
Neurodevelopmental Disorders Associated with PSD-95 and Its Interaction Partners




Abstract
1. Introduction
2. PSD-95 and the Synapse
3. PSD-95 Binding Partners and Their Involvement in Neurodevelopmental Disorders
3.1. Shaker-Type Voltage-Gated Potassium Channels
3.2. AMPA Receptors
3.3. NMDA Receptors
3.4. NLGN1
3.5. ADAM22 and LGI1
4. PSD-95 and Neurodevelopmental Disorders
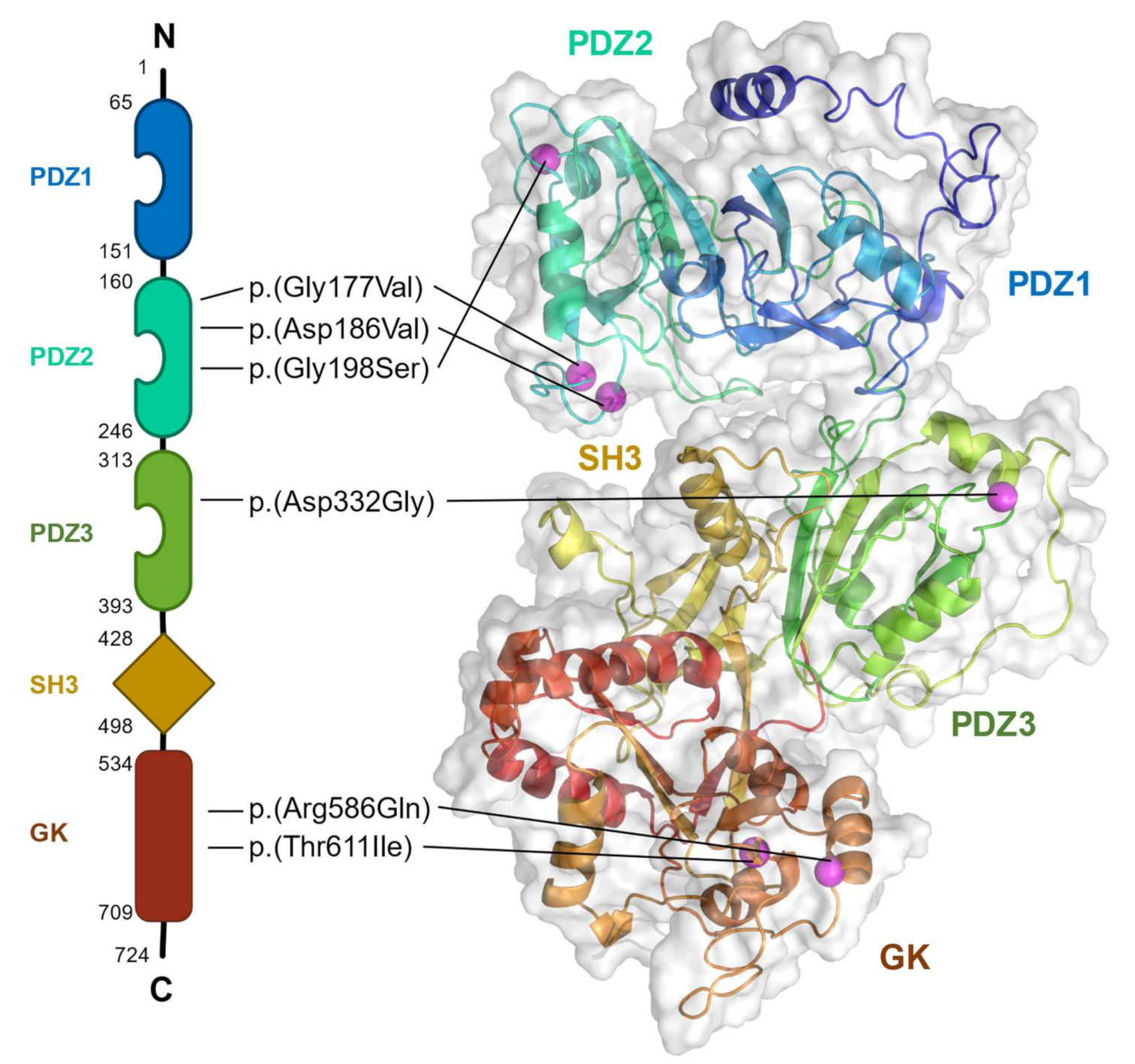
5. The Pathogenesis of DLG4 Missense Variants
6. Final Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sheng, M.; Kim, E. The Postsynaptic Organization of Synapses. Cold Spring Harb. Perspect. Biol. 2011, 3, a005678. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Shang, Y.; Zhang, M. Mechanistic basis of MAGUK-organized complexes in synaptic development and signaling. Nat. Rev. Neurosci. 2016, 17, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Fukata, Y.; Hirano, Y.; Miyazaki, Y.; Yokoi, N.; Fukata, M. Trans-synaptic LGI1–ADAM22–MAGUK in AMPA and NMDA receptor regulation. Neuropharmacology 2021, 194, 108628. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Niethammer, M.; Rothschild, A.; Jan, Y.N.; Sheng, M. Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature 1995, 378, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.-H.; Chen, H.; Li, T.P.; Metzbower, S.R.; MacGillavry, H.D.; Blanpied, T.A. A trans-synaptic nanocolumn aligns neurotransmitter release to receptors. Nature 2016, 536, 210–214. [Google Scholar] [CrossRef]
- Fukata, Y.; Chen, X.; Chiken, S.; Hirano, Y.; Yamagata, A.; Inahashi, H.; Sanbo, M.; Sano, H.; Goto, T.; Hirabayashi, M.; et al. LGI1–ADAM22–MAGUK configures transsynaptic nanoalignment for synaptic transmission and epilepsy prevention. Proc. Natl. Acad. Sci. USA 2021, 118, e2022580118. [Google Scholar] [CrossRef]
- Haas, K.T.; Compans, B.; Letellier, M.; Bartol, T.M.; Grillo-Bosch, D.; Sejnowski, T.J.; Sainlos, M.; Choquet, D.; Thoumine, O.; Hosy, E. Pre-post synaptic alignment through neuroligin-1 tunes synaptic transmission efficiency. eLife 2018, 7, e31755. [Google Scholar] [CrossRef]
- Oliva, C.; Escobedo, P.; Astorga, C.; Molina, C.; Sierralta, J. Role of the maguk protein family in synapse formation and function. Dev. Neurobiol. 2012, 72, 57–72. [Google Scholar] [CrossRef]
- Feng, W.; Zhang, M. Organization and dynamics of PDZ-domain-related supramodules in the postsynaptic density. Nat. Rev. Neurosci. 2009, 10, 87–99. [Google Scholar] [CrossRef]
- Won, S.; Levy, J.M.; Nicoll, R.A.; Roche, K.W. MAGUKs: Multifaceted synaptic organizers. Curr. Opin. Neurobiol. 2017, 43, 94–101. [Google Scholar] [CrossRef]
- Rodríguez-Palmero, A.; Boerrigter, M.M.; Gómez-Andrés, D.; Aldinger, K.A.; Íñigo, M.-A.; Popp, B.; Everman, D.B.; Lovgren, A.K.; Arpin, S.; Bahrambeigi, V.; et al. DLG4-related synaptopathy: A new rare brain disorder. Genet. Med. 2021, 23, 888–899. [Google Scholar] [CrossRef] [PubMed]
- McCann, J.J.; Zheng, L.; Rohrbeck, D.; Felekyan, S.; Kühnemuth, R.; Sutton, R.B.; Seidel, C.A.M.; Bowen, M.E. Supertertiary structure of the synaptic MAGuK scaffold proteins is conserved. Proc. Natl. Acad. Sci. USA 2012, 109, 15775–15780. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lewis, S.M.; Kuhlman, B.; Lee, A.L. Supertertiary Structure of the MAGUK Core from PSD-95. Structure 2013, 21, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Weng, J.; Zhang, X.; Liu, M.; Zhang, M. Creating Conformational Entropy by Increasing Interdomain Mobility in Ligand Binding Regulation: A Revisit to N-Terminal Tandem PDZ Domains of PSD-95. J. Am. Chem. Soc. 2009, 131, 787–796. [Google Scholar] [CrossRef]
- Lee, H.-J.; Zheng, J.J. PDZ domains and their binding partners: Structure, specificity, and modification. Cell Commun. Signal. 2010, 8, 8. [Google Scholar] [CrossRef]
- Luck, K.; Charbonnier, S.; Travé, G. The emerging contribution of sequence context to the specificity of protein interactions mediated by PDZ domains. FEBS Lett. 2012, 586, 2648–2661. [Google Scholar] [CrossRef]
- Seabold, G.K.; Burette, A.; Lim, I.A.; Weinberg, R.J.; Hell, J.W. Interaction of the Tyrosine Kinase Pyk2 with the N-Methyl-D-Aspartate Receptor Complex via the Src Homology 3 Domains of PSD-95 and SAP102. J. Biol. Chem. 2003, 278, 15040–15048. [Google Scholar] [CrossRef]
- Kim, J.H.; Liao, D.; Lau, L.-F.; Huganir, R.L. SynGAP: A Synaptic RasGAP that Associates with the PSD-95/SAP90 Protein Family. Neuron 1998, 20, 683–691. [Google Scholar] [CrossRef]
- Colledge, M.; Dean, R.A.; Scott, G.K.; Langeberg, L.K.; Huganir, R.L.; Scott, J.D. Targeting of PKA to Glutamate Receptors through a MAGUK-AKAP Complex. Neuron 2000, 27, 107–119. [Google Scholar] [CrossRef]
