
K+-Dependent Na+/Ca2+ Exchanger Isoform 2, Nckx2, Takes Part in the Neuroprotection Elicited by Ischemic Preconditioning in Brain Ischemia


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
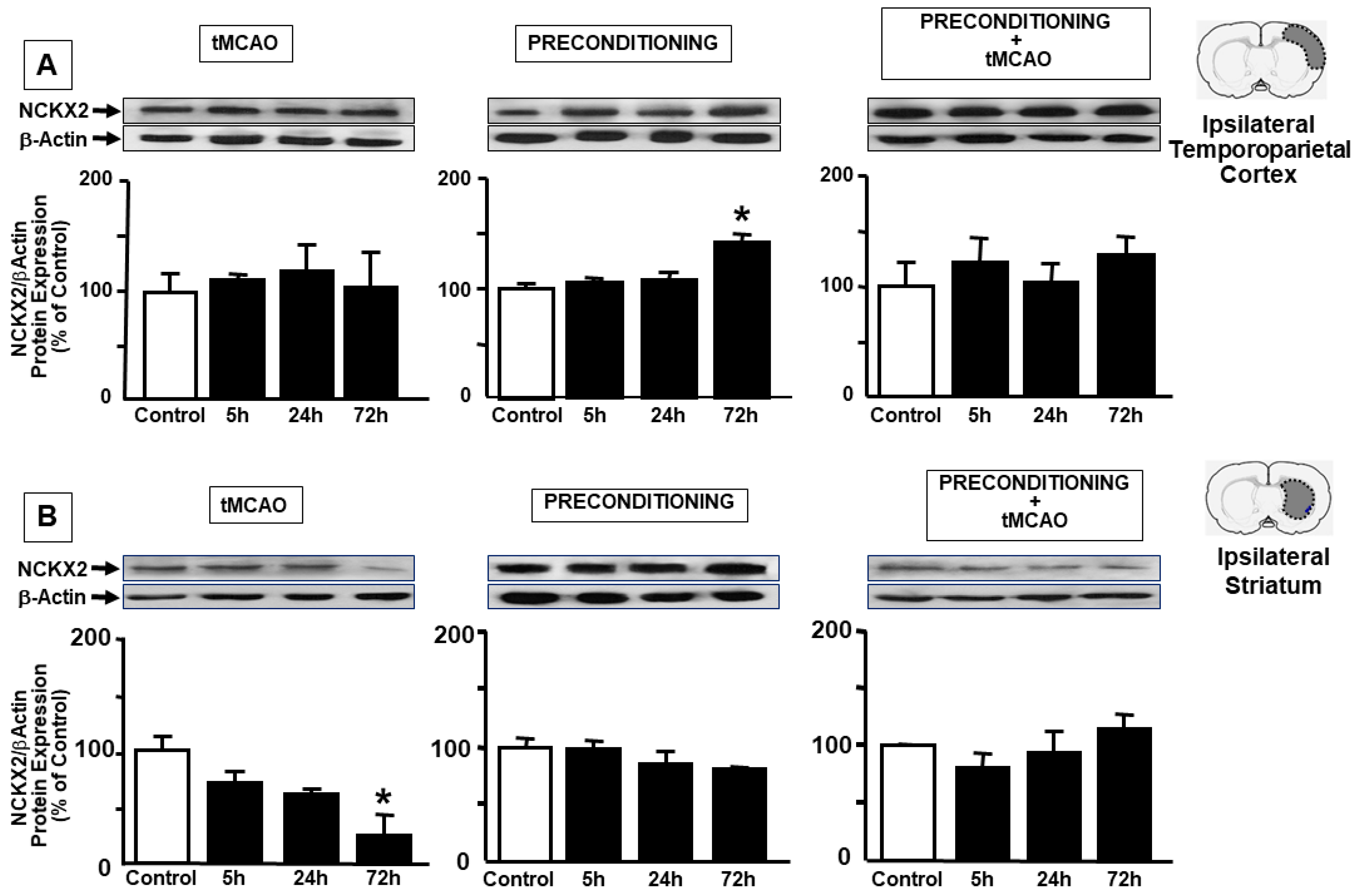
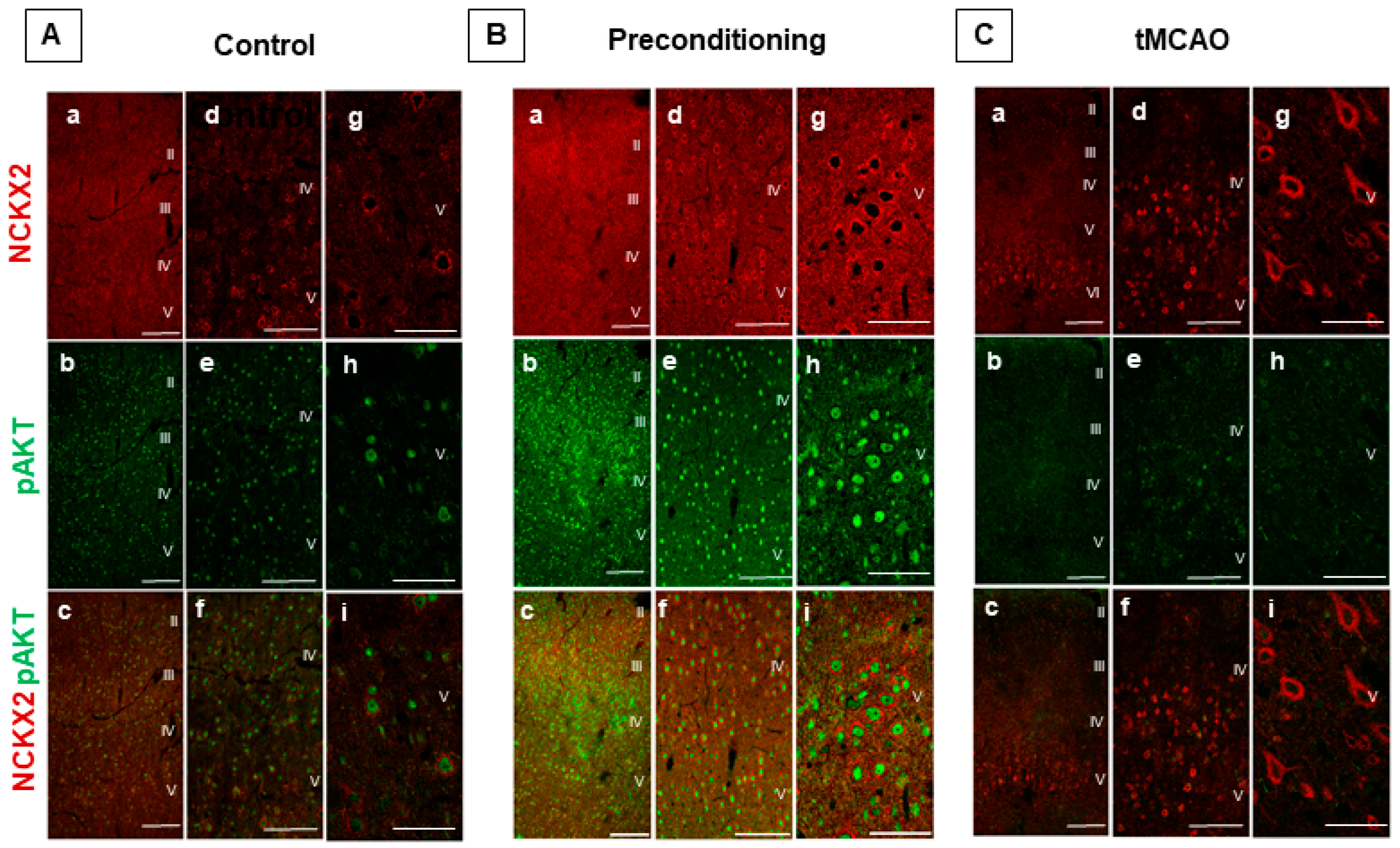
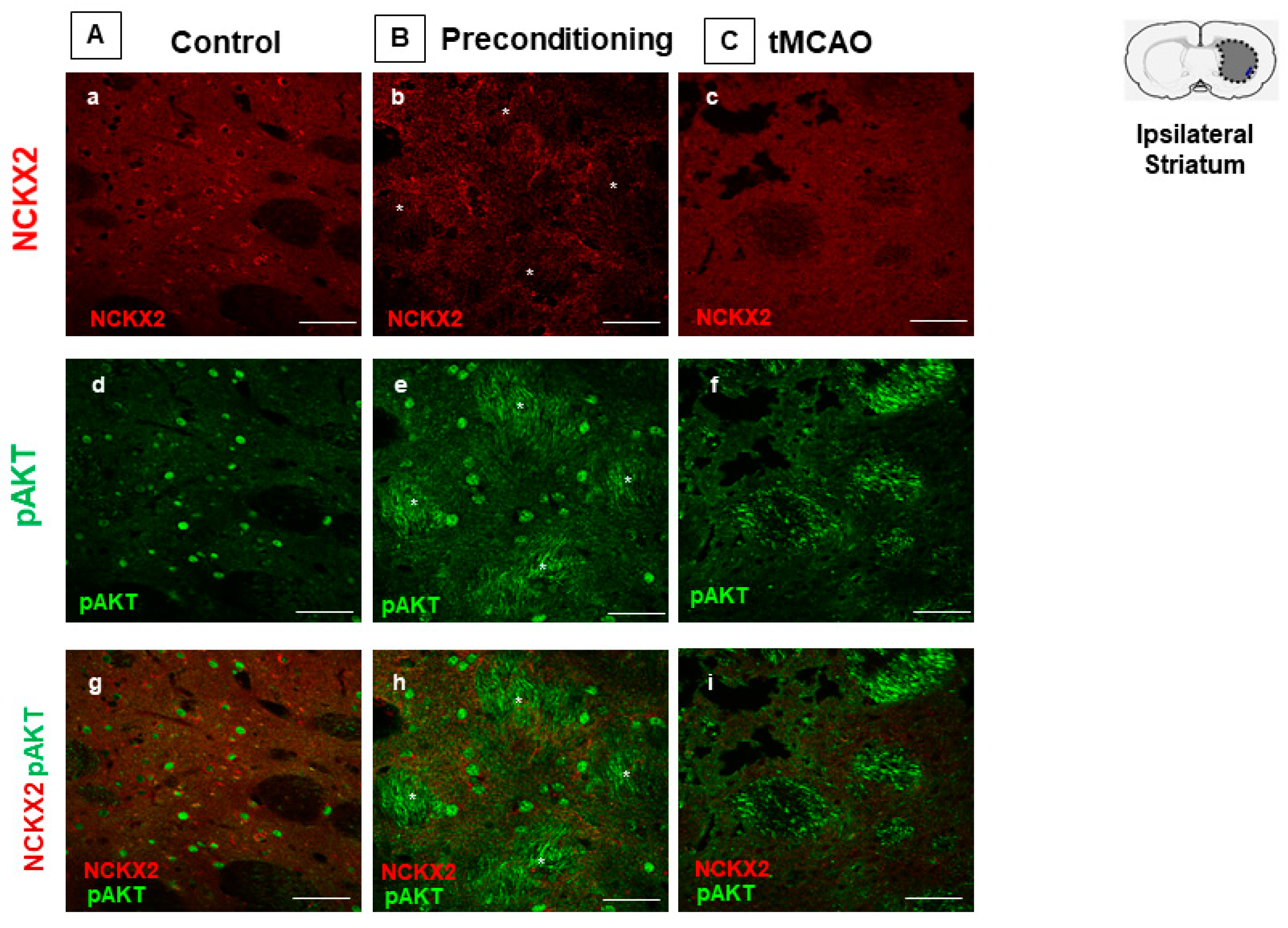
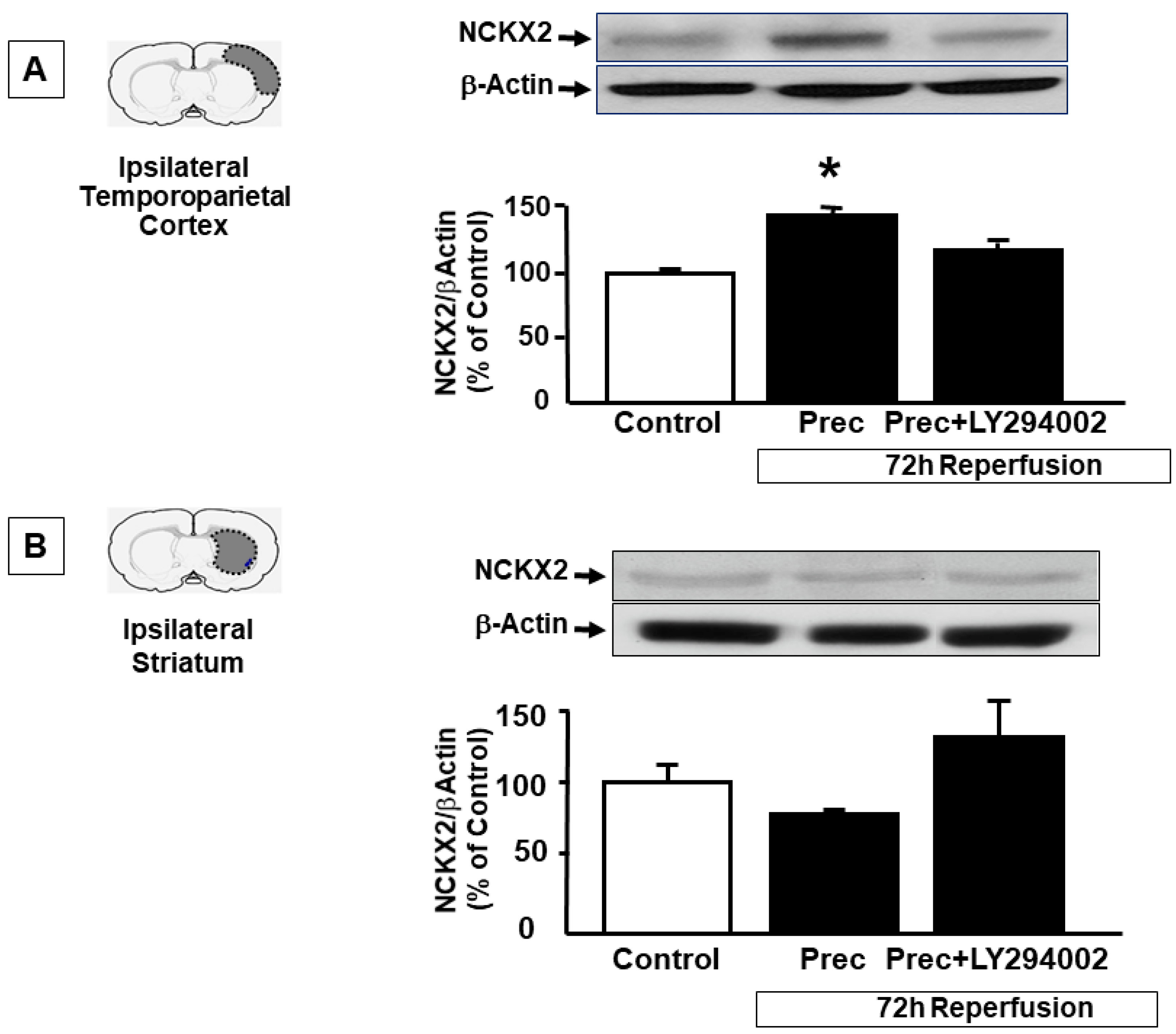
2.1. Ischemic Preconditioning Induces NCKX2 Overexpression in the Peri-Ischemic Temporoparietal Cortex and Prevents NCKX2 Downregulation in the Striatum of Ischemic Rats
2.2. p-AKT Inhibitor LY294002 Prevents NCKX2 Overexpression Induced by Preconditioning in the Peri-Ischemic Temporoparietal Cortex
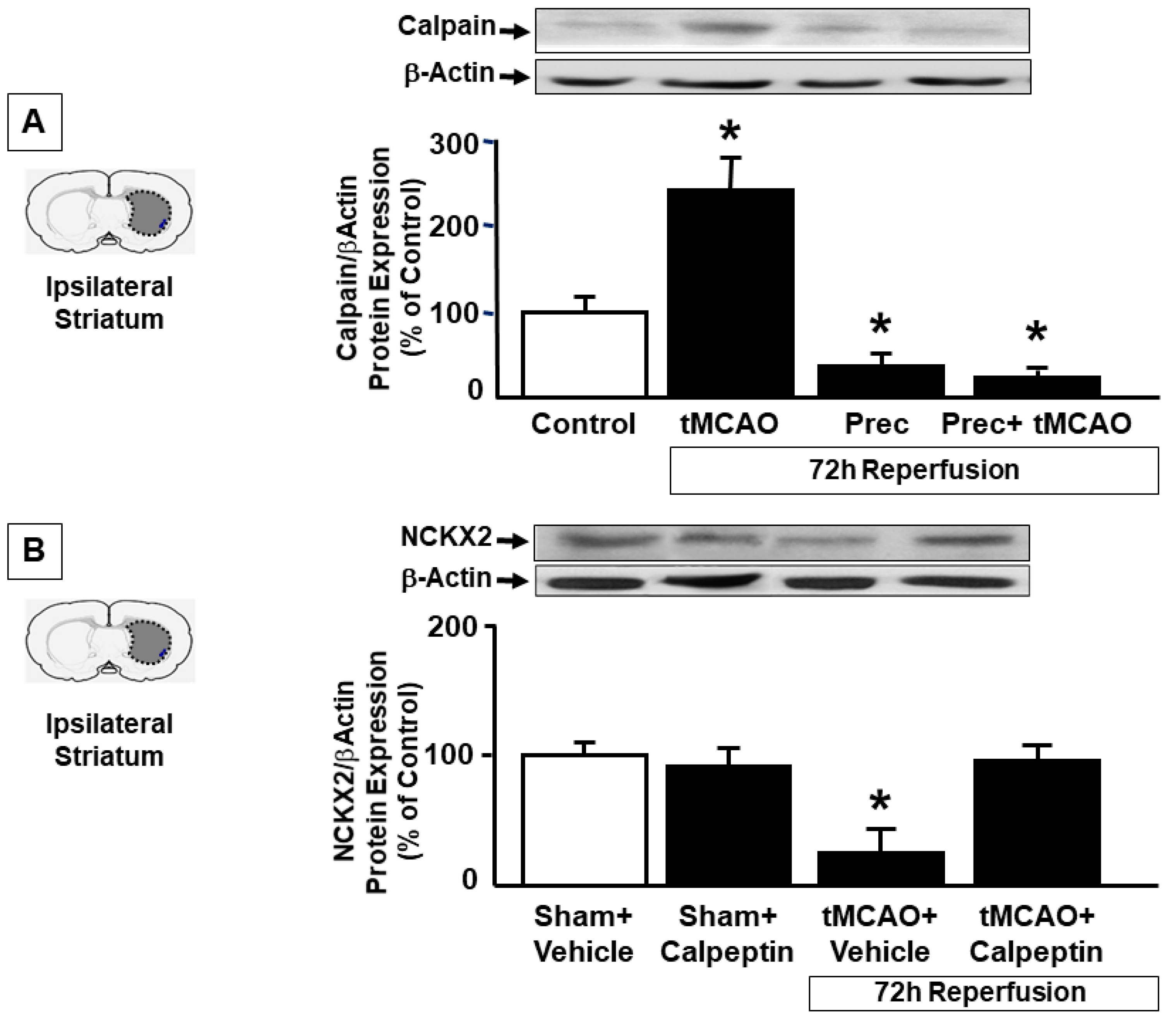
2.3. Preconditioning Prevents Calpain Activation Responsible for NCKX2 Downregulation in the Striatum of Ischemic Rats
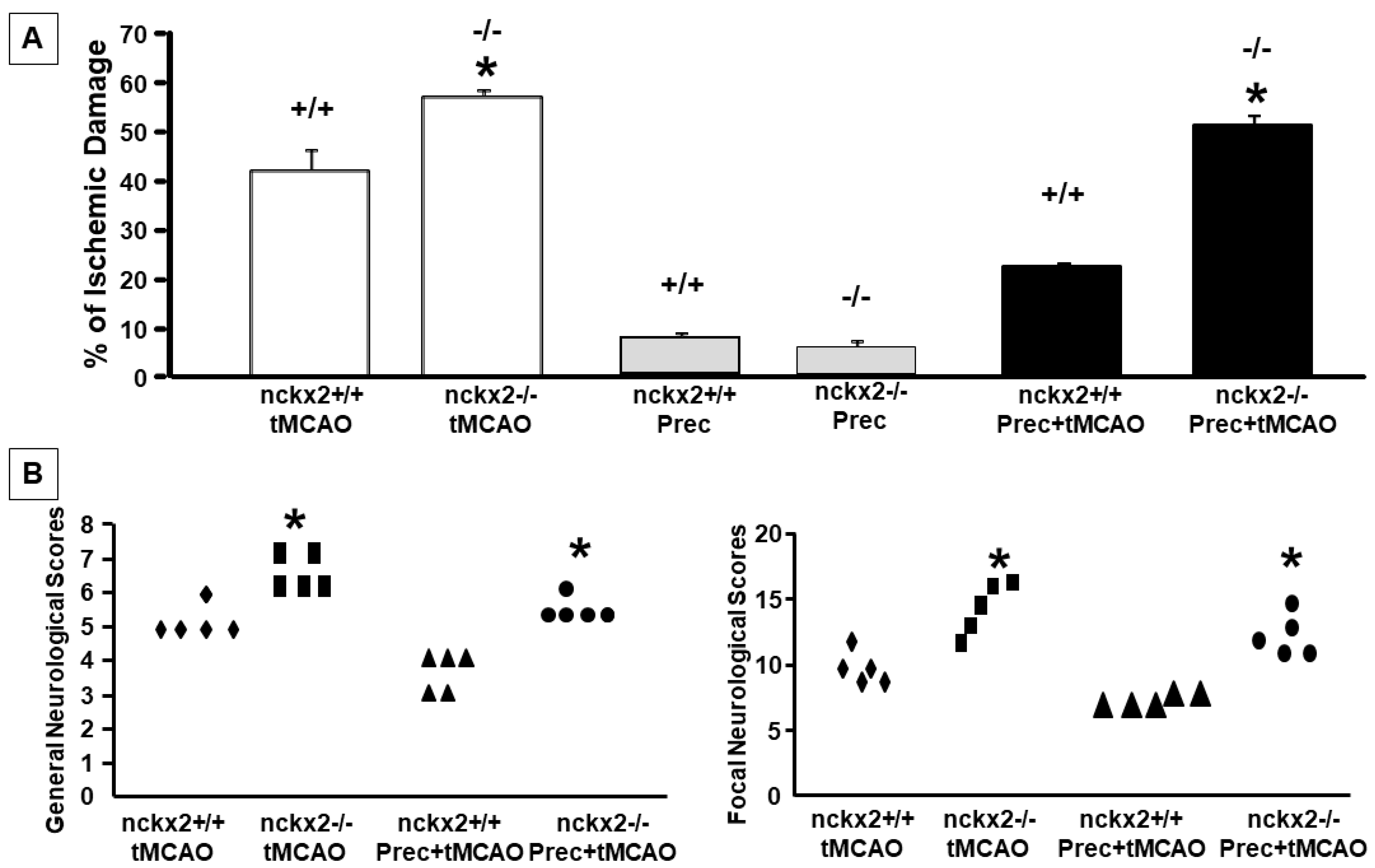
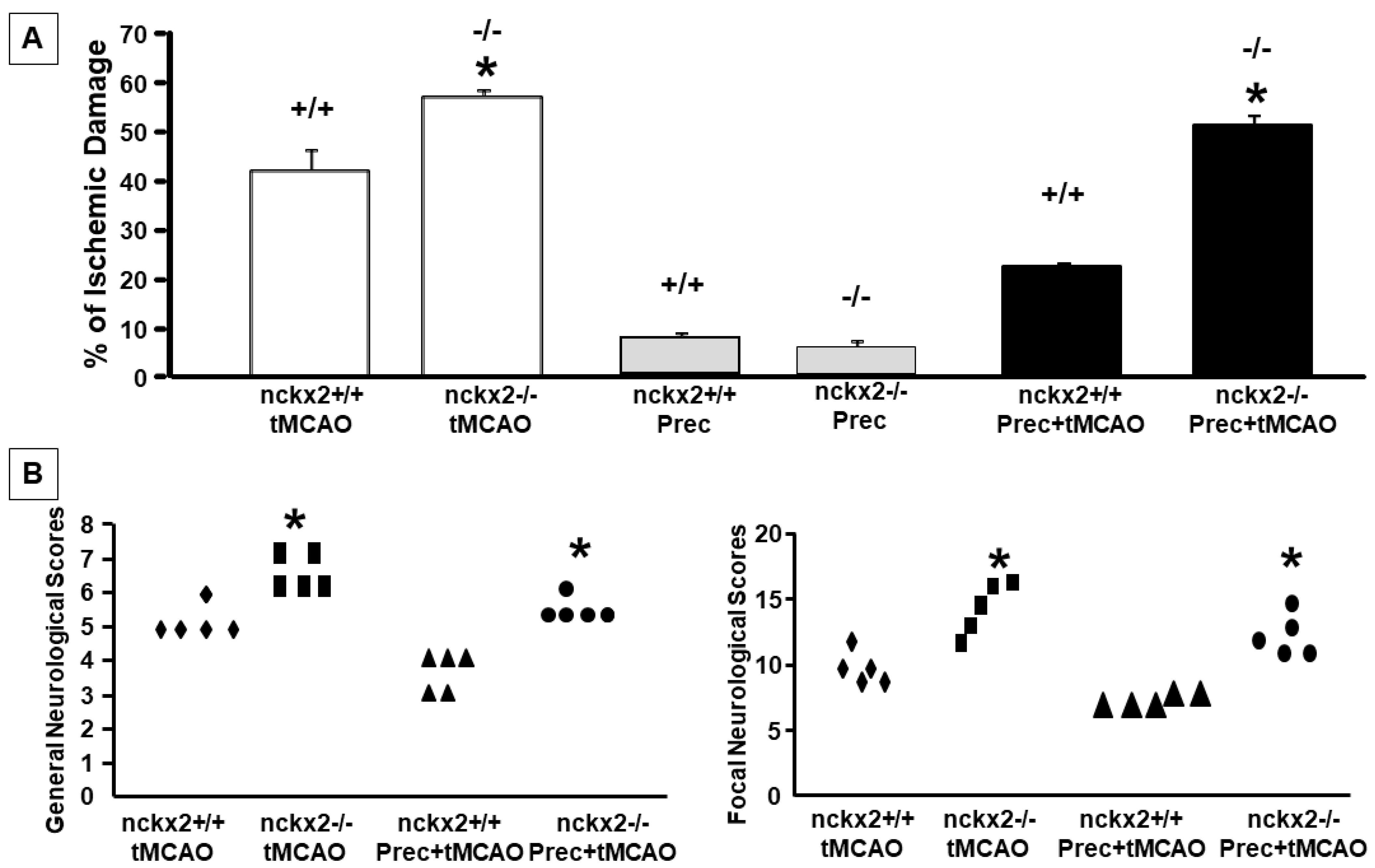
2.4. NCKX2 Knocking-out Prevents the Neuroprotective Effect on Brain Ischemia Induced by Ischemic Preconditioning
3. Discussion
4. Materials and Methods
4.1. Experimental Groups
4.2. Focal Ischemia
