
ErbB3-Targeting Oncolytic Adenovirus Causes Potent Tumor Suppression by Induction of Apoptosis in Cancer Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
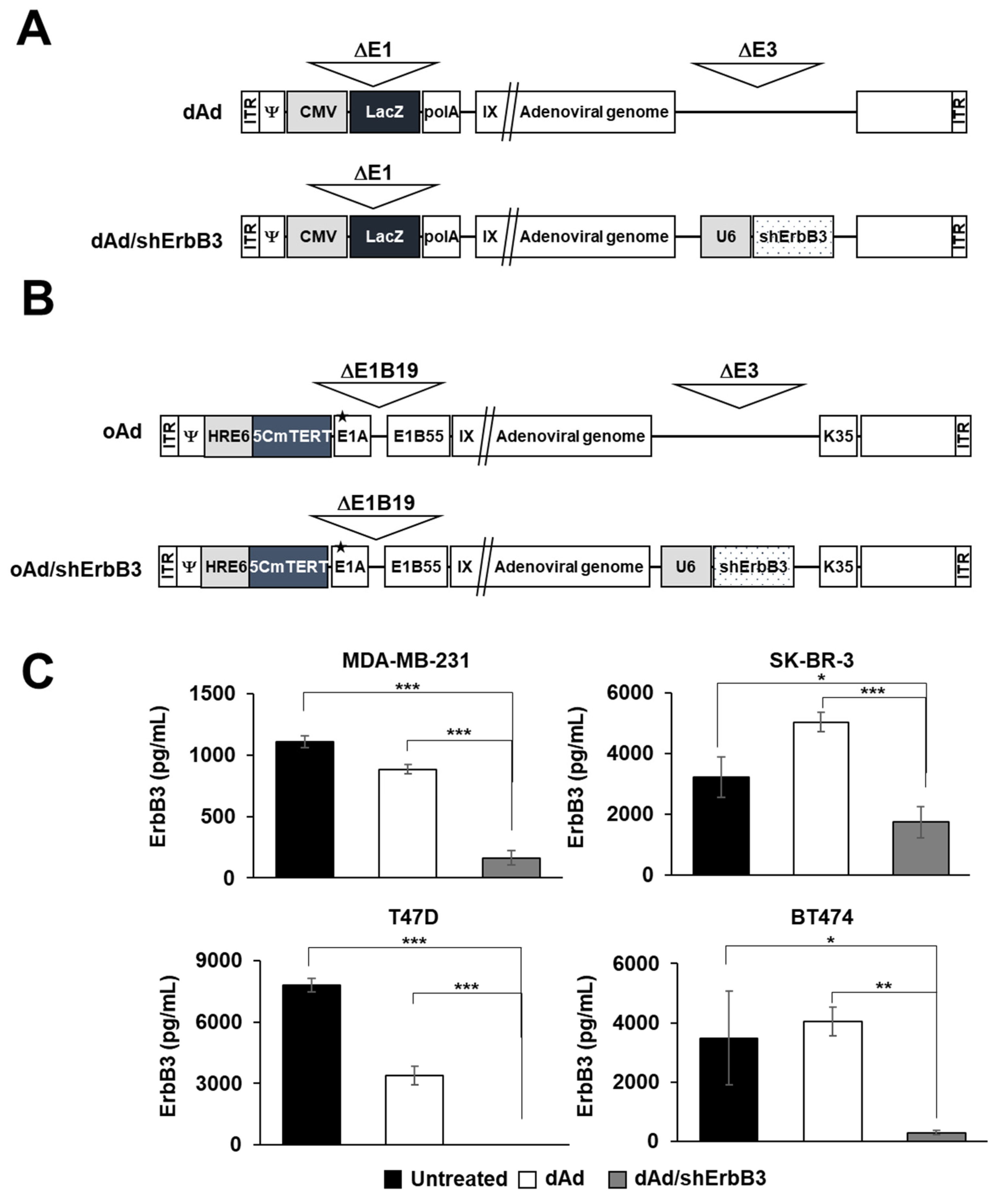
2.1. ErbB3 Silencing by Replication-Incompetent Adenoviral Vector, dAd/shErbB3
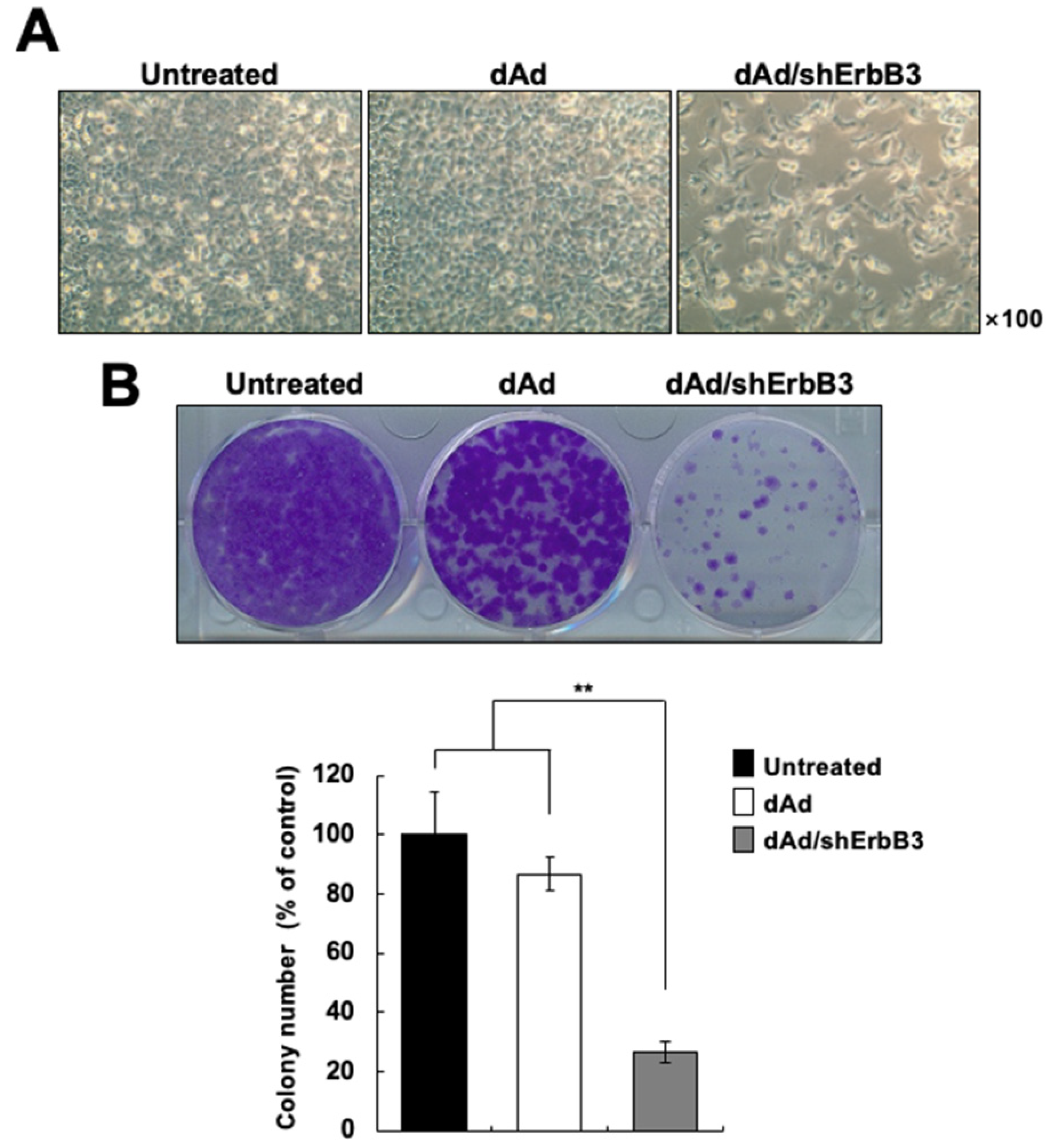
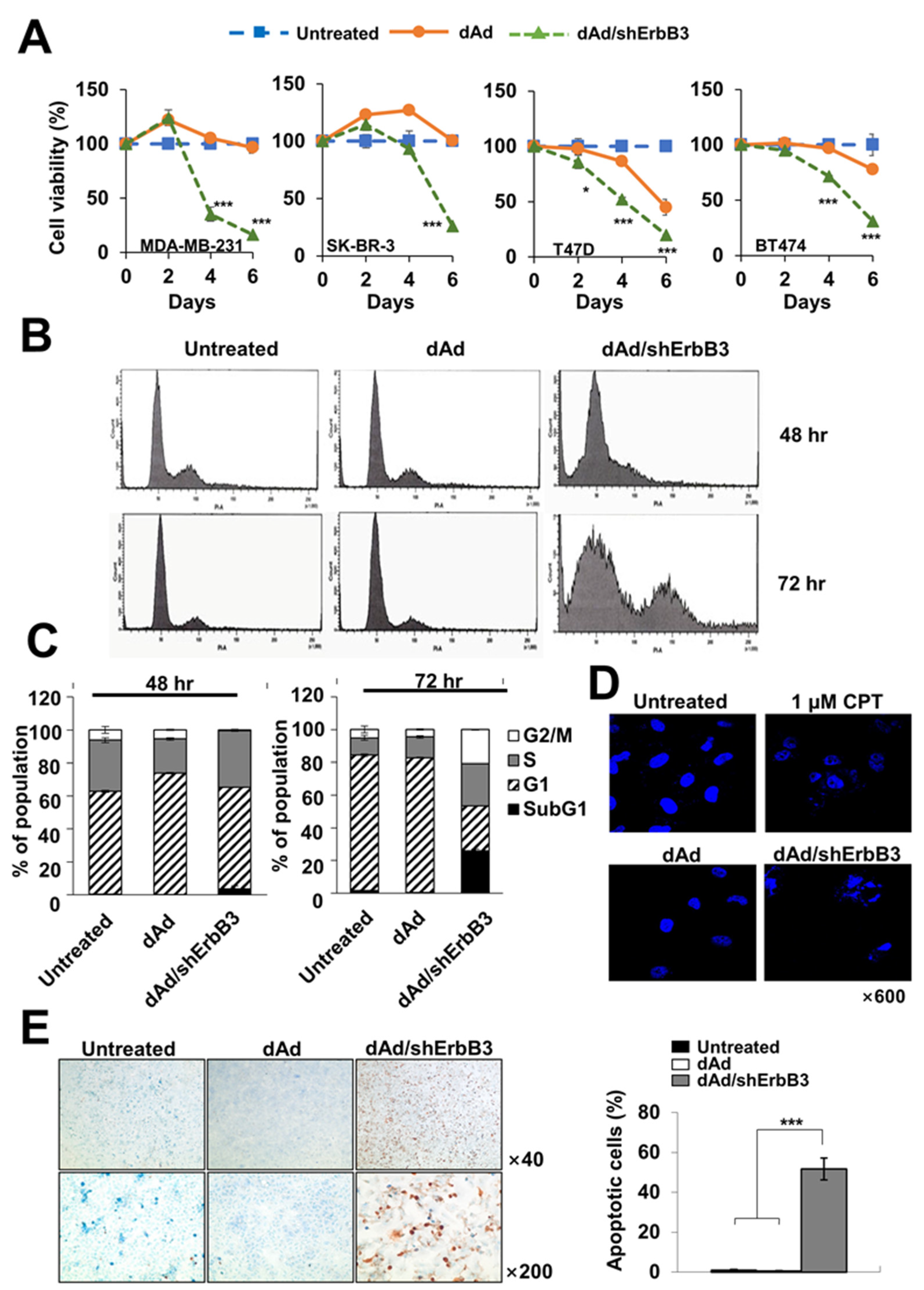
2.2. ErbB3 Silencing Caused Attenuation of Cell Proliferation
2.3. ErbB3 Silencing Caused Induction of Apoptosis
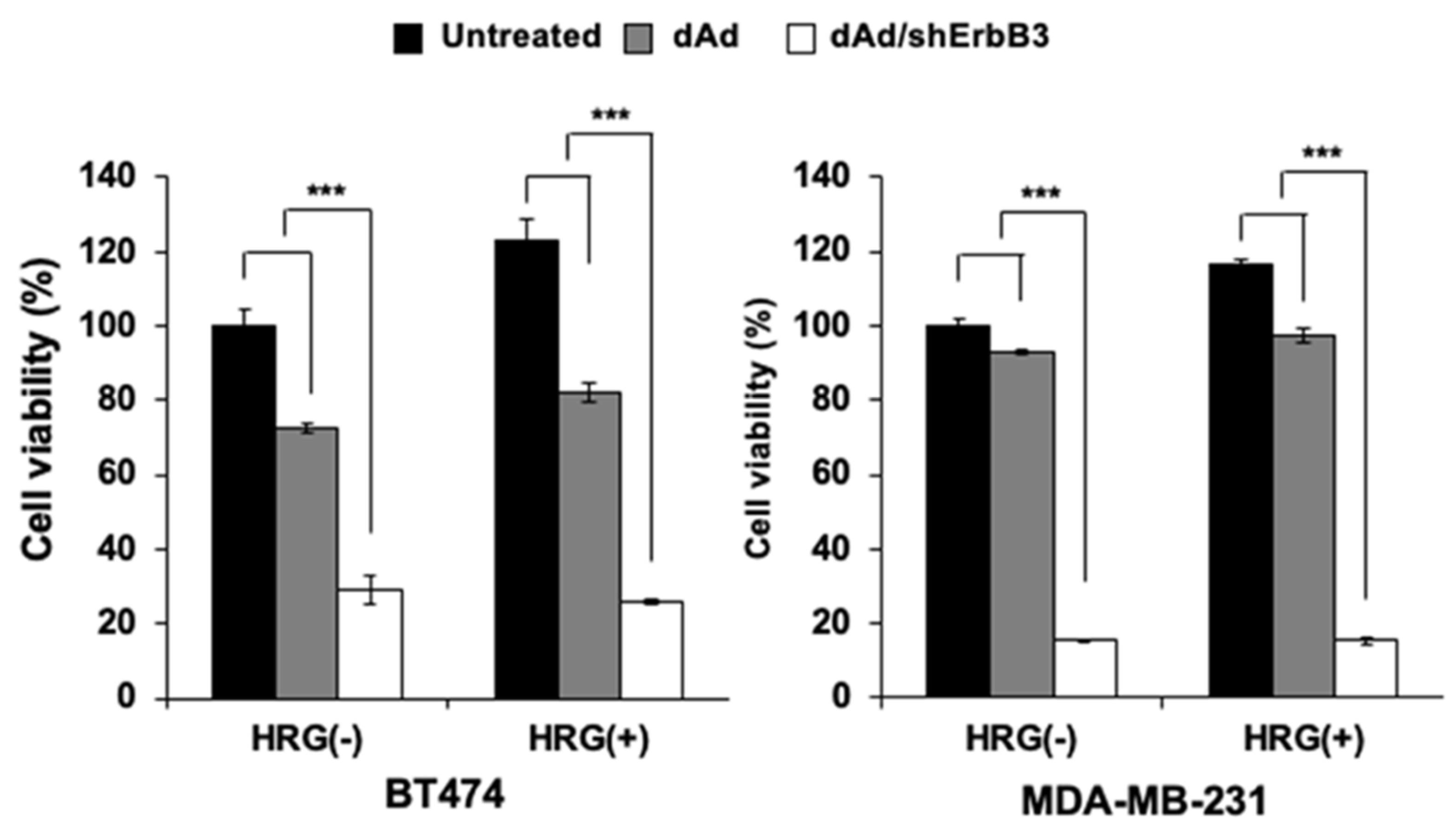
2.4. Inhibition of Heregulin-Dependent and -Independent Cell Proliferation
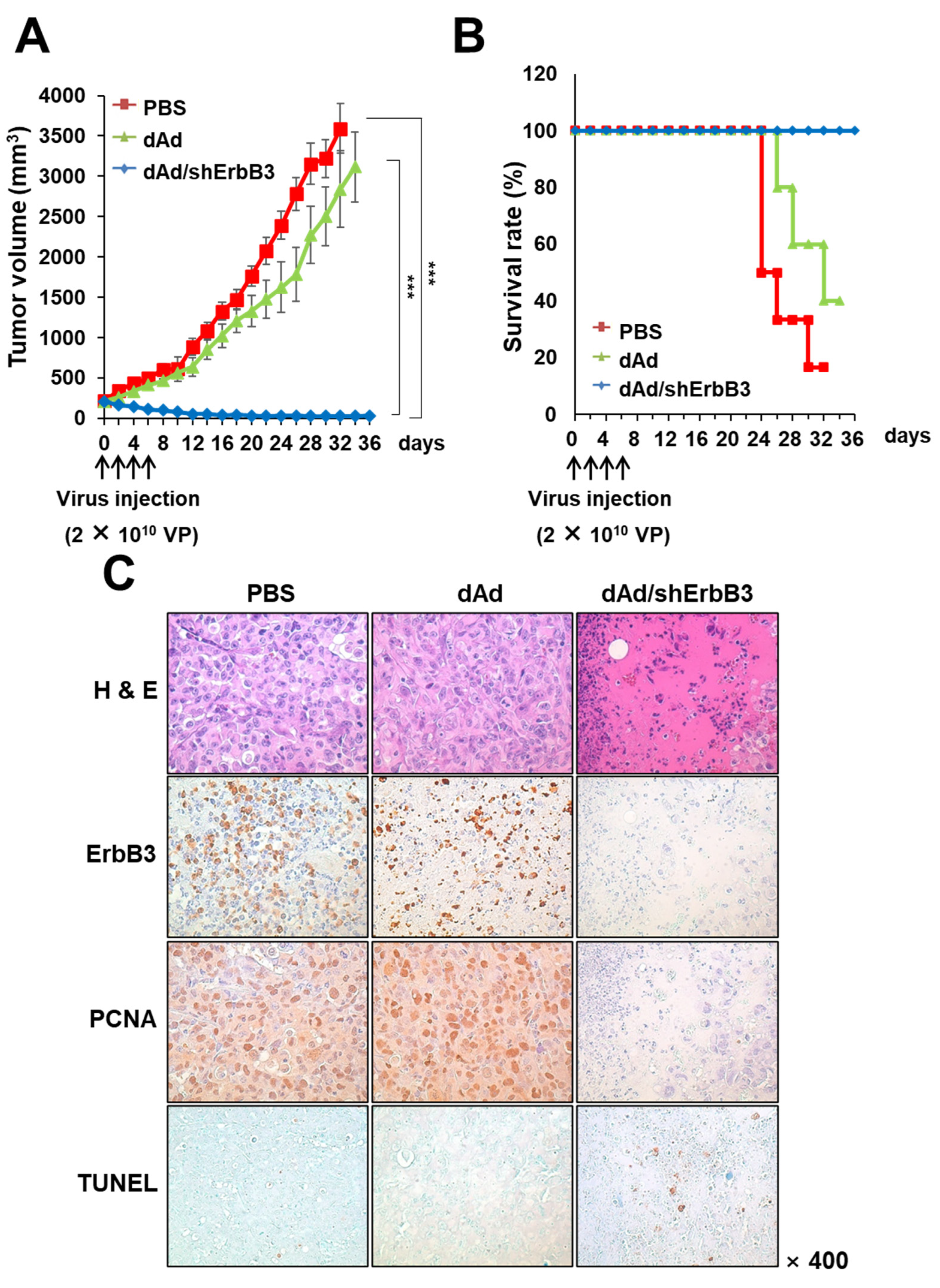
2.5. dAd/shErbB3 Caused Potent Antitumor Effect
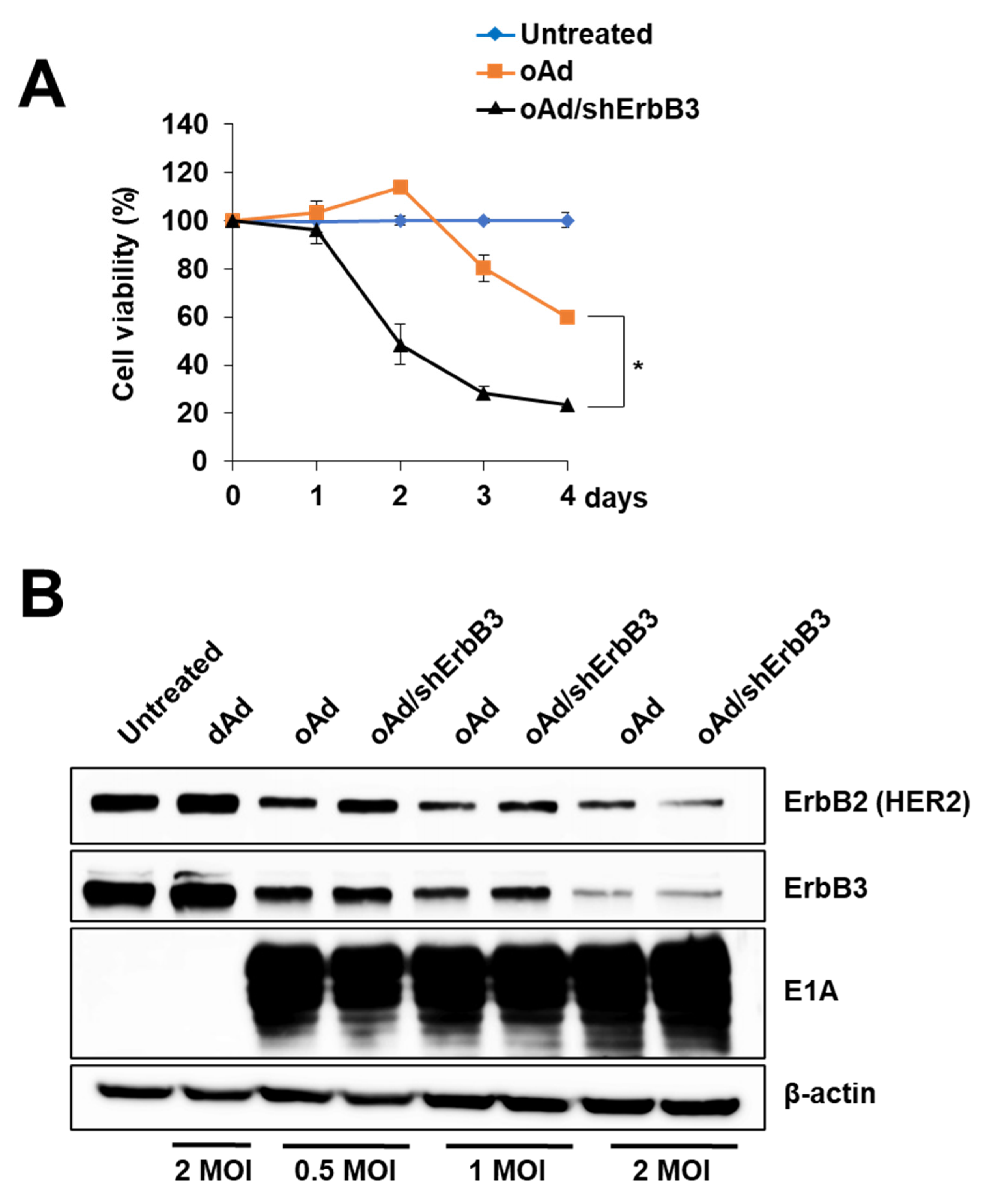
2.6. ErbB3 Silencing by Replication-Competent Oncolytic Ad, oAd/shErbB3
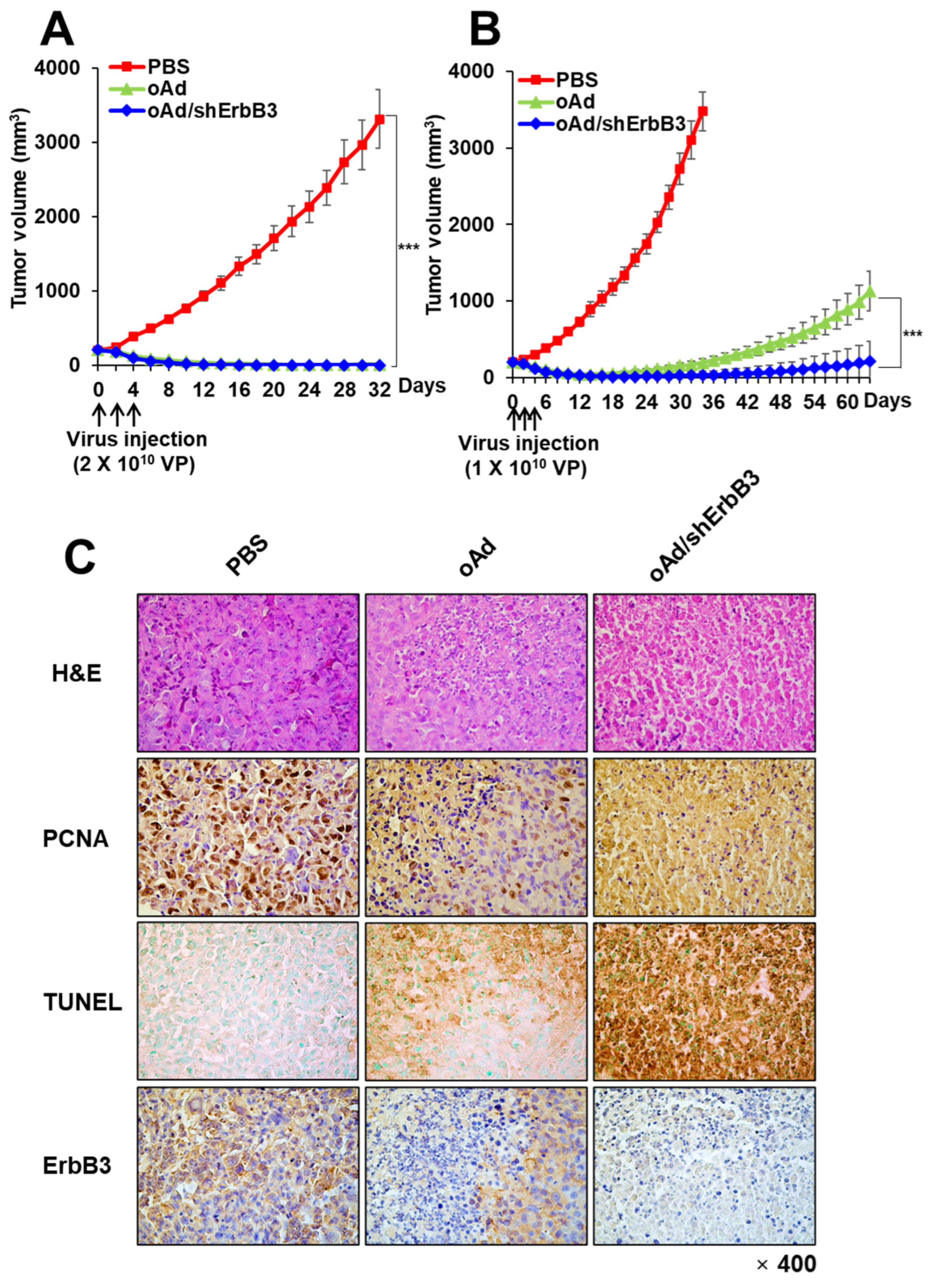
2.7. oAd/shErbB3 Showed Stronger Antitumor Activity
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Construction and Generation of an Ad-Expressing ErbB3-Specific shRNA
4.3. ErbB3 ELISA
4.4. Colony Formation Assay
4.5. MTT Assay
4.6. Cell Cycle Analysis
4.7. Hoechst Staining
4.8. Immunoblotting Analysis
4.9. The Antitumor Effect in a Human Breast Cancer Xenograft Model
4.10. Histology and Immunohistochemistry
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Yen, L.; Cao, Z.; Wu, X.; Ingalla, E.R.Q.; Baron, C.; Young, L.J.T.; Gregg, J.P.; Cardiff, R.D.; Borowsky, A.D.; Sweeney, C.; et al. Loss of Nrdp1 Enhances ErbB2/ErbB3–Dependent Breast Tumor Cell Growth. Cancer Res. 2006, 66, 11279–11286. [Google Scholar] [CrossRef] [PubMed]
- Sibilia, M.; Kroismayr, R.; Lichtenberger, B.M.; Natarajan, A.; Hecking, M.; Holcmann, M. The epidermal growth factor receptor: From development to tumorigenesis. Differentiation 2007, 75, 770–787. [Google Scholar] [CrossRef]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [PubMed]
- Rusnak, D.W.; Affleck, K.; Cockerill, S.G.; Stubberfield, C.; Harris, R.; Page, M.; Smith, K.J.; Guntrip, S.B.; Carter, M.C.; Shaw, R.J.; et al. The characterization of novel, dual ErbB-2/EGFR, tyrosine kinase inhibitors: Potential therapy for cancer. Cancer Res. 2001, 61, 7196–7203. [Google Scholar]
- Lopez, S.; Cocco, E.; Black, J.; Bellone, S.; Bonazzoli, E.; Predolini, F.; Ferrari, F.; Schwab, C.L.; English, D.P.; Ratner, E.; et al. Dual HER2/PIK3CA Targeting Overcomes Single-Agent Acquired Resistance in HER2-Amplified Uterine Serous Carcinoma Cell Lines In Vitro and In Vivo. Mol. Cancer Ther. 2015, 14, 2519–2526. [Google Scholar] [CrossRef]