- Kim, E.; Naisbitt, S.; Hsueh, Y.-P.; Rao, A.; Rothschild, A.; Craig, A.M.; Sheng, M. GKAP, a Novel Synaptic Protein That Interacts with the Guanylate Kinase-like Domain of the PSD-95/SAP90 Family of Channel Clustering Molecules. J. Cell Biol. 1997, 136, 669–678. [Google Scholar] [CrossRef]
- Takeuchi, M.; Hata, Y.; Hirao, K.; Toyoda, A.; Irie, M.; Takai, Y. SAPAPs. A Family of PSD-95/SAP90-Associated Proteins Localized at Postsynaptic Densit. J. Biol. Chem. 1997, 272, 11943–11951. [Google Scholar] [CrossRef] [PubMed]
- Naisbitt, S.; Kim, E.; Tu, J.C.; Xiao, B.; Sala, C.; Valtschanoff, J.; Weinberg, R.J.; Worley, P.F.; Sheng, M. Shank, a Novel Family of Postsynaptic Density Proteins that Binds to the NMDA Receptor/PSD-95/GKAP Complex and Cortactin. Neuron 1999, 23, 569–582. [Google Scholar] [CrossRef]
- Chen, X.; Nelson, C.D.; Li, X.; Winters, C.A.; Azzam, R.; Sousa, A.A.; Leapman, R.D.; Gainer, H.; Sheng, M.; Reese, T.S. PSD-95 Is Required to Sustain the Molecular Organization of the Postsynaptic Density. J. Neurosci. 2011, 31, 6329–6338. [Google Scholar] [CrossRef]
- Chen, X.; Levy, J.M.; Hou, A.; Winters, C.; Azzam, R.; Sousa, A.A.; Leapman, R.D.; Nicoll, R.A.; Reese, T.S. PSD-95 family MAGUKs are essential for anchoring AMPA and NMDA receptor complexes at the postsynaptic density. Proc. Natl. Acad. Sci. USA 2015, 112, E6983–E6992. [Google Scholar] [CrossRef]
- Elias, G.M.; Funke, L.; Stein, V.; Grant, S.G.; Bredt, D.S.; Nicoll, R.A. Synapse-Specific and Developmentally Regulated Targeting of AMPA Receptors by a Family of MAGUK Scaffolding Proteins. Neuron 2006, 52, 307–320. [Google Scholar] [CrossRef]
- Vallejo, D.; Codocedo, J.F.; Inestrosa, N.C. Posttranslational Modifications Regulate the Postsynaptic Localization of PSD-95. Mol. Neurobiol. 2017, 54, 1759–1776. [Google Scholar] [CrossRef]
- El-Husseini, A.E.-D.; Schnell, E.; Dakoji, S.; Sweeney, N.; Zhou, Q.; Prange, O.; Gauthier-Campbell, C.; Aguilera-Moreno, A.; Nicoll, R.A.; Bredt, D.S. Synaptic Strength Regulated by Palmitate Cycling on PSD-95. Cell 2002, 108, 849–863. [Google Scholar] [CrossRef]
- Fukata, Y.; Dimitrov, A.; Boncompain, G.; Vielemeyer, O.; Perez, F.; Fukata, M. Local palmitoylation cycles define activity-regulated postsynaptic subdomains. J. Cell Biol. 2013, 202, 145–161. [Google Scholar] [CrossRef]
- Jeyifous, O.; Lin, E.I.; Chen, X.; Antinone, S.E.; Mastro, R.; Drisdel, R.; Reese, T.S.; Green, W.N. Palmitoylation regulates glutamate receptor distributions in postsynaptic densities through control of PSD95 conformation and orientation. Proc. Natl. Acad. Sci. USA 2016, 113, E8482–E8491. [Google Scholar] [CrossRef]
- Parsons, M.P.; Kang, R.; Buren, C.; Dau, A.; Southwell, A.L.; Doty, C.N.; Sanders, S.S.; Hayden, M.R.; Raymond, L.A. Bidirectional Control of Postsynaptic Density-95 (PSD-95) Clustering by Huntingtin. J. Biol. Chem. 2014, 289, 3518–3528. [Google Scholar] [CrossRef]
- Yoshii, A.; Constantine-Paton, M. Postsynaptic localization of PSD-95 is regulated by all three pathways downstream of TrkB signaling. Front. Synaptic Neurosci. 2014, 6, 6. [Google Scholar] [CrossRef]
- Zhang, Y.; Matt, L.; Patriarchi, T.; Malik, Z.A.; Chowdhury, D.; Park, D.K.; Renieri, A.; Ames, J.B.; Hell, J.W. Capping of the N-terminus of PSD-95 by calmodulin triggers its postsynaptic release. EMBO J. 2014, 33, 1341–1353. [Google Scholar] [CrossRef]
- Pedersen, S.W.; Albertsen, L.; Moran, G.E.; Levesque, B.; Pedersen, S.B.; Bartels, L.; Wapenaar, H.; Ye, F.; Zhang, M.; Bowen, M.E.; et al. Site-Specific Phosphorylation of PSD-95 PDZ Domains Reveals Fine-Tuned Regulation of Protein–Protein Interactions. ACS Chem. Biol. 2017, 12, 2313–2323. [Google Scholar] [CrossRef]
- Vacher, H.; Trimmer, J.S. Shaker Family Kv1 Voltage-Gated Potassium Channels in Mammalian Brain Neurons. In Structure, Function, and Modulation of Neuronal Voltagegated Ion Channels; Gribkoff, V.K., Kaczmarek, L.K., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 127–154. [Google Scholar]
- Gutman, G.A.; Chandy, K.G.; Grissmer, S.; Lazdunski, M.; McKinnon, D.; Pardo, L.A.; Robertson, G.A.; Rudy, B.; Sanguinetti, M.C.; Stühmer, W.; et al. International Union of Pharmacology. LIII. Nomenclature and Molecular Relationships of Voltage-Gated Potassium Channels. Pharmacol. Rev. 2005, 57, 473–508. [Google Scholar] [CrossRef]
- Tiffany, A.M.; Manganas, L.N.; Kim, E.; Hsueh, Y.-P.; Sheng, M.; Trimmer, J.S. PSD-95 and SAP97 Exhibit Distinct Mechanisms for Regulating K+ Channel Surface Expression and Clustering. J. Cell Biol. 2000, 148, 147–157. [Google Scholar] [CrossRef]