4.3. Preconditioning Experimental Protocol
4.4. Evaluation of the Infarct Volume
4.5. Western Blotting Analysis
4.6. Immunocytochemistry
4.7. p-AKT and Calpain Inhibition
4.8. Statistic Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gladstone, D.J.; Black, S.E.; Hakim, A.M. Toward wisdom from failure: Lessons from neuroprotective stroke trials and new therapeutic directions. Stroke 2002, 33, 2123–2136. [Google Scholar] [CrossRef]
- Dirnagl, U.; Simon, R.P.; Hallenbeck, J.M. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003, 26, 248–254. [Google Scholar] [CrossRef]
- Gidday, J.M. Cerebral preconditioning and ischaemic tolerance. Nat. Rev. Neurosci. 2006, 7, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Kirino, T. Ischemic tolerance. J. Cereb. Blood Flow Metab. 2002, 22, 1283–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignataro, G.; Meller, R.; Inoue, K.; Ordonez, A.N.; Ashley, M.D.; Xiong, Z.; Gala, R.; Simon, R.P. In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: Ischemic postconditioning. J. Cereb. Blood Flow Metab. 2008, 28, 232–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H. Ischemic postconditioning as a novel avenue to protect against brain injury after stroke. J. Cereb. Blood Flow Metab. 2009, 29, 873–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignataro, G.; Cuomo, O.; Vinciguerra, A.; Sirabella, R.; Esposito, E.; Boscia, F.; Di Renzo, G.; Annunziato, L. NCX as a key player in the neuroprotection exerted by ischemic preconditioning and postconditioning. Adv. Exp. Med. Biol. 2013, 961, 223–240. [Google Scholar]
- Cuomo, O.; Vinciguerra, A.; Cerullo, P.; Anzilotti, S.; Brancaccio, P.; Bilo, L.; Scorziello, A.; Molinaro, P.; Di Renzo, G.; Pignataro, G. Ionic homeostasis in brain conditioning. Front. Neurosci. 2015, 9, 277. [Google Scholar] [CrossRef] [Green Version]
- Pignataro, G.; Esposito, E.; Cuomo, O.; Sirabella, R.; Boscia, F.; Guida, N.; Di Renzo, G.F.; Annunziato, L. The NCX3 isoform of the Na+/Ca2+ exchanger contributes to neuroprotection elicited by ischemic postconditioning. J. Cereb. Blood Flow Metab. 2011, 31, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Pignataro, G.; Boscia, F.; Esposito, E.; Sirabella, R.; Cuomo, O.; Vinciguerra, A.; Di Renzo, G.F.; Annunziato, L. NCX1 and NCX3: Two new effectors of delayed preconditioning in brain ischemia. Neurobio. Dis. 2012, 45, 616–623. [Google Scholar] [CrossRef]
- Formisano, L.; Saggese, M.; Secondo, A.; Sirabella, R.; Vito, P.; Valsecchi, V.; Molinaro, P.; Di Renzo, G.; Annunziato, L. The two isoforms of the Na+/Ca2+ exchanger, NCX1 and NCX3, constitute novel additional targets for the prosurvival action of Akt/protein kinase B pathway. Mol. Pharm. 2008, 73, 727–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuomo, O.; Gala, R.; Pignataro, G.; Boscia, F.; Secondo, A.; Scorziello, A.; Pannaccione, A.; Viggiano, D.; Adornetto, A.; Molinaro, P.; et al. A Critical Role for the Potassium-Dependent Sodium/Calcium Exchanger NCKX2 in Protection Against Focal Ischemic Brain Damage. J. Neurosci. 2008, 28, 2053–2063. [Google Scholar] [CrossRef]
- Mies, G.; Ishimaru, S.; Xie, Y.; Seo, K.; Hossmann, K.A. Ischemic Thresholds of Cerebral Protein Synthesis and Energy State Following Middle Cerebral Artery Occlusion in Rat. J. Cereb. Blood Flow Metab. 