- Clarke, R.; Leonessa, F.; Welch, J.N.; Skaar, T.C. Cellular and molecular pharmacology of antiestrogen action and resistance. Pharmacol. Rev. 2001, 53, 25–71. [Google Scholar]
- Stewart, E.L.; Tan, S.Z.; Liu, G.; Tsao, M.-S. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations—A review. Transl. Lung Cancer Res. 2015, 4, 67–81. [Google Scholar]
- Nahta, R.; Yu, D.; Hung, M.C.; Hortobagyi, G.N.; Esteva, F.J. Mechanisms of disease: Understanding resistance to HER2-targeted therapy in human breast cancer. Nat. Clin. Pract. Oncol. 2006, 3, 269–280. [Google Scholar] [CrossRef]
- Weickhardt, A.J.; Price, T.J.; Chong, G.; Gebski, V.; Pavlakis, N.; Johns, T.G.; Azad, A.; Skrinos, E.; Fluck, K.; Dobrovic, A.; et al. Dual Targeting of the Epidermal Growth Factor Receptor Using the Combination of Cetuximab and Erlotinib: Preclinical Evaluation and Results of the Phase II DUX Study in Chemotherapy-Refractory, Advanced Colorectal Cancer. J. Clin. Oncol. 2012, 30, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Wheler, J.J.; Tsimberidou, A.M.; Falchook, G.S.; Zinner, R.G.; Hong, D.S.; Fok, J.Y.; Fu, S.; Piha-Paul, S.A.; Naing, A.; Kurzrock, R. Combining erlotinib and cetuximab is associated with activity in patients with non-small cell lung cancer (including squamous cell carcinomas) and wild-type EGFR or resistant mutations. Mol. Cancer Ther. 2013, 12, 2167–2175. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.M.; Li, H.; Yu, J.P.; Ren, X.B.; Wang, C.L. Combined Erlotinib and Cetuximab overcome the acquired resistance to epidermal growth factor receptors tyrosine kinase inhibitor in non-small-cell lung cancer. J. Cancer Res. Clin. Oncol. 2012, 138, 2069–2077. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.E.; Lee, S.; McDowell, A.L.; Erskine, M.; Loh, Q.T.M.; Grice, O.; Argyle, D.J.; Bergkvist, G.T. Dual targeting of EGFR and ERBB2 pathways produces a synergistic effect on cancer cell proliferation and migration in vitro. Vet. Comp. Oncol. 2017, 15, 890–909. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Huang, S.; Li, C.; Armstrong, E.A.; Peet, C.R.; Saker, J.; Amler, L.C.; Sliwkowski, M.X.; Harari, P.M. Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res. 2013, 73, 824–833. [Google Scholar] [CrossRef]