- Lim, I.A.; Hall, D.D.; Hell, J.W. Selectivity and Promiscuity of the First and Second PDZ Domains of PSD-95 and Synapse-associated Protein 102. J. Biol. Chem. 2002, 277, 21697–21711. [Google Scholar] [CrossRef]
- Kaya, N.; Alsagob, M.; D’Adamo, M.C.; Al-Bakheet, A.; Hasan, S.; Muccioli, M.; Almutairi, F.B.; Almass, R.; Aldosary, M.; Monies, D.; et al. KCNA4deficiency leads to a syndrome of abnormal striatum, congenital cataract and intellectual disability. J. Med. Genet. 2016, 53, 786–792. [Google Scholar] [CrossRef]
- Paulhus, K.; Ammerman, L.; Glasscock, E. Clinical Spectrum of KCNA1 Mutations: New Insights into Episodic Ataxia and Epilepsy Comorbidity. Int. J. Mol. Sci. 2020, 21, 2802. [Google Scholar] [CrossRef]
- Döring, J.H.; Schröter, J.; Jüngling, J.; Biskup, S.; Klotz, K.A.; Bast, T.; Dietel, T.; Korenke, G.C.; Christoph, S.; Brennenstuhl, H.; et al. Refining Genotypes and Phenotypes in KCNA2-Related Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 2824. [Google Scholar] [CrossRef]
- Kamalova, A.; Nakagawa, T. AMPA receptor structure and auxiliary subunits. J. Physiol. 2021, 599, 453–469. [Google Scholar] [CrossRef]
- Petrini, E.M.; Lu, J.; Cognet, L.; Lounis, B.; Ehlers, M.D.; Choquet, D. Endocytic Trafficking and Recycling Maintain a Pool of Mobile Surface AMPA Receptors Required for Synaptic Potentiation. Neuron 2009, 63, 92–105. [Google Scholar] [CrossRef]
- Makino, H.; Malinow, R. AMPA Receptor Incorporation into Synapses during LTP: The Role of Lateral Movement and Exocytosis. Neuron 2009, 64, 381–390. [Google Scholar] [CrossRef]
- Rossmann, M.; Sukumaran, M.; Penn, A.C.; Veprintsev, D.B.; Babu, M.M.; Greger, I.H. Subunit-selective N-terminal domain associations organize the formation of AMPA receptor heteromers. EMBO J. 2011, 30, 959–971. [Google Scholar] [CrossRef]
- Watson, J.F.; Ho, H.; Greger, I.H. Synaptic transmission and plasticity require AMPA receptor anchoring via its N-terminal domain. eLife 2017, 6, e23024. [Google Scholar] [CrossRef]
- Sia, G.-M.; Béïque, J.-C.; Rumbaugh, G.; Cho, R.; Worley, P.F.; Huganir, R.L. Interaction of the N-Terminal Domain of the AMPA Receptor GluR4 Subunit with the Neuronal Pentraxin NP1 Mediates GluR4 Synaptic Recruitment. Neuron 2007, 55, 87–102. [Google Scholar] [CrossRef]
- Armstrong, N.; Gouaux, E. Mechanisms for Activation and Antagonism of an AMPA-Sensitive Glutamate Receptor: Crystal Structures of the GluR2 Ligand Binding Core. Neuron 2000, 28, 165–181. [Google Scholar] [CrossRef]
- Twomey, E.C.; Yelshanskaya, M.V.; Grassucci, R.A.; Frank, J.; Sobolevsky, A.I. Channel opening and gating mechanism in AMPA-subtype glutamate receptors. Nature 2017, 549, 60–65. [Google Scholar] [CrossRef]
- Diering, G.H.; Huganir, R.L. The AMPA Receptor Code of Synaptic Plasticity. Neuron 2018, 100, 314–329. [Google Scholar] [CrossRef]
- Schwenk, J.; Baehrens, D.; Haupt, A.; Bildl, W.; Boudkkazi, S.; Roeper, J.; Fakler, B.; Schulte, U. Regional Diversity and Developmental Dynamics of the AMPA-Receptor Proteome in the Mammalian Brain. Neuron 2014, 84, 41–54. [Google Scholar] [CrossRef]
- Italia, M.; Ferrari, E.; Di Luca, M.; Gardoni, F. GluA3-containing AMPA receptors: From physiology to synaptic dysfunction in brain disorders. Neurobiol. Dis. 2021, 161, 105539. [Google Scholar] [CrossRef]
- Shi, S.-H.; Hayashi, Y.; Esteban, J.A.; Malinow, R. Subunit-Specific Rules Governing AMPA Receptor Trafficking to Synapses in Hippocampal Pyramidal Neurons. Cell 2001, 105, 331–343. [Google Scholar] [CrossRef]
- Wollmuth, L.P. Ion permeation in ionotropic glutamate receptors: Still dynamic after all these years. Curr. Opin. Physiol. 2018, 2, 36–41. [Google Scholar] [CrossRef]
- Tomita, S. Regulation of Ionotropic Glutamate Receptors by Their Auxiliary Subunits. Physiology 2010, 25, 41–49. [Google Scholar] [CrossRef]
- Schnell, E.; Sizemore, M.; Karimzadegan, S.; Chen, L.; Bredt, D.S.; Nicoll, R.A. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc. Natl. Acad. Sci. USA 2002, 99, 13902–13907. [Google Scholar] [CrossRef]
- Nair, D.; Hosy, E.; Petersen, J.D.; Constals, A.; Giannone, G.; Choquet, D.; Sibarita, J.-B. Super-Resolution Imaging Reveals That AMPA Receptors Inside Synapses Are Dynamically Organized in Nanodomains Regulated by PSD95. J. Neurosci. 2013, 33, 13204–13224. [Google Scholar] [CrossRef]
- Bats, C.; Groc, L.; Choquet, D. The Interaction between Stargazin and PSD-95 Regulates AMPA Receptor Surface Trafficking. Neuron 2007, 53, 719–734. [Google Scholar] [CrossRef]
- Opazo, P.; Choquet, D. A three-step model for the synaptic recruitment of AMPA receptors. Mol. Cell. Neurosci. 2011, 46, 1–8. [Google Scholar] [CrossRef]