1991, 11, 753–761. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Kim, M.H.; Park, K.H.; Earm, Y.E.; Ho, W.K. K+-dependent Na+/Ca2+ exchange is a major Ca2+ clearance mechanism in axon terminals of rat neurohypophysis. J. Neurosci. 2002, 22, 6891–6899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visser, F.; Valsecchi, V.; Annunziato, L.; Lytton, J. Exchangers NCKX2, NCKX3, and NCKX4: Identification of Thr-551 as a key residue in defining the apparent K+ affinity of NCKX2. J. Biol. Chem. 2007, 282, 4453–4462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gidö, G.; Kristián, T.; Siesjö, B.K. Extracellular potassium in a neocortical core area after transient focal ischemia. Stroke 1997, 28, 206–210. [Google Scholar] [CrossRef]
- Guida, N.; Laudati, G.; Mascolo, L.; Cuomo, O.; Anzilotti, S.; Sirabella, R.; Santopaolo, M.; Galgani, M.; Montuori, P.; Renzo, G.F.; et al. MC1568 inhibits thimerosal-induced apoptotic cell death by preventing HDAC4 up-regulation in neuronal cells and in rat prefrontal cortex. Toxicol. Sci. 2016, 154, 227–240. [Google Scholar] [CrossRef] [Green Version]
- Pignataro, G.; Ziaco, B.; Tortiglione, A.; Gala, R.; Cuomo, O.; Vinciguerra, A.; Lapi, D.; Mastantuono, T.; Anzilotti, S.; D’Andrea, L.D.; et al. Cataldi, L Neuroprotective Effect of VEGF-Mimetic Peptide QK in Experimental Brain Ischemia Induced in Rat by Middle Cerebral Artery Occlusion ACS. Chem. Neurosci. 2015, 16, 1517–1525. [Google Scholar] [CrossRef]
- Valsecchi, V.; Pignataro, G.; Del Prete, A.; Sirabella, R.; Matrone, C.; Boscia, F.; Scorziello, A.; Sisalli, M.J.; Esposito, R.; Zambrano, N.; et al. Annunziato L NCX1 is a novel target gene for hypoxia-inducible factor-1 in ischemic brain preconditioning. Stroke 2011, 42, 754–763. [Google Scholar] [CrossRef] [Green Version]
- Bederson, J.B.; Pitts, L.H.; Germano, S.M.; Nishimura, M.C.; Davis, R.L.; Bartkowski, H.M. Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke 1986, 17, 1304–1308. [Google Scholar] [CrossRef] [Green Version]
- Pignataro, G.; Gala, R.; Cuomo, O.; Tortiglione, A.; Giaccio, L.; Castaldo, P.; Sirabella, R.; Matrone, C.; Canitano, A.; Amoroso, S.; et al. Two Sodium/Calcium Exchanger Gene Products, NCX1 and NCX3, Play a Major Role in the Development of Permanent Focal Cerebral Ischemia. Stroke 2004, 35, 2566–2570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiolino, M.; Castaldo, P.; Lariccia, V.; Piccirillo, S.; Amoroso, S.; Magi, S. Essential role of the Na+-Ca2+ exchanger (NCX) in glutamate-enhanced cell survival in cardiac cells exposed to hypoxia/reoxygenation. Sci. Rep. 2017, 7, 13073. [Google Scholar] [CrossRef] [PubMed]
- Boscia, F.; Gala, R.; Pannaccione, A.; Secondo, A.; Scorziello, A.; Di Renzo, G.; Annunziato, L. NCX1 expression and functional activity increase in microglia invading the infarct core. Stroke 2009, 40, 3608–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Academic Press: New York, NY, USA, 1997. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuomo, O.; Sirabella, R.; Boscia, F.; Casamassa, A.; Lytton, J.; Annunziato, L.; Pignataro, G. K+-Dependent Na+/Ca2+ Exchanger Isoform 2, Nckx2, Takes Part in the Neuroprotection Elicited by Ischemic Preconditioning in Brain Ischemia. Int. J. Mol. Sci. 2022, 23, 7128. https://doi.org/10.3390/ijms23137128
Cuomo O, Sirabella R, Boscia F, Casamassa A, Lytton J, Annunziato L, Pignataro G. K+-Dependent Na+/Ca2+ Exchanger Isoform 2, Nckx2, Takes Part in the Neuroprotection Elicited by Ischemic Preconditioning in Brain Ischemia. International Journal of Molecular Sciences. 2022; 23(13):7128. https://doi.org/10.3390/ijms23137128
Chicago/Turabian StyleCuomo, Ornella, Rossana Sirabella, Francesca Boscia, Antonella Casamassa, Jonathan Lytton, Lucio Annunziato, and Giuseppe Pignataro. 2022. "K+-Dependent Na+/Ca2+ Exchanger Isoform 2, Nckx2, Takes Part in the Neuroprotection Elicited by Ischemic Preconditioning in Brain Ischemia" International Journal of Molecular Sciences 23, no. 13: 7128. https://doi.org/10.3390/ijms23137128
APA StyleCuomo, O., Sirabella, R., Boscia, F., Casamassa, A., Lytton, J., Annunziato, L., & Pignataro, G. (2022). K+-Dependent Na+/Ca2+ Exchanger Isoform 2, Nckx2, Takes Part in the Neuroprotection Elicited by Ischemic Preconditioning in Brain Ischemia. International Journal of Molecular Sciences, 23(13), 7128. https://doi.org/10.3390/ijms23137128