
Recent Advances in Renal Medullary Carcinoma


{kind=link}
{kind=link}
Abstract
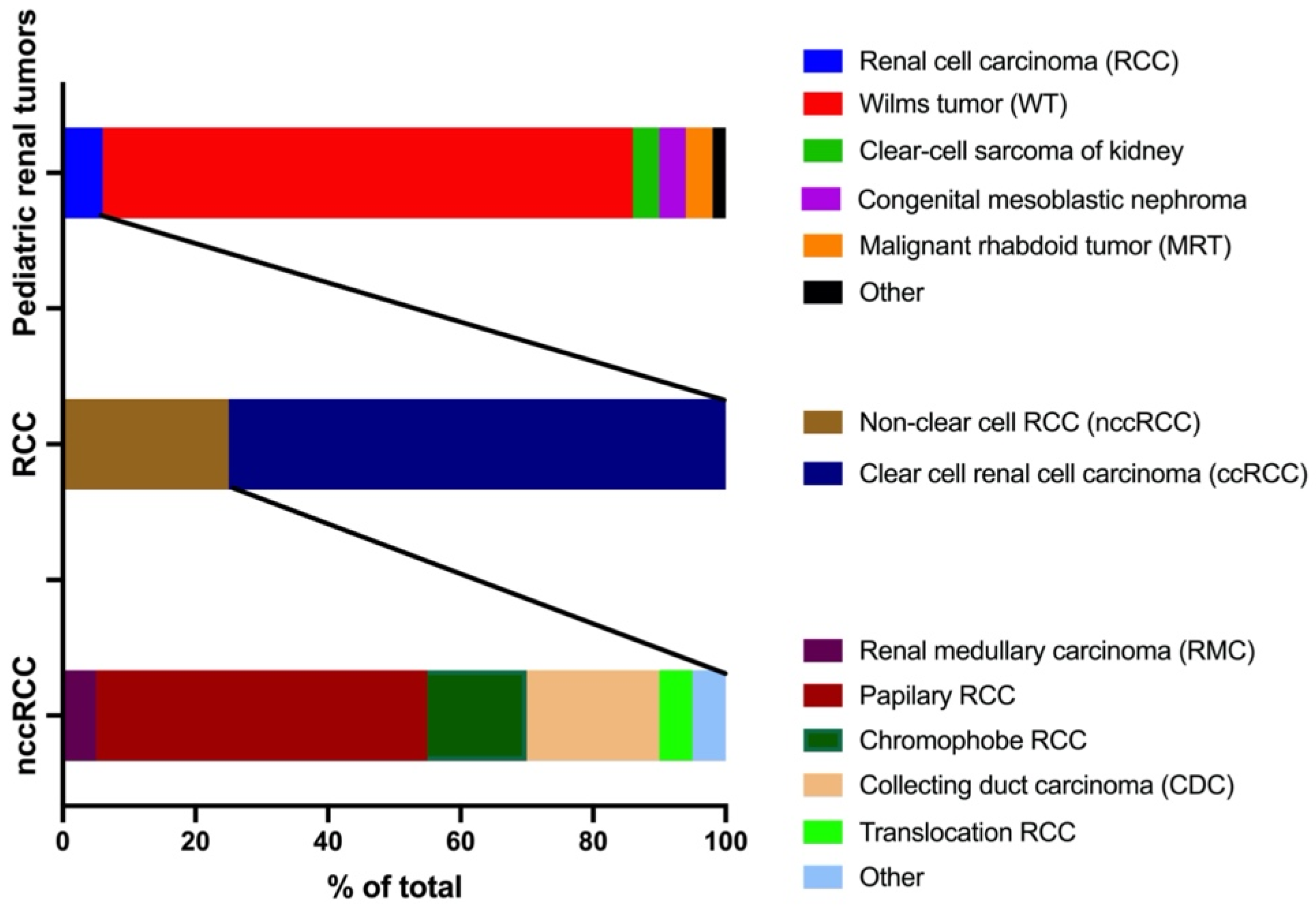
1. Introduction
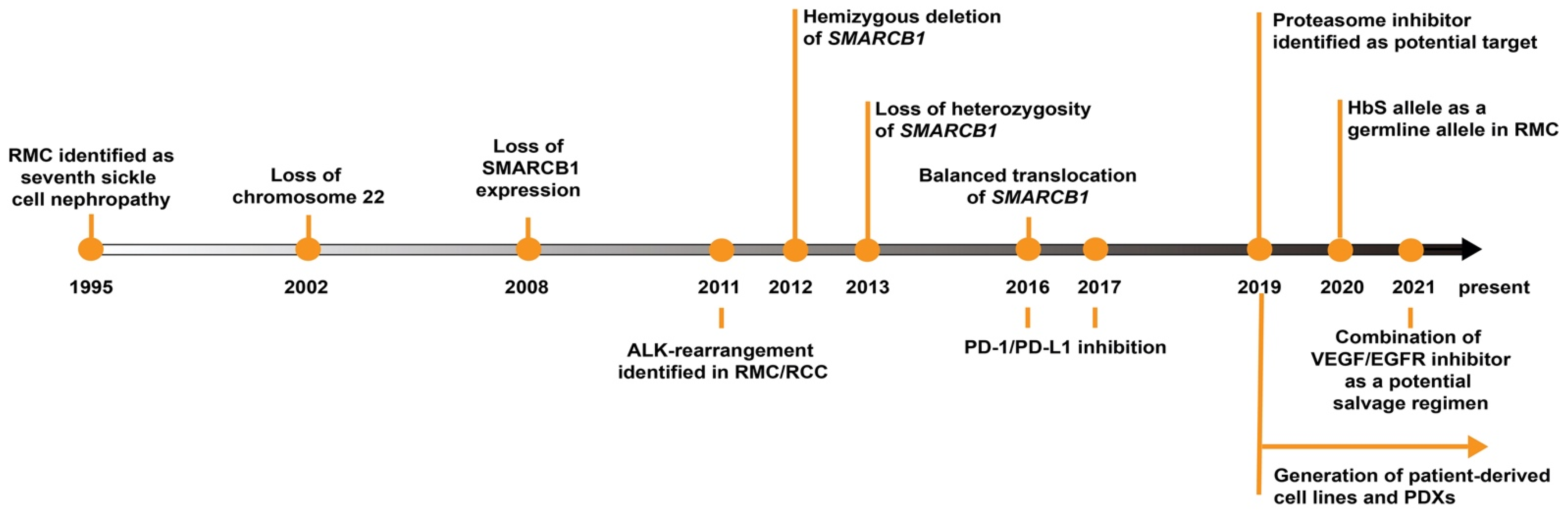
2. SMARCB1 Loss as the Primary Driver in RMC
3. Connection between RMC and Sickle Hemoglobinopathies
4. Therapeutic Considerations from Biology
4.1. Role of Proteasome Inhibitors
4.2. Other Potential Targets
5. Outstanding Questions in RMC Research
5.1. Similar Entities of RMC (ALK Rearranged RCC)
5.2. Unclassified Renal Cell Carcinoma with Medullary Phenotype
5.3. Limited Mouse Models for RMC
5.4. Distinguishing RMC from CDC
5.5. Modifiable Risk Factors in RMC
6. Discussion
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- D’Angio, G.J. The National Wilms Tumor Study: A 40 Year Perspective. Lifetime Data Anal. 2007, 13, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Kaatsch, P. Epidemiology of Childhood Cancer. Cancer Treat. Rev. 2010, 36, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Jain, J.; Sutton, K.S.; Hong, A.L. Progress Update in Pediatric Renal Tumors. Curr. Oncol. Rep. 2021, 23, 33. [Google Scholar] [CrossRef]
- He, M.; Cai, J.; Zhu, K.; Gu, W.; Li, M.; Xiong, J.; Guan, Z.; Wang, J.; Shu, Q. Renal Cell Carcinoma in Children and Adolescents: Single-Center Experience and Literature Review. Medicine 2021, 100, e23717. [Google Scholar] [CrossRef]
- Gao, H.; Cheng, Q.-Y.; Zhao, Q.; Tao, L.-X.; Zhang, C. Childhood Clear Cell Sarcoma of Kidney: Incidence and Survival. Front. Pediatr. 2021, 9, 448. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, G.T.; Cheng, L. Neoplasms of the kidney. In Essentials of Anatomic Pathology; Springer: Berlin/Heidelberg, Germany, 2016; pp. 1645–1679. [Google Scholar]
- Li, J.; Zhang, W.; Hu, H.; Zhang, Y.; Wang, Y.; Gu, H.; Huang, D. Case Analysis of 14 Children with Malignant Rhabdoid Tumor of the Kidney. Cancer Manag. Res. 2021, 13, 4865. [Google Scholar] [CrossRef] [PubMed]
- Treece, A.L. Pediatric Renal Tumors: Updates in the Molecular Era. Surg. Pathol. Clin. 2020, 13, 695–718. [Google Scholar] [CrossRef]
- Ridge, C.A.; Pua, B.B.; Madoff, D.C. Epidemiology and Staging of Renal Cell Carcinoma, Seminars in Interventional Radiology; Thieme Medical Publishers: New York, NY, USA, 2014; pp. 003–008. [Google Scholar]