- Kang, W.S.; Park, J.K.; Kim, S.K.; Park, H.J.; Lee, S.M.; Song, J.Y.; Chung, J.-H.; Kim, J.W. Genetic variants of GRIA1 are associated with susceptibility to schizophrenia in Korean population. Mol. Biol. Rep. 2012, 39, 10697–10703. [Google Scholar] [CrossRef]
- Bygrave, A.M.; Jahans-Price, T.; Wolff, A.R.; Sprengel, R.; Kullmann, D.M.; Bannerman, D.M.; Kätzel, D. Hippocampal–prefrontal coherence mediates working memory and selective attention at distinct frequency bands and provides a causal link between schizophrenia and its risk gene GRIA1. Transl. Psychiatry 2019, 9, 142. [Google Scholar] [CrossRef]
- Magri, C.; Gardella, R.; Barlati, S.D.; Podavini, D.; Iatropoulos, P.; Bonomi, S.; Valsecchi, P.; Sacchetti, E.; Barlati, S. Glutamate AMPA receptor subunit 1 gene (GRIA1) and DSM-IV-TR schizophrenia: A pilot case-control association study in an Italian sample. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2006, 141, 287–293. [Google Scholar] [CrossRef]
- O’Connor, J.A.; Hemby, S.E. Elevated GRIA1 mRNA expression in Layer II/III and V pyramidal cells of the DLPFC in schizophrenia. Schizophr. Res. 2007, 97, 277–288. [Google Scholar] [CrossRef]
- De Ligt, J.; Willemsen, M.H.; Van Bon, B.W.M.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; De Vries, P.; Gilissen, C.; et al. Diagnostic Exome Sequencing in Persons with Severe Intellectual Disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef]
- Geisheker, M.R.; Heymann, G.; Wang, T.; Coe, B.P.; Turner, T.N.; Stessman, H.A.F.; Hoekzema, K.; Kvarnung, M.; Shaw, M.; Friend, K.; et al. Hotspots of missense mutation identify neurodevelopmental disorder genes and functional domains. Nat. Neurosci. 2017, 20, 1043–1051. [Google Scholar] [CrossRef]
- Poot, M.; Eleveld, M.J.; Van ’T Slot, R.; Ploos Van Amstel, H.K.; Hochstenbach, R. Recurrent copy number changes in mentally retarded children harbour genes involved in cellular localization and the glutamate receptor complex. Eur. J. Hum. Genet. 2010, 18, 39–46. [Google Scholar] [CrossRef]
- Hackmann, K.; Matko, S.; Gerlach, E.-M.; Von Der Hagen, M.; Klink, B.; Schrock, E.; Rump, A.; Di Donato, N. Partial deletion of GLRB and GRIA2 in a patient with intellectual disability. Eur. J. Hum. Genet. 2013, 21, 112–114. [Google Scholar] [CrossRef][Green Version]
- Tzschach, A.; Menzel, C.; Erdogan, F.; Istifli, E.S.; Rieger, M.; Ovens-Raeder, A.; Macke, A.; Ropers, H.-H.; Ullmann, R.; Kalscheuer, V. Characterization of an interstitial 4q32 deletion in a patient with mental retardation and a complex chromosome rearrangement. Am. J. Med. Genet. Part A 2010, 152, 1008–1012. [Google Scholar] [CrossRef]
- Ramanathan, S.; Woodroffe, A.; Flodman, P.L.; Mays, L.Z.; Hanouni, M.; Modahl, C.B.; Steinberg-Epstein, R.; Bocian, M.E.; Spence, M.A.; Smith, M. A case of autism with an interstitial deletion on 4q leading to hemizygosity for genes encoding for glutamine and glycine neurotransmitter receptor sub-units (AMPA 2, GLRA3, GLRB) and neuropeptide receptors NPY1R, NPY5R. BMC Med. Genet. 2004, 5, 10. [Google Scholar] [CrossRef][Green Version]
- Salpietro, V.; Dixon, C.L.; Guo, H.; Bello, O.D.; Vandrovcova, J.; Efthymiou, S.; Maroofian, R.; Heimer, G.; Burglen, L.; Valence, S.; et al. AMPA receptor GluA2 subunit defects are a cause of neurodevelopmental disorders. Nat. Commun. 2019, 10, 3094. [Google Scholar] [CrossRef]
- Trivisano, M.; Santarone, M.E.; Micalizzi, A.; Ferretti, A.; Dentici, M.L.; Novelli, A.; Vigevano, F.; Specchio, N. GRIA3 missense mutation is cause of an x-linked developmental and epileptic encephalopathy. Seizure 2020, 82, 1–6. [Google Scholar] [CrossRef]
- Sun, J.-H.; Chen, J.; Valenzuela, F.E.A.; Brown, C.; Masser-Frye, D.; Jones, M.; Romero, L.P.; Rinaldi, B.; Li, W.L.; Li, Q.-Q.; et al. X-linked neonatal-onset epileptic encephalopathy associated with a gain-of-function variant p.R660T in GRIA3. PLoS Genet. 2021, 17, e1009608. [Google Scholar] [CrossRef]
- Wu, Y.; Arai, A.C.; Rumbaugh, G.; Srivastava, A.K.; Turner, G.; Hayashi, T.; Suzuki, E.; Jiang, Y.; Zhang, L.; Rodriguez, J.; et al. Mutations in ionotropic AMPA receptor 3 alter channel properties and are associated with moderate cognitive impairment in humans. Proc. Natl. Acad. Sci. USA 2007, 104, 18163–18168. [Google Scholar] [CrossRef]
- Martin, S.; Chamberlin, A.; Shinde, D.N.; Hempel, M.; Strom, T.M.; Schreiber, A.; Johannsen, J.; Ousager, L.B.; Larsen, M.J.; Hansen, L.K.; et al. De Novo Variants in GRIA4 Lead to Intellectual Disability with or without Seizures and Gait Abnormalities. Am. J. Hum. Genet. 2017, 101, 1013–1020. [Google Scholar] [CrossRef]
- Beyer, B.; Deleuze, C.; Letts, V.A.; Mahaffey, C.L.; Boumil, R.M.; Lew, T.A.; Huguenard, J.R.; Frankel, W.N. Absence seizures in C3H/HeJ and knockout mice caused by mutation of the AMPA receptor subunit Gria4. Hum. Mol. Genet. 2008, 17, 1738–1749. [Google Scholar] [CrossRef]
- Paz, J.T.; Bryant, A.S.; Peng, K.; Fenno, L.; Yizhar, O.; Frankel, W.N.; Deisseroth, K.; Huguenard, J.R. A new mode of corticothalamic transmission revealed in the Gria4−/− model of absence epilepsy. Nat. Neurosci. 2011, 14, 1167–1173. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Gauthier, J.; Araki, Y.; Lin, D.-T.; Yoshizawa, Y.; Higashi, K.; Park, A.-R.; Spiegelman, D.; Dobrzeniecka, S.; Piton, A.; et al. Excess of De Novo Deleterious Mutations in Genes Associated with Glutamatergic Systems in Nonsyndromic Intellectual Disability. Am. J. Hum. Genet. 2011, 88, 306–316. [Google Scholar] [CrossRef]
- Brandler, W.M.; Antaki, D.; Gujral, M.; Noor, A.; Rosanio, G.; Chapman, T.R.; Barrera, D.J.; Lin, G.N.; Malhotra, D.; Watts, A.C.; et al. Frequency and Complexity of De Novo Structural Mutation in Autism. Am. J. Hum. Genet. 2016, 98, 667–679. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef]
- Volianskis, A.; France, G.; Jensen, M.S.; Bortolotto, Z.A.; Jane, D.E.; Collingridge, G.L. Long-term potentiation and the role of N -methyl- d -aspartate receptors. Brain Res. 2015, 1621, 5–16. [Google Scholar] [CrossRef]
- Citri, A.; Malenka, R.C. Synaptic Plasticity: Multiple Forms, Functions, and Mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef]
- Ishchenko, Y.; Carrizales, M.G.; Koleske, A.J. Regulation of the NMDA receptor by its cytoplasmic domains: (How) is the tail wagging the dog? Neuropharmacology 2021, 195, 108634. [Google Scholar] [CrossRef] [PubMed]
- Cousins, S.L.; Stephenson, F.A. Identification of N-Methyl-d-aspartic Acid (NMDA) Receptor Subtype-specific Binding Sites That Mediate Direct Interactions with Scaffold Protein PSD-95. J. Biol. Chem. 2012, 287, 13465–13476. [Google Scholar] [CrossRef]
- Turic, D.; Langley, K.; Mills, S.; Stephens, M.; Lawson, D.; Govan, C.; Williams, N.; Bree, M.V.D.; Craddock, N.; Kent, L.; et al. Follow-up of genetic linkage findings on chromosome 16p13: Evidence of association of N-methyl-D aspartate glutamate receptor 2A gene polymorphism with ADHD. Mol. Psychiatry 2004, 9, 169–173. [Google Scholar] [CrossRef]
- Dorval, K.M.; Wigg, K.G.; Crosbie, J.; Tannock, R.; Kennedy, J.L.; Ickowicz, A.; Pathare, T.; Malone, M.; Schachar, R.; Barr, C.L. Association of the glutamate receptor subunit gene GRIN2B with attention-deficit/hyperactivity disorder. Genes Brain Behav. 2007, 6, 444–452. [Google Scholar] [CrossRef]
- Endele, S.; Rosenberger, G.; Geider, K.; Popp, B.; Tamer, C.; Stefanova, I.; Milh, M.; Kortüm, F.; Fritsch, A.; Pientka, F.K.; et al. Mutations in GRIN2A and GRIN2B encoding regulatory subunits of NMDA receptors cause variable neurodevelopmental phenotypes. Nat. Genet. 2010, 42, 1021–1026. [Google Scholar] [CrossRef]
- Reutlinger, C.; Helbig, I.; Gawelczyk, B.; Subero, J.I.M.; Tönnies, H.; Muhle, H.; Finsterwalder, K.; Vermeer, S.; Pfundt, R.; Sperner, J.; et al. Deletions in 16p13 including GRIN2A in patients with intellectual disability, various dysmorphic features, and seizure disorders of the rolandic region. Epilepsia 2010, 51, 1870–1873. [Google Scholar] [CrossRef]
- Zhang, Y.; Kong, W.; Gao, Y.; Liu, X.; Gao, K.; Xie, H.; Wu, Y.; Zhang, Y.; Wang, J.; Gao, F.; et al. Gene Mutation Analysis in 253 Chinese Children with Unexplained Epilepsy and Intellectual/Developmental Disabilities. PLoS ONE 2015, 10, e0141782. [Google Scholar] [CrossRef]
- Santos-Gómez, A.; Miguez-Cabello, F.; García-Recio, A.; Locubiche-Serra, S.; García-Díaz, R.; Soto-Insuga, V.; Guerrero-López, R.; Juliá-Palacios, N.; Ciruela, F.; García-Cazorla, À.; et al. Disease-associated GRIN protein truncating variants trigger NMDA receptor loss-of-function. Hum. Mol. Genet. 2021, 29, 3859–3871. [Google Scholar] [CrossRef]
- Gai, X.; Xie, H.M.; Perin, J.C.; Takahashi, N.; Murphy, K.; Wenocur, A.S.; D’Arcy, M.; O’Hara, R.J.; Goldmuntz, E.; Grice, D.E.; et al. Rare structural variation of synapse and neurotransmission genes in autism. Mol. Psychiatry 2012, 17, 402–411. [Google Scholar] [CrossRef]
- McRae, J.F.; Clayton, S.; Fitzgerald, T.W.; Kaplanis, J.; Prigmore, E.; Rajan, D.; Sifrim, A.; Aitken, S.; Akawi, N.; Alvi, M.; et al. Prevalence, Phenotype and Architecture of Developmental Disorders Caused by de Novo Mutation. bioRxiv, 2016; Submitted. [Google Scholar] [CrossRef]
- Strehlow, V.; Heyne, H.O.; Vlaskamp, D.R.M.; Marwick, K.F.M.; Rudolf, G.; de Bellescize, J.; Biskup, S.; Brilstra, E.H.; Brouwer, O.F.; Callenbach, P.M.C.; et al. GRIN2A-related disorders: Genotype and functional consequence predict phenotype. Brain 2019, 142, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhou, L.; Yuan, H.; Vieira, M.; Sanz-Clemente, A.; Badger, J.D.; Lu, W.; Traynelis, S.F.; Roche, K.W. A Rare Variant Identified Within the GluN2B C-Terminus in a Patient with Autism Affects NMDA Receptor Surface Expression and Spine Density. J. Neurosci. 2017, 37, 4093–4102. [Google Scholar] [CrossRef] [PubMed]
- Platzer, K.; Yuan, H.; Schütz, H.; Winschel, A.; Chen, W.; Hu, C.; Kusumoto, H.; Heyne, H.O.; Helbig, K.L.; Tang, S.; et al. GRIN2B encephalopathy: Novel findings on phenotype, variant clustering, functional consequences and treatment aspects. J. Med. Genet. 2017, 54, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marmiesse, A.; Kusumoto, H.; Rekarte, S.; Roca, I.; Zhang, J.; Myers, S.J.; Traynelis, S.F.; Couce, M.L.; Gutiérrez-Solana, L.; Yuan, H. A novel missense mutation in GRIN2A causes a nonepileptic neurodevelopmental disorder. Mov. Disord. 2018, 33, 992–999. [Google Scholar] [CrossRef]
- XiangWei, W.; Jiang, Y.; Yuan, H. De novo mutations and rare variants occurring in NMDA receptors. Curr. Opin. Physiol. 2018, 2, 27–35. [Google Scholar] [CrossRef]
- Amin, J.B.; Moody, G.R.; Wollmuth, L.P. From bedside-to-bench: What disease-associated variants are teaching us about the NMDA receptor. J. Physiol. 2021, 599, 397–416. [Google Scholar] [CrossRef]
- Turner, S.J.; Mayes, A.K.; Verhoeven, A.; Mandelstam, S.A.; Morgan, A.T.; Scheffer, I.E. GRIN2A: An aptly named gene for speech dysfunction. Neurology 2015, 84, 586–593. [Google Scholar] [CrossRef]
- Carvill, G.L.; Regan, B.M.; Yendle, S.C.; O’Roak, B.J.; Lozovaya, N.; Bruneau, N.; Burnashev, N.; Khan, A.; Cook, J.; Geraghty, E.; et al. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat. Genet. 2013, 45, 1073–1076. [Google Scholar] [CrossRef]
- Conroy, J.; McGettigan, P.A.; McCreary, D.; Shah, N.; Collins, K.; Parry-Fielder, B.; Moran, M.; Hanrahan, D.; Deonna, T.W.; Korff, C.M.; et al. Towards the identification of a genetic basis for Landau-Kleffner syndrome. Epilepsia 2014, 55, 858–865. [Google Scholar] [CrossRef]
- Yu, Y.; Lin, Y.; Takasaki, Y.; Wang, C.; Kimura, H.; Xing, J.; Ishizuka, K.; Toyama, M.; Kushima, I.; Mori, D.; et al. Rare loss of function mutations in N-methyl-d-aspartate glutamate receptors and their contributions to schizophrenia susceptibility. Transl. Psychiatry 2018, 8, 12. [Google Scholar] [CrossRef]
- Li, D.; Yuan, H.; Ortiz-Gonzalez, X.R.; Marsh, E.D.; Tian, L.; McCormick, E.M.; Kosobucki, G.J.; Chen, W.; Schulien, A.J.; Chiavacci, R.; et al. GRIN2D Recurrent De Novo Dominant Mutation Causes a Severe Epileptic Encephalopathy Treatable with NMDA Receptor Channel Blockers. Am. J. Hum. Genet. 2016, 99, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Xiangwei, W.; Kannan, V.; Xu, Y.; Kosobucki, G.J.; Schulien, A.J.; Kusumoto, H.; El Achkar, C.M.; Bhattacharya, S.; Lesca, G.; Nguyen, S.; et al. Heterogeneous clinical and functional features of GRIN2D-related developmental and epileptic encephalopathy. Brain 2019, 142, 3009–3027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tang, W.; Bhatia, N.K.; Xu, Y.; Paudyal, N.; Liu, D.; Kim, S.; Song, R.; XiangWei, W.; Shaulsky, G.; et al. A de novo GRIN1 Variant Associated With Myoclonus and Developmental Delay: From Molecular Mechanism to Rescue Pharmacology. Front. Genet. 2021, 12, 694312. [Google Scholar] [CrossRef]
- Allen, A.S.; Berkovic, S.F.; Cossette, P.; Delanty, N.; Dlugos, D.; Eichler, E.E.; Epstein, M.P.; Glauser, T.; Goldstein, D.B.; Han, Y.; et al. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Wu, K.; Pandey, S.; Lehr, A.W.; Li, Y.; Bemben, M.A.; Badger, J.D.; Lauzon, J.L.; Wang, T.; Zaghloul, K.A.; et al. A Cluster of Autism-Associated Variants on X-Linked NLGN4X Functionally Resemble NLGN4Y. Neuron 2020, 106, 759–768.e7. [Google Scholar] [CrossRef] [PubMed]
- Bemben, M.A.; Shipman, S.L.; Nicoll, R.A.; Roche, K.W. The cellular and molecular landscape of neuroligins. Trends Neurosci. 2015, 38, 496–505. [Google Scholar] [CrossRef]
- Niescier, R.F.; Lin, Y.-C. The Potential Role of AMPA Receptor Trafficking in Autism and Other Neurodevelopmental Conditions. Neuroscience 2021, 479, 180–191. [Google Scholar] [CrossRef]
- Varoqueaux, F.; Aramuni, G.; Rawson, R.L.; Mohrmann, R.; Missler, M.; Gottmann, K.; Zhang, W.; Südhof, T.C.; Brose, N. Neuroligins Determine Synapse Maturation and Function. Neuron 2006, 51, 741–754. [Google Scholar] [CrossRef]
- Heine, M.; Thoumine, O.; Mondin, M.; Tessier, B.; Giannone, G.; Choquet, D. Activity-independent and subunit-specific recruitment of functional AMPA receptors at neurexin/neuroligin contacts. Proc. Natl. Acad. Sci. USA 2008, 105, 20947–20952. [Google Scholar] [CrossRef]
- Irie, M.; Hata, Y.; Takeuchi, M.; Ichtchenko, K.; Toyoda, A.; Hirao, K.; Takai, Y.; Rosahl, T.W.; Südhof, T.C. Binding of Neuroligins to PSD-95. Science 1997, 277, 1511–1515. [Google Scholar] [CrossRef]
- Futai, K.; Kim, M.J.; Hashikawa, T.; Scheiffele, P.; Sheng, M.; Hayashi, Y. Retrograde modulation of presynaptic release probability through signaling mediated by PSD-95–neuroligin. Nat. Neurosci. 2007, 10, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Prange, O.; Wong, T.P.; Gerrow, K.; Wang, Y.T.; El-Husseini, A. A balance between excitatory and inhibitory synapses is controlled by PSD-95 and neuroligin. Proc. Natl. Acad. Sci. USA 2004, 101, 13915–13920. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Pandey, S.; Li, Y.; Badger, J.D.; Lu, W.; Roche, K.W. PSD-95 binding dynamically regulates NLGN1 trafficking and function. Proc. Natl. Acad. Sci. USA 2019, 116, 12035–12044. [Google Scholar] [CrossRef] [PubMed]
- Glessner, J.T.; Wang, K.; Cai, G.; Korvatska, O.; Kim, C.E.; Wood, S.; Zhang, H.; Estes, A.; Brune, C.W.; Bradfield, J.P.; et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009, 459, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Millson, A.; Lagrave, D.; Willis, M.J.H.; Rowe, L.R.; Lyon, E.; South, S.T. Chromosomal loss of 3q26.3-3q26.32, involving a partial neuroligin 1 deletion, identified by genomic microarray in a child with microcephaly, seizure disorder, and severe intellectual disability. Am. J. Med Genet. Part A 2012, 158A, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Nomura, J.; Ji, X.; Tamada, K.; Arai, T.; Takahashi, E.; Bućan, M.; Takumi, T. Functional significance of rare neuroligin 1 variants found in autism. PLoS Genet. 2017, 13, e1006940. [Google Scholar] [CrossRef]
- Tejada, M.-I.; Elcoroaristizabal, X.; Ibarluzea, N.; Botella, M.-P.; De La Hoz, A.-B.; Ocio, I. A novel nonsense homozygous variant in the NLGN1 gene found in a pair of monozygotic twin brothers with intellectual disability and autism. Clin. Genet. 2019, 95, 339–340. [Google Scholar] [CrossRef]
- Hsia, H.-E.; Tüshaus, J.; Brummer, T.; Zheng, Y.; Scilabra, S.D.; Lichtenthaler, S.F. Functions of ‘A disintegrin and metalloproteases (ADAMs)’ in the mammalian nervous system. Cell. Mol. Life Sci. 2019, 76, 3055–3081. [Google Scholar] [CrossRef]
- Fukata, Y.; Yokoi, N.; Miyazaki, Y.; Fukata, M. The LGI1–ADAM22 protein complex in synaptic transmission and synaptic disorders. Neurosci. Res. 2017, 116, 39–45. [Google Scholar] [CrossRef]
- Fukata, Y.; Adesnik, H.; Iwanaga, T.; Bredt, D.S.; Nicoll, R.A.; Fukata, M. Epilepsy-Related Ligand/Receptor Complex LGI1 and ADAM22 Regulate Synaptic Transmission. Science 2006, 313, 1792–1795. [Google Scholar] [CrossRef]
- Lovero, K.L.; Fukata, Y.; Granger, A.J.; Fukata, M.; Nicoll, R.A. The LGI1–ADAM22 protein complex directs synapse maturation through regulation of PSD-95 function. Proc. Natl. Acad. Sci. USA 2015, 112, E4129–E4137. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, A.; Fukai, S. Insights into the mechanisms of epilepsy from structural biology of LGI1–ADAM22. Cell. Mol. Life Sci. 2020, 77, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, B.; Brilstra, E.H.; Lindhout, D.; Baulac, S.; De Haan, G.-J.; Van Kempen, M. Hyperactive behavior in a family with autosomal dominant lateral temporal lobe epilepsy caused by a mutation in the LGI1/epitempin gene. Epilepsy Behav. 2013, 28, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Morante-Redolat, J.M.; Gorostidi-Pagola, A.; Sirerol, M.S.; Saenz, A.; Poza, J.J.; Galán, J.; Gesk, S.; Sarafidou, T.; Mautner, V.-F.; Binelli, S.; et al. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum. Mol. Genet. 2002, 11, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, A.; Miyazaki, Y.; Yokoi, N.; Shigematsu, H.; Sato, Y.; Goto-Ito, S.; Maeda, A.; Goto, T.; Sanbo, M.; Hirabayashi, M.; et al. Structural basis of epilepsy-related ligand–receptor complex LGI1-ADAM22. Nat. Commun. 2018, 9, 1546. [Google Scholar] [CrossRef] [PubMed]
- Muona, M.; Fukata, Y.; Anttonen, A.-K.; Laari, A.; Palotie, A.; Pihko, H.; Lönnqvist, T.; Valanne, L.; Somer, M.; Fukata, M.; et al. Dysfunctional ADAM22 implicated in progressive encephalopathy with cortical atrophy and epilepsy. Neurol. Genet. 2016, 2, e46. [Google Scholar] [CrossRef]
- Maddirevula, S.; Alzahrani, F.; Al-Owain, M.; Al Muhaizea, M.A.; Kayyali, H.R.; AlHashem, A.; Rahbeeni, Z.; Al-Otaibi, M.; Alzaidan, H.I.; Balobaid, A.; et al. Autozygome and high throughput confirmation of disease genes candidacy. Genet. Med. 2019, 21, 736–742. [Google Scholar] [CrossRef]