- Wang, Y.C.; Morrison, G.; Gillihan, R.; Guo, J.; Ward, R.M.; Fu, X.; Botero, M.F.; Healy, N.A.; Hilsenbeck, S.G.; Phillips, G.L.; et al. Different mechanisms for resistance to trastuzumab versus lapatinib in HER2-positive breast cancers--role of estrogen receptor and HER2 reactivation. Breast Cancer Res. 2011, 13, R121. [Google Scholar] [CrossRef]
- D’Amato, V.; Raimondo, L.; Formisano, L.; Giuliano, M.; De Placido, S.; Rosa, R.; Bianco, R. Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treat. Rev. 2015, 41, 877–883. [Google Scholar] [CrossRef]
- Shi, H.; Zhang, W.; Zhi, Q.; Jiang, M. Lapatinib resistance in HER2+ cancers: Latest findings and new concepts on molecular mechanisms. Tumour Biol. 2016, 37, 15411–15431. [Google Scholar] [CrossRef]
- Sun, Z.; Shi, Y.; Shen, Y.; Cao, L.; Zhang, W.; Guan, X. Analysis of different HER-2 mutations in breast cancer progression and drug resistance. J. Cell. Mol. Med. 2015, 19, 2691–2701. [Google Scholar] [CrossRef]
- Holbro, T.; Beerli, R.R.; Maurer, F.; Koziczak, M.; Barbas, C.F., 3rd; Hynes, N.E. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2003, 100, 8933–8938. [Google Scholar] [CrossRef] [PubMed]
- van Lengerich, B.; Agnew, C.; Puchner, E.M.; Huang, B.; Jura, N. EGF and NRG induce phosphorylation of HER3/ERBB3 by EGFR using distinct oligomeric mechanisms. Proc. Natl. Acad. Sci. USA 2017, 114, E2836–E2845. [Google Scholar] [CrossRef] [PubMed]
- Amin, D.N.; Sergina, N.; Ahuja, D.; McMahon, M.; Blair, J.A.; Wang, D.; Hann, B.; Koch, K.M.; Shokat, K.M.; Moasser, M.M. Resiliency and vulnerability in the HER2-HER3 tumorigenic driver. Sci. Transl. Med. 2010, 2, 16ra7. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR family: Not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar] [CrossRef]
- Fraguas, S.; Barberan, S.; Cebria, F. EGFR signaling regulates cell proliferation, differentiation and morphogenesis during planarian regeneration and homeostasis. Dev. Biol. 2011, 354, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Way, T.D.; Lin, J.K. Role of HER2/HER3 co-receptor in breast carcinogenesis. Future Oncol. 2005, 1, 841–849. [Google Scholar] [CrossRef]
- Perez-Nadales, E.; Lloyd, A.C. Essential function for ErbB3 in breast cancer proliferation. Breast Cancer Res. 2004, 6, 137–139. [Google Scholar] [CrossRef]