- Breen, D.J.; King, A.J.; Patel, N.; Lockyer, R.; Hayes, M. Image-Guided Cryoablation for Sporadic Renal Cell Carcinoma: Three-and 5-Year Outcomes in 220 Patients with Biopsy-Proven Renal Cell Carcinoma. Radiology 2018, 289, 554–561. [Google Scholar] [CrossRef]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs—Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef]
- Sepe, P.; Ottini, A.; Pircher, C.C.; Franza, A.; Claps, M.; Guadalupi, V.; Verzoni, E.; Procopio, G. Characteristics and Treatment Challenges of Non-Clear Cell Renal Cell Carcinoma. Cancers 2021, 13, 3807. [Google Scholar] [CrossRef]
- Lipworth, L.; Morgans, A.K.; Edwards, T.L.; Barocas, D.A.; Chang, S.S.; Herrell, S.D.; Penson, D.F.; Resnick, M.J.; Smith, J.A.; Clark, P.E. Renal Cell Cancer Histological Subtype Distribution Differs by Race and Sex. BJU Int. 2016, 117, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Avery, R.A.; Harris, J.E.; Davis Jr, C.J.; Borgaonkar, D.S.; Byrd, J.C.; Weiss, R.B. Renal Medullary Carcinoma: Clinical and Therapeutic Aspects of a Newly Described Tumor. Cancer 1996, 78, 128–132. [Google Scholar] [CrossRef]
- Davis, C.J., Jr.; Mostofi, F.; Sesterhenn, I.A. Renal Medullary Carcinoma. The Seventh Sickle Cell Nephropathy. Am. J. Surg. Pathol. 1995, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Maroja Silvino, M.C.; Venchiarutti Moniz, C.M.; Munhoz Piotto, G.H.; Siqueira, S.; Galapo Kann, A.; Dzik, C. Renal medullary carcinoma response to chemotherapy: A referral center experience in Brazil. Rare Tumors 2013, 5, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.-D.; Gaashan, M.; Haddad, H. Atypical Presentation of Renal Medullary Carcinoma: A Case Report and Review of the Literature. Urol. Case Rep. 2019, 22, 8. [Google Scholar]
- Johnson, R.P.; Krauland, K.; Owens, N.M.; Peckham, S. Renal Medullary Carcinoma Metastatic to the Scalp. Am. J. Dermatopathol. 2011, 33, 11–13. [Google Scholar] [CrossRef]
- Walsh, A.M.; Fiveash, J.B.; Reddy, A.T.; Friedman, G.K. Response to radiation in renal medullary carcinoma. Rare Tumors 2011, 3, 100–103. [Google Scholar] [CrossRef]
- Zdinak, L.A.; Nik, N.A.; Hidayat, A.A.; Hargett, N.A. Renal medullary carcinoma metastatic to the orbit: A clinicopathologic report. Ophthal. Plast. Reconstr. Surg. 2004, 20, 322–325. [Google Scholar] [CrossRef]