- Rauch, A.; Wieczorek, D.; Graf, E.; Wieland, T.; Endele, S.; Schwarzmayr, T.; Albrecht, B.; Bartholdi, D.; Beygo, J.; Di Donato, N.; et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: An exome sequencing study. Lancet 2012, 380, 1674–1682. [Google Scholar] [CrossRef]
- Lelieveld, S.H.; Reijnders, M.R.F.; Pfundt, R.; Yntema, H.G.; Kamsteeg, E.-J.; de Vries, P.; de Vries, B.B.A.; Willemsen, M.H.; Kleefstra, T.; Löhner, K.; et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 2016, 19, 1194–1196. [Google Scholar] [CrossRef]
- Fitzgerald, T.W.; Gerety, S.S.; Jones, W.D.; Van Kogelenberg, M.; King, D.A.; McRae, J.; Morley, K.I.; Parthiban, V.; Al-Turki, S.; Ambridge, K.; et al. Large-Scale Discovery of Novel Genetic Causes of Developmental Disorders. Nature 2015, 519, 223–228. [Google Scholar] [CrossRef]
- Moutton, S.; Bruel, A.-L.; Assoum, M.; Chevarin, M.; Sarrazin, E.; Goizet, C.; Guerrot, A.-M.; Charollais, A.; Charles, P.; Heron, D.; et al. Truncating variants of the DLG4 gene are responsible for intellectual disability with marfanoid features. Clin. Genet. 2018, 93, 1172–1178. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, T.; Wu, H.; Long, M.; Coe, B.P.; Li, H.; Xun, G.; Ou, J.; Chen, B.; Duan, G.; et al. Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol. Autism 2018, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Roos, J.L.; Levy, S.; Van Rensburg, E.J.; Gogos, J.A.; Karayiorgou, M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 2008, 40, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Kirov, G.; Pocklington, A.J.; Holmans, P.; Ivanov, D.; Ikeda, M.; Ruderfer, D.; Moran, J.; Chambert, K.; Toncheva, D.; Georgieva, L.; et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signaling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 2012, 17, 142–153. [Google Scholar] [CrossRef]
- Tarpey, P.; Parnau, J.; Blow, M.; Woffendin, H.; Bignell, G.; Cox, C.; Cox, J.; Davies, H.; Edkins, S.; Holden, S.; et al. Mutations in the DLG3 Gene Cause Nonsyndromic X-Linked Mental Retardation. Am. J. Hum. Genet. 2004, 75, 318–324. [Google Scholar] [CrossRef]
- Philips, A.K.; Sirén, A.; Avela, K.; Somer, M.; Peippo, M.; Ahvenainen, M.; Doagu, F.; Arvio, M.; Kääriäinen, H.; Van Esch, H.; et al. X-exome sequencing in Finnish families with Intellectual Disability-four novel mutations and two novel syndromic phenotypes. Orphanet J. Rare Dis. 2014, 9, 49. [Google Scholar] [CrossRef]
- Shao, C.Y.; Mirra, S.S.; Sait, H.B.R.; Sacktor, T.C.; Sigurdsson, E.M. Postsynaptic degeneration as revealed by PSD-95 reduction occurs after advanced Aβ and tau pathology in transgenic mouse models of Alzheimer’s disease. Acta Neuropathol. 2011, 122, 285–292. [Google Scholar] [CrossRef]
- Dore, K.; Carrico, Z.; Alfonso, S.; Marino, M.; Koymans, K.; Kessels, H.W.; Malinow, R. PSD-95 protects synapses from β-amyloid. Cell Rep. 2021, 35, 109194. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Group | Gene | Protein | Alternative Names |
|---|---|---|---|
| DLG subfamily | DLG1 | SAP97 | |
| DLG2 | PSD-93 | ||
| DLG3 | SAP102 | ||
| DLG4 | PSD-95 | SAP90 | |
| Kv1 channel subunits | KCNA1 | Kv1.1 | |
| KCNA2 | Kv1.2 | ||
| KCNA4 | Kv1.3 | ||
| NMDA receptor subunits | GRIN1 | GluN1 | NR1 |
| GRIN2A | GluN2A | NR2 | |
| GRIN2B | GluN2B | ||
| GRIN2C | GluN2C | ||
| GRIN2D | GluN2D | ||
| GRIN3A | GluN3A | NR3 | |
| GRIN3B | GluN3B | ||
| AMPA receptor subunits | GRIA1 | GluA1 | GluR1 |
| GRIA2 | GluA2 | GluR2 | |
| GRIA3 | GluA3 | GluR3 | |
| GRIA4 | GluA4 | GluRA-D2 | |
| CACNG2 | Stargazin | TARP γ2 | |
| Neuroligins | NLGN1 | NLGN1 | |
| NLGN2 | NLGN2 | ||
| NLGN3 | NLGN3 | ||
| NLGN4 | NLGN4 | NLGN4X | |
| NLGN4Y | NLGN4Y | NLGN5 | |
| LGI1 and ADAM22 | LGI1 | LGI1 | Epitempin |
| ADAM22 | ADAM22 | Mdc2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levy, A.M.; Gomez-Puertas, P.; Tümer, Z. Neurodevelopmental Disorders Associated with PSD-95 and Its Interaction Partners. Int. J. Mol. Sci. 2022, 23, 4390. https://doi.org/10.3390/ijms23084390
Levy AM, Gomez-Puertas P, Tümer Z. Neurodevelopmental Disorders Associated with PSD-95 and Its Interaction Partners. International Journal of Molecular Sciences. 2022; 23(8):4390. https://doi.org/10.3390/ijms23084390
Chicago/Turabian StyleLevy, Amanda M., Paulino Gomez-Puertas, and Zeynep Tümer. 2022. "Neurodevelopmental Disorders Associated with PSD-95 and Its Interaction Partners" International Journal of Molecular Sciences 23, no. 8: 4390. https://doi.org/10.3390/ijms23084390
APA StyleLevy, A. M., Gomez-Puertas, P., & Tümer, Z. (2022). Neurodevelopmental Disorders Associated with PSD-95 and Its Interaction Partners. International Journal of Molecular Sciences, 23(8), 4390. https://doi.org/10.3390/ijms23084390