- van der Horst, E.H.; Murgia, M.; Treder, M.; Ullrich, A. Anti-HER-3 MAbs inhibit HER-3-mediated signaling in breast cancer cell lines resistant to anti-HER-2 antibodies. Int. J. Cancer 2005, 115, 519–527. [Google Scholar] [CrossRef]
- Sithanandam, G.; Fornwald, L.W.; Fields, J.; Anderson, L.M. Inactivation of ErbB3 by siRNA promotes apoptosis and attenuates growth and invasiveness of human lung adenocarcinoma cell line A549. Oncogene 2005, 24, 1847–1859. [Google Scholar] [CrossRef]
- Izquierdo, M. Short interfering RNAs as a tool for cancer gene therapy. Cancer Gene Ther. 2005, 12, 217. [Google Scholar] [CrossRef]
- Kanasty, R.; Dorkin, J.R.; Vegas, A.; Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, J.-H.; Shin, H.-Y.; Lee, H.; Yang, J.M.; Kim, J.; Sohn, J.-H.; Kim, H.; Yun, C.-O. Ad-mTERT-Δ19, a conditional replication-competent adenovirus driven by the human telomerase promoter, selectively replicates in and elicits cytopathic effect in a cancer cell-specific manner. Hum. Gene Ther. 2003, 14, 1415–1428. [Google Scholar] [CrossRef] [PubMed]
- Kasala, D.; Choi, J.W.; Kim, S.W.; Yun, C.O. Utilizing adenovirus vectors for gene delivery in cancer. Expert Opin. Drug Deliv. 2014, 11, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cho, J.Y.; Joo-Hang, K.; Jung, K.C.; Yun, C.-O. Evaluation of E1B gene-attenuated replicating adenoviruses for cancer gene therapy. Cancer Gene Ther. 2002, 9, 725. [Google Scholar] [CrossRef][Green Version]
- Yoo, J.Y.; Kim, J.H.; Kim, J.; Huang, J.H.; Zhang, S.N.; Kang, Y.A.; Kim, H.; Yun, C.O. Short hairpin RNA-expressing oncolytic adenovirus-mediated inhibition of IL-8: Effects on antiangiogenesis and tumor growth inhibition. Gene Ther. 2008, 15, 635–651. [Google Scholar] [CrossRef]
- Lee, J.-S.; Oh, E.; Yoo, J.Y.; Choi, K.S.; Yoon, M.J.; Yun, C.-O. Adenovirus expressing dual c-Met-specific shRNA exhibits potent antitumor effect through autophagic cell death accompanied by senescence-like phenotypes in glioblastoma cells. Oncotarget 2015, 6, 4051–4065. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Kim, J.H.; Kwon, Y.G.; Kim, E.C.; Kim, N.K.; Choi, H.J.; Yun, C.O. VEGF-specific short hairpin RNA-expressing oncolytic adenovirus elicits potent inhibition of angiogenesis and tumor growth. Mol. Ther. 2007, 15, 295–302. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef]
- Liu, B.; Ordonez-Ercan, D.; Fan, Z.; Edgerton, S.M.; Yang, X.; Thor, A.D. Downregulation of erbB3 abrogates erbB2-mediated tamoxifen resistance in breast cancer cells. Int. J. Cancer 2007, 120, 1874–1882. [Google Scholar] [CrossRef]
- Falls, D.L. Neuregulins: Functions, forms, and signaling strategies. Exp. Cell Res. 2003, 284, 14–30. [Google Scholar] [CrossRef]
- Jeong, H.; Kim, J.; Lee, Y.; Seo, J.H.; Hong, S.R.; Kim, A. Neuregulin-1 induces cancer stem cell characteristics in breast cancer cell lines. Oncol. Rep. 2014, 32, 1218–1224. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jia, S.-F.; Hung, M.-C.; Kleinerman, E.S. E1A Sensitizes HER2/neu-overexpressing Ewing’s Sarcoma Cells to Topoisomerase II-targeting Anticancer Drugs. Cancer Res. 2001, 61, 3394–3398. [Google Scholar] [PubMed]
- Ueno, N.T.; Bartholomeusz, C.; Xia, W.; Anklesaria, P.; Bruckheimer, E.M.; Mebel, E.; Paul, R.; Li, S.; Yo, G.H.; Huang, L.; et al. Systemic gene therapy in human xenograft tumor models by liposomal delivery of the E1A gene. Cancer Res. 2002, 62, 6712–6716. [Google Scholar] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Jebelli, A.; Baradaran, B.; Mosafer, J.; Baghbanzadeh, A.; Mokhtarzadeh, A.; Tayebi, L. Recent developments in targeting genes and pathways by RNAi-based approaches in colorectal cancer. Med. Res. Rev. 2021, 41, 395–434. [Google Scholar] [CrossRef]
- Wang, L.; Li, F.; Dang, L.; Liang, C.; Wang, C.; He, B.; Liu, J.; Li, D.; Wu, X.; Xu, X.; et al. In Vivo Delivery Systems for Therapeutic Genome Editing. Int. J. Mol. Sci. 2016, 17, 26. [Google Scholar] [CrossRef]
- Ildefonso, C.J.; Lewin, A.S. Adeno-Associated Virus Delivery of Viral Serpins for Ocular Diseases: Design and Validation. Methods Mol. Biol. 2018, 1826, 237–254. [Google Scholar]
- Hromic-Jahjefendic, A.; Lundstrom, K. Viral Vector-Based Melanoma Gene Therapy. Biomedicines 2020, 8, 60. [Google Scholar] [CrossRef]
- Wold, W.S.; Toth, K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef]
- Zamir, G.; Zeira, E.; Gelman, A.E.; Shaked, A.; Olthoff, K.M.; Eid, A.; Galun, E. Replication-deficient adenovirus induces host topoisomerase I activity: Implications for adenovirus-mediated gene expression. Mol. Ther. 2007, 15, 772–781. [Google Scholar] [CrossRef]
- Hemminki, O.; Dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.K.; Yun, C.O. Recent developments in oncolytic adenovirus-based immunotherapeutic agents for use against metastatic cancers. Cancer Gene Ther. 2013, 20, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.J.; Kim, J.H.; Lee, Y.S.; Kim, J.; Suh, B.S.; Kim, H.; Cho, S.; Sohn, J.H.; Kim, G.E.; Yun, C.O. Concurrent delivery of GM-CSF and B7-1 using an oncolytic adenovirus elicits potent antitumor effect. Gene Ther. 2006, 13, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Miska, J.; Young, J.S.; Rashidi, A.; Kane, J.R.; Panek, W.K.; Kanojia, D.; Han, Y.; Balyasnikova, I.V.; Lesniak, M.S. A Comparative Study of Replication-Incompetent and -Competent Adenoviral Therapy-Mediated Immune Response in a Murine Glioma Model. Mol. Ther. Oncolytics 2017, 5, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sun, S.; Liu, G.; Yan, B.; Niu, J. Nrdp1 inhibits metastasis of colorectal cancer cells by EGFR signaling-dependent MMP7 modulation. Tumour Biol. 2015, 36, 1129–1133. [Google Scholar] [CrossRef]
- Ding, X.; Tong, C.; Chen, R.; Wang, X.; Gao, D.; Zhu, L. Systematic molecular profiling of inhibitor response to the clinical missense mutations of ErbB family kinases in human gastric cancer. J. Mol. Graph. Model. 2020, 96, 107526. [Google Scholar] [CrossRef]
- Kiavue, N.; Cabel, L.; Melaabi, S.; Bataillon, G.; Callens, C.; Lerebours, F.; Pierga, J.Y.; Bidard, F.C. ERBB3 mutations in cancer: Biological aspects, prevalence and therapeutics. Oncogene 2020, 39, 487–502. [Google Scholar] [CrossRef]
- Claus, J.; Patel, G.; Autore, F.; Colomba, A.; Weitsman, G.; Soliman, T.N.; Roberts, S.; Zanetti-Domingues, L.C.; Hirsch, M.; Collu, F.; et al. Inhibitor-induced HER2-HER3 heterodimerisation promotes proliferation through a novel dimer interface. Elife 2018, 7, e32271. [Google Scholar] [CrossRef]
- Al-Akhrass, H.; Conway, J.R.W.; Poulsen, A.S.A.; Paatero, I.; Kaivola, J.; Padzik, A.; Andersen, O.M.; Ivaska, J. A feed-forward loop between SorLA and HER3 determines heregulin response and neratinib resistance. Oncogene 2021, 40, 1300–1317. [Google Scholar] [CrossRef]
- Hafeez, U.; Parslow, A.C.; Gan, H.K.; Scott, A.M. New insights into ErbB3 function and therapeutic targeting in cancer. Expert Rev. Anticancer Ther. 2020, 20, 1057–1074. [Google Scholar] [CrossRef]
- Dan, V.M.; Raveendran, R.S.; Baby, S. Resistance to Intervention: Paclitaxel in Breast Cancer. Mini Rev. Med. Chem. 2021, 21, 1237–1268. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.A.; Chang, Y.W.; Tseng, C.F.; Chiu, C.F.; Hong, C.C.; Wang, W.; Wang, M.Y.; Hsiao, M.; Ma, J.T.; Chen, C.H.; et al. E1A-mediated inhibition of HSPA5 suppresses cell migration and invasion in triple-negative breast cancer. Ann. Surg Oncol. 2015, 22, 889–898. [Google Scholar] [CrossRef]
- Chang, Y.W.; Hung, M.C.; Su, J.L. The anti-tumor activity of E1A and its implications in cancer therapy. Arch. Immunol. Ther. Exp. 2014, 62, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Kobayashi, N.; Nishikawa, M.; Takakura, Y. Sequence-specific suppression of mdr1a/1b expression in mice via RNA interference. Pharm. Res. 2005, 22, 2091–2098. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, Y.; Wang, J.; Wang, J.; Yang, J.; Zhai, Y.; Li, B. Dual silencing of Bcl-2 and Survivin by HSV-1 vector shows better antitumor efficacy in higher PKR phosphorylation tumor cells in vitro and in vivo. Cancer Gene Ther. 2015, 22, 380–386. [Google Scholar] [CrossRef]
- Brachtlova, T.; van Ginkel, J.W.; Luinenburg, M.J.; de Menezes, R.X.; Koppers-Lalic, D.; Pegtel, D.M.; Dong, W.; de Gruijl, T.D.; van Beusechem, V.W. Expression of Oncolytic Adenovirus-Encoded RNAi Molecules Is Most Effective in a pri-miRNA Precursor Format. Mol. Ther. Oncolytics 2020, 19, 332–343. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Ryu, J.; Gao, R.; Yaguchi, T.; Kaul, S.C.; Wadhwa, R.; Yun, C.O. Tumor suppression by apoptotic and anti-angiogenic effects of mortalin-targeting adeno-oncolytic virus. J. Gene Med. 2010, 12, 586–595. [Google Scholar] [CrossRef]
- Anesti, A.M.; Simpson, G.R.; Price, T.; Pandha, H.S.; Coffin, R.S. Expression of RNA interference triggers from an oncolytic herpes simplex virus results in specific silencing in tumour cells in vitro and tumours in vivo. BMC Cancer 2010, 10, 486. [Google Scholar] [CrossRef]
- Ryu, J.; Kaul, Z.; Yoon, A.R.; Liu, Y.; Yaguchi, T.; Na, Y.; Ahn, H.M.; Gao, R.; Choi, I.K.; Yun, C.O.; et al. Identification and functional characterization of nuclear mortalin in human carcinogenesis. J. Biol. Chem. 2014, 289, 24832–24844. [Google Scholar] [CrossRef]
- Yun, C.O.; Bhargava, P.; Na, Y.; Lee, J.S.; Ryu, J.; Kaul, S.C.; Wadhwa, R. Relevance of mortalin to cancer cell stemness and cancer therapy. Sci. Rep. 2017, 7, 42016. [Google Scholar] [CrossRef]
- Yun, C.O.; Kim, E.; Koo, T.; Kim, H.; Lee, Y.S.; Kim, J.H. ADP-overexpressing adenovirus elicits enhanced cytopathic effect by induction of apoptosis. Cancer Gene Ther. 2005, 12, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Hong, J.; Kwon, O.J.; Yun, C.O. A hypoxia- and telomerase-responsive oncolytic adenovirus expressing secretable trimeric TRAIL triggers tumour-specific apoptosis and promotes viral dispersion in TRAIL-resistant glioblastoma. Sci. Rep. 2018, 8, 1420. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.K.; Oh, E.; Hong, J.; Lee, Y.; Park, K.D.; Yun, C.O. A hydrogel matrix prolongs persistence and promotes specific localization of an oncolytic adenovirus in a tumor by restricting nonspecific shedding and an antiviral immune response. Biomaterials 2017, 147, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Chung, H.K.; Kang, J.H.; Kim, K.I.; Jeon, Y.H.; Jin, Y.N.; Yun, C.O.; Chung, J.K. Tumor-targeted radionuclide imaging and therapy based on human sodium iodide symporter gene driven by a modified telomerase reverse transcriptase promoter. Hum. Gene Ther. 2008, 19, 951–957. [Google Scholar] [CrossRef]
- Na, Y.; Choi, J.W.; Kasala, D.; Hong, J.; Oh, E.; Li, Y.; Jung, S.J.; Kim, S.W.; Yun, C.O. Potent antitumor effect of neurotensin receptor-targeted oncolytic adenovirus co-expressing decorin and Wnt antagonist in an orthotopic pancreatic tumor model. J. Control. Release 2015, 220, 766–782. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jung, B.-K.; Kim, Y.J.; Hong, J.; Chang, H.-G.; Yoon, A.-R.; Yun, C.-O. ErbB3-Targeting Oncolytic Adenovirus Causes Potent Tumor Suppression by Induction of Apoptosis in Cancer Cells. Int. J. Mol. Sci. 2022, 23, 7127. https://doi.org/10.3390/ijms23137127
Jung B-K, Kim YJ, Hong J, Chang H-G, Yoon A-R, Yun C-O. ErbB3-Targeting Oncolytic Adenovirus Causes Potent Tumor Suppression by Induction of Apoptosis in Cancer Cells. International Journal of Molecular Sciences. 2022; 23(13):7127. https://doi.org/10.3390/ijms23137127
Chicago/Turabian StyleJung, Bo-Kyeong, Young Jun Kim, JinWoo Hong, Han-Gyu Chang, A-Rum Yoon, and Chae-Ok Yun. 2022. "ErbB3-Targeting Oncolytic Adenovirus Causes Potent Tumor Suppression by Induction of Apoptosis in Cancer Cells" International Journal of Molecular Sciences 23, no. 13: 7127. https://doi.org/10.3390/ijms23137127
APA StyleJung, B.-K., Kim, Y. J., Hong, J., Chang, H.-G., Yoon, A.-R., & Yun, C.-O. (2022). ErbB3-Targeting Oncolytic Adenovirus Causes Potent Tumor Suppression by Induction of Apoptosis in Cancer Cells. International Journal of Molecular Sciences, 23(13), 7127. https://doi.org/10.3390/ijms23137127