- Ibilibor, C.; Medway, A.; Nelius, T. Renal Medullary Carcinoma with an Ophthalmic Metastasis. Urol. Ann. 2017, 9, 184. [Google Scholar] [CrossRef]
- Heuermann, K.; Romero, J.; Abromowitch, M.; Gordon, B.G.; Gross, T. Fatal Coagulase-Negative Staphylococci Infection after Bone Marrow Transplantation in a Patient with Persistent Adverse Reactions to Vancomycin. J. Pediatr. Hematol. Oncol. 1999, 21, 80–81. [Google Scholar] [CrossRef]
- Watanabe, I.C.; Billis, A.; Guimaraes, M.S.; Alvarenga, M.; Matos, A.C.; Cardinalli, I.A.; Filippi, R.Z.; Castro, M.G.; Suzigan, S. Renal medullary carcinoma: Report of seven cases from Brazil. Mod. Pathol. 2007, 20, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.X.; Tretiakova, M.; Gong, C.; Mandal, S.; Krausz, T.; Taxy, J.B. Renal Medullary Carcinoma: Rhabdoid Features and the Absence of INI1 Expression as Markers of Aggressive Behavior. Mod. Pathol. 2008, 21, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.G.; Roberts, C.W. SWI/SNF Nucleosome Remodellers and Cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, T.J.; Hornick, J.L. INI1-Deficient Tumors: Diagnostic Features and Molecular Genetics. Am. J. Surg. Pathol. 2011, 35, e47–e63. [Google Scholar] [CrossRef]
- Pawel, B.R. SMARCB1-Deficient Tumors of Childhood: A Practical Guide. Pediatr. Dev. Pathol. 2018, 21, 6–28. [Google Scholar] [CrossRef]
- Stahlschmidt, J.; Cullinane, C.; Roberts, P.; Picton, S.V. Renal Medullary Carcinoma: Prolonged Remission with Chemotherapy, Immunohistochemical Characterisation and Evidence of Bcr/Abl Rearrangement. Med. Pediatr. Oncol. 1999, 33, 551–557. [Google Scholar] [CrossRef]
- Calderaro, J.; Moroch, J.; Pierron, G.; Pedeutour, F.; Grison, C.; Maillé, P.; Soyeux, P.; Taille, A.; Couturier, J.; Vieillefond, A. SMARCB1/INI1 Inactivation in Renal Medullary Carcinoma. Histopothology 2012, 61, 428–435. [Google Scholar] [CrossRef]
- Liu, Q.; Galli, S.; Srinivasan, R.; Linehan, W.M.; Tsokos, M.; Merino, M.J. Renal Medullary Carcinoma: Molecular, Immunohistochemistry, and Morphologic Correlation. Am. J. Surg. Pathol. 2013, 37, 368–374. [Google Scholar] [CrossRef]
- Calderaro, J.; Masliah-Planchon, J.; Richer, W.; Maillot, L.; Maille, P.; Mansuy, L.; Bastien, C.; Taille, A.; Boussion, H.; Charpy, C. Balanced Translocations Disrupting SMARCB1 Are Hallmark Recurrent Genetic Alterations in Renal Medullary Carcinomas. Eur. Urol. 2016, 69, 1055–1061. [Google Scholar] [CrossRef]
