z.; Li, M. Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res. 2008, 36, 3025–3030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Sun, S.; Hu, C.; Yao, Y.; Yan, Y.; Zhang, Y. DeepPPI: Boosting Prediction of Protein–Protein Interactions with Deep Neural Networks. J. Chem. Inf. Model. 2017, 57, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Tuncbag, N.; Gursoy, A.; Nussinov, R.; Keskin, O. Predicting protein-protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using PRISM. Nat. Protoc. 2011, 6, 1341–1354. [Google Scholar] [CrossRef]
- Zhang, L.V.; Wong, S.L.; King, O.D.; Roth, F.P. Predicting co-complexed protein pairs using genomic and proteomic data integration. BMC Bioinform. 2004, 5, 38. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zhu, F.; Ling, X.; Liu, Q. Protein Interaction Network Reconstruction through Ensemble Deep Learning with Attention Mechanism. Front. Bioeng. Biotechnol. 2020, 8, 390. [Google Scholar] [CrossRef]
- Armean, I.M.; Lilley, K.S.; Trotter, M.W.B.; Pilkington, N.C.V.; Holden, S.B. Co-complex protein membership evaluation using Maximum Entropy on GO ontology and InterPro annotation. Bioinformatics 2018, 34, 1884–1892. [Google Scholar] [CrossRef] [Green Version]
- Hooper, C.M.; Castleden, I.R.; Tanz, S.K.; Grasso, S.V.; Millar, A.H. Subcellular Proteomics as a Unified Approach of Experimental Localizations and Computed Prediction Data for Arabidopsis and Crop Plants. Adv. Exp. Med. Biol. 2021, 1346, 67–89. [Google Scholar] [CrossRef]
- Johnson, K.L.; Qi, Z.; Yan, Z.; Wen, X.; Nguyen, T.C.; Zaleta-Rivera, K.; Chen, C.-J.; Fan, X.; Sriram, K.; Wan, X.; et al. Revealing protein-protein interactions at the transcriptome scale by sequencing. Mol. Cell 2021, 81, 4091–4103.e9. [Google Scholar] [CrossRef]
- Ying, K.-C.; Lin, S.-W. Maximizing cohesion and separation for detecting protein functional modules in protein-protein interaction networks. PLoS ONE 2020, 15, e0240628. [Google Scholar] [CrossRef]
- Bern, M.; King, A.; Applewhite, D.A.; Ritz, A. Network-based prediction of polygenic disease genes involved in cell motility. BMC Bioinform. 2019, 20, 313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Jiang, Q.; Song, Y.; He, Z.; Zhang, H.; Song, M.; Zhang, X.; Dai, Y.; Karalay, O.; Dieterich, C.; et al. Ageing induces tissue-specific transcriptomic changes in Caenorhabditis elegans. EMBO J. 2022, 41, e109633. [Google Scholar] [CrossRef] [PubMed]
- Izgi, H.; Han, D.; Isildak, U.; Huang, S.; Kocabiyik, E.; Khaitovich, P.; Somel, M.; Dönertaş, H.M. Inter-tissue convergence of gene expression during ageing suggests age-related loss of tissue and cellular identity. eLife 2022, 11, e68048. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; He, J. DPPIN: A Biological Repository of Dynamic Protein-Protein Interaction Network Data. arXiv 2017, 02168. [Google Scholar] [CrossRef]
- Zhang, L.; Lu, Q.; Chang, C. Epigenetics in Health and Disease. Adv. Exp. Med. Biol. 2020, 1253, 3–55. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Hawkins, R.D. Three-dimensional genome architecture and emerging technologies: Looping in disease. Genome Med. 2017, 9, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babraham Bioinformatics. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 January 2022).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra; O’Reilly Media: Sebastopol, CA, USA, 2020. [Google Scholar]
- Cooke, D.P.; Wedge, D.C.; Lunter, G. A unified haplotype-based method for accurate and comprehensive variant calling. Nat. Biotechnol. 2021, 39, 885–892. [Google Scholar] [CrossRef]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Ebert, D.; Muruganujan, A.; Mills, C.; Albou, L.-P.; Mushayamaha, T.; Thomas, P.D. PANTHER version 16: A revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 2020, 49, D394–D403. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genotype-Tissue Expression (GTEx) Project. Available online: https://gtexportal.org (accessed on 15 January 2022).
- Bahl, E.; Koomar, T.; Michaelson, J. cerebroViz: An R package for anatomical visu-alization of spatiotemporal brain data. Bioinformatics 2016, 33, 762–763. [Google Scholar] [CrossRef] [PubMed]
- Smedley, D.; Haider, S.; Ballester, B.; Holland, R.; London, D.; Thorisson, G.; Kasprzyk, A. BioMart—biological queries made easy. BMC Genom. 2009, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org (accessed on 15 January 2022).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IMPACT | Total Number | CADD > 20 | %CADD > 20 |
|---|---|---|---|
| HIGH | 36,753 | 32,207 | 87.63 |
| LOW | 7010 | 800 | 11.41 |
| MODERATE | 884,264 | 513,406 | 58.06 |
| MODIFIER | 13,027 | 1536 | 11.79 |
| 941,054 | 547,949 | 58.23 |
| IMPACT | Pathogenic | Likely Pathogenic | VUS 1 | Likely Benign | Benign |
|---|---|---|---|---|---|
| HIGH | 11 | 11 | 65 | 14 | 119 |
| LOW | - | 4 | 118 | 388 | 1284 |
| MODERATE | - | 7 | 186 | 284 | 722 |
| MODIFIER | - | - | 1368 | 476 | 3842 |
| 11 | 22 | 1737 | 1162 | 5967 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez, G.; Yubero, D.; Palau, F.; Armstrong, J. Molecular Modelling Hurdle in the Next-Generation Sequencing Era. Int. J. Mol. Sci. 2022, 23, 7176. https://doi.org/10.3390/ijms23137176
Fernandez G, Yubero D, Palau F, Armstrong J. Molecular Modelling Hurdle in the Next-Generation Sequencing Era. International Journal of Molecular Sciences. 2022; 23(13):7176. https://doi.org/10.3390/ijms23137176
Chicago/Turabian StyleFernandez, Guerau, Dèlia Yubero, Francesc Palau, and Judith Armstrong. 2022. "Molecular Modelling Hurdle in the Next-Generation Sequencing Era" International Journal of Molecular Sciences 23, no. 13: 7176. https://doi.org/10.3390/ijms23137176
APA StyleFernandez, G., Yubero, D., Palau, F., & Armstrong, J. (2022). Molecular Modelling Hurdle in the Next-Generation Sequencing Era. International Journal of Molecular Sciences, 23(13), 7176. https://doi.org/10.3390/ijms23137176