- Jia, L.; Carlo, M.I.; Khan, H.; Nanjangud, G.J.; Rana, S.; Cimera, R.; Zhang, Y.; Hakimi, A.A.; Verma, A.K.; Al-Ahmadie, H.A. Distinctive mechanisms underlie the loss of SMARCB1 protein expression in renal medullary carcinoma: Morphologic and molecular analysis of 20 cases. Mod. Pathol. 2019, 32, 1329–1343. [Google Scholar] [CrossRef]
- Hong, A.L.; Tseng, Y.-Y.; Wala, J.A.; Kim, W.-J.; Kynnap, B.D.; Doshi, M.B.; Kugener, G.; Sandoval, G.J.; Howard, T.P.; Li, J. Renal Medullary Carcinomas Depend upon SMARCB1 Loss and Are Sensitive to Proteasome Inhibition. Elife 2019, 8, 44161. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Malouf, G.G.; Su, X.; Yao, H.; Tripathi, D.N.; Soeung, M.; Gao, J.; Rao, P.; Coarfa, C.; Creighton, C.J. Comprehensive Molecular Characterization Identifies Distinct Genomic and Immune Hallmarks of Renal Medullary Carcinoma. Cancer Cell 2020, 37, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Tannir, N.M.; Walker, C.L. A Model Linking Sickle Cell Hemoglobinopathies and SMARCB1 Loss in Renal Medullary Carcinoma. Clin. Cancer Res. 2018, 24, 2044–2049. [Google Scholar] [CrossRef] [PubMed]
- Gatalica, Z.; Lilleberg, S.L.; Monzon, F.A.; Koul, M.S.; Bridge, J.A.; Knezetic, J.; Legendre, B.; Sharma, P.; McCue, P.A. Renal Medullary Carcinomas: Histopathologic Phenotype Associated with Diverse Genotypes. Hum. Pathol. 2011, 42, 1979–1988. [Google Scholar] [CrossRef]
- Tan, K.-T.; Kim, H.; Carrot-Zhang, J.; Zhang, Y.; Kim, W.J.; Kugener, G.; Wala, J.A.; Howard, T.P.; Chi, Y.-Y.; Beroukhim, R. Haplotype-Resolved Germline and Somatic Alterations in Renal Medullary Carcinomas. Genome Med. 2021, 13, 1–13. [Google Scholar] [CrossRef]
- Zheng, G.X.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M. Haplotyping Germline and Cancer Genomes with High-Throughput Linked-Read Sequencing. Nat. Biotechnol. 2016, 34, 303–311. [Google Scholar] [CrossRef]
- Marks, P.; Garcia, S.; Barrio, A.M.; Belhocine, K.; Bernate, J.; Bharadwaj, R.; Bjornson, K.; Catalanotti, C.; Delaney, J.; Fehr, A. Resolving the Full Spectrum of Human Genome Variation Using Linked-Reads. Genome Res. 2019, 29, 635–645. [Google Scholar] [CrossRef]
- Cheng, H.; Jarvis, E.D.; Fedrigo, O.; Koepfli, K.-P.; Urban, L.; Gemmell, N.J.; Li, H. Haplotype-Resolved Assembly of Diploid Genomes without Parental Data. Nat. Biotechnol. 2022, 1–4. [Google Scholar] [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A. The Complete Sequence of a Human Genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef]
- Wiele, A.J.; Surasi, D.S.; Rao, P.; Sircar, K.; Su, X.; Bathala, T.K.; Shah, A.Y.; Jonasch, E.; Cataldo, V.D.; Genovese, G. Efficacy and Safety of Bevacizumab plus Erlotinib in Patients with Renal Medullary Carcinoma. Cancers 2021, 13, 2170. [Google Scholar] [CrossRef]
- Rathmell, W.K.; Monk, J.P. High-Dose-Intensity MVAC for Advanced Renal Medullary Carcinoma: Report of Three Cases and Literature Review. Urology 2008, 72, 659–663. [Google Scholar] [CrossRef]
- Anne, M.; Sammartino, D.; Chaudhary, S.; Bhuiya, T.; Mehrotra, B. Renal Medullary Carcinoma Masquerading as Bilateral Breast Carcinoma Category: Case Report. World J. Oncol. 2013, 4, 169. [Google Scholar] [CrossRef]
- Walsh, A.; Kelly, D.R.; Vaid, Y.N.; Hilliard, L.M.; Friedman, G.K. Complete Response to Carboplatin, Gemcitabine, and Paclitaxel in a Patient with Advanced Metastatic Renal Medullary Carcinoma. Pediatr. Blood Cancer 2010, 55, 1217–1220. [Google Scholar] [CrossRef] [PubMed]
- Strouse, J.J.; Spevak, M.; Mack, A.K.; Arceci, R.J.; Small, D.; Loeb, D.M. Significant Responses to Platinum-based Chemotherapy in Renal Medullary Carcinoma. Pediatr. Blood Cancer 2005, 44, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Goenaga-Vázquez, Y.; Colón, G.; Barrios, N.; Correa, M. Renal Medullary Carcinoma: A Nearly Fatal Malignancy Specifically Affecting Patients with a so-Called Benign Condition. CEN Case Rep. 2018, 7, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Blas, L.; Roberti, J.; Petroni, J.; Reniero, L.; Cicora, F. Renal Medullary Carcinoma: A Report of the Current Literature. Curr. Urol. Rep. 2019, 20, 4. [Google Scholar] [CrossRef]
- Chauhan, D.; Tian, Z.; Zhou, B.; Kuhn, D.; Orlowski, R.; Raje, N.; Richardson, P.; Anderson, K.C. In Vitro and in Vivo Selective Antitumor Activity of a Novel Orally Bioavailable Proteasome Inhibitor MLN9708 against Multiple Myeloma Cells. Clin. Cancer 2011, 17, 5311–5321. [Google Scholar] [CrossRef]
- Seo, A.N.; Yoon, G.; Ro, J.Y. Clinicopathologic and Molecular Pathology of Collecting Duct Carcinoma and Related Renal Cell Carcinomas. Adv. Anat. Pathol. 2017, 24, 65–77. [Google Scholar] [CrossRef]
- Carugo, A.; Minelli, R.; Sapio, L.; Soeung, M.; Carbone, F.; Robinson, F.S.; Tepper, J.; Chen, Z.; Lovisa, S.; Svelto, M. P53 Is a Master Regulator of Proteostasis in SMARCB1-Deficient Malignant Rhabdoid Tumors. Cancer Cell 2019, 35, 204–220. [Google Scholar] [CrossRef]
- Ryan, A.; Tawagi, K.; VanderVeen, N.; Matrana, M.; Vasquez, R. Combination Therapy with Bortezomib in Renal Medullary Carcinoma: A Case Series. Clin. Genitourin. Cancer 2021, 19, e395–e400. [Google Scholar] [CrossRef]
- Knutson, S.K.; Warholic, N.M.; Wigle, T.J.; Klaus, C.R.; Allain, C.J.; Raimondi, A.; Scott, M.P.; Chesworth, R.; Moyer, M.P.; Copeland, R.A. Durable Tumor Regression in Genetically Altered Malignant Rhabdoid Tumors by Inhibition of Methyltransferase EZH2. Proc. Natl. Acad. Sci. USA 2013, 110, 7922–7927. [Google Scholar] [CrossRef] [PubMed]
- Gall Trošelj, K.; Novak Kujundzic, R.; Ugarkovic, D. Polycomb Repressive Complex’s Evolutionary Conserved Function: The Role of EZH2 Status and Cellular Background. Clin. Epigenetics 2016, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Beckermann, K.E.; Jolly, P.C.; Kim, J.Y.; Bordeaux, J.; Puzanov, I.; Rathmell, W.K.; Johnson, D.B. Clinical and Immunologic Correlates of Response to PD-1 Blockade in a Patient with Metastatic Renal Medullary Carcinoma. J. Immunother. Cancer 2017, 5, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sodji, Q.; Klein, K.; Sravan, K.; Parikh, J. Predictive Role of PD-L1 Expression in the Response of Renal Medullary Carcinoma to PD-1 Inhibition. J. Immunother. Cancer 2017, 5, 1–6. [Google Scholar] [CrossRef]
- Leruste, A.; Tosello, J.; Ramos, R.N.; Tauziède-Espariat, A.; Brohard, S.; Han, Z.-Y.; Beccaria, K.; Andrianteranagna, M.; Caudana, P.; Nikolic, J. Clonally Expanded T Cells Reveal Immunogenicity of Rhabdoid Tumors. Cancer Cell 2019, 36, 597–612. [Google Scholar] [CrossRef]
- Lipkin, J.S.; Rizvi, S.M.; Gatalica, Z.; Sarwani, N.E.; Holder, S.L.; Kaag, M.; Drabick, J.J.; Joshi, M. Therapeutic Approach Guided by Genetic Alteration: Use of MTOR Inhibitor in Renal Medullary Carcinoma with Loss of PTEN Expression. Cancer Biol. Ther. 2015, 16, 28–33. [Google Scholar] [CrossRef]
- Templeton, A.J.; Dutoit, V.; Cathomas, R.; Rothermundt, C.; Bärtschi, D.; Dröge, C.; Gautschi, O.; Borner, M.; Fechter, E.; Stenner, F. Phase 2 Trial of Single-Agent Everolimus in Chemotherapy-Naive Patients with Castration-Resistant Prostate Cancer (SAKK 08/08). Eur. Urol. 2013, 64, 150–158. [Google Scholar] [CrossRef]
- Voss, M.H.; Molina, A.M.; Chen, Y.-B.; Woo, K.M.; Chaim, J.L.; Coskey, D.T.; Redzematovic, A.; Wang, P.; Lee, W.; Selcuklu, S.D. Phase II Trial and Correlative Genomic Analysis of Everolimus plus Bevacizumab in Advanced Non–Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 2016, 34, 3846. [Google Scholar] [CrossRef]
- Msaouel, P.; Walker, C.L.; Genovese, G.; Tannir, N.M. Molecular Hallmarks of Renal Medullary Carcinoma: More to c-MYC than Meets the Eye. Mol. Cell. Oncol. 2020, 7, 1777060. [Google Scholar] [CrossRef]
- Amin, M.B.; Smith, S.C.; Agaimy, A.; Argani, P.; Compérat, E.M.; Delahunt, B.; Epstein, J.I.; Eble, J.N.; Grignon, D.J.; Hartmann, A. Collecting Duct Carcinoma versus Renal Medullary Carcinoma: An Appeal for Nosologic and Biological Clarity. Am. J. Surg. Pathol. 2014, 38, 871–874. [Google Scholar] [CrossRef]
- Elwood, H.; Chaux, A.; Schultz, L.; Illei, P.B.; Baydar, D.E.; Billis, A.; Sharma, R.; Argani, P.; Epstein, J.I.; Netto, G.J. Immunohistochemical Analysis of SMARCB1/INI-1 Expression in Collecting Duct Carcinoma. CEN Case Rep. 2011, 78, 474.e1–474.e5. [Google Scholar] [CrossRef] [PubMed]
- Bratslavsky, G.; Gleicher, S.; Jacob, J.M.; Sanford, T.H.; Shapiro, O.; Bourboulia, D.; Gay, L.M.; Elvin, J.A.; Vergilio, J.-A.; Suh, J. Comprehensive Genomic Profiling of Metastatic Collecting Duct Carcinoma, Renal Medullary Carcinoma, and Clear Cell Renal Cell Carcinoma. Elsevier 2021, 39, 367.e1–367.e5. [Google Scholar] [CrossRef] [PubMed]
- Mariño-Enríquez, A.; Ou, W.; Weldon, C.B.; Fletcher, J.A.; Pérez-Atayde, A.R. ALK Rearrangement in Sickle Cell Trait-associated Renal Medullary Carcinoma. Genes Chromosom. Cancer 2011, 50, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Cajaiba, M.M.; Jennings, L.J.; Rohan, S.M.; Perez-Atayde, A.R.; Marino-Enriquez, A.; Fletcher, J.A.; Geller, J.I.; Leuer, K.M.; Bridge, J.A.; Perlman, E.J. ALK-rearranged Renal Cell Carcinomas in Children. Genes Chromosom. Cancer 2016, 55, 442–451. [Google Scholar] [CrossRef]
- Yu, W.; Wang, Y.; Jiang, Y.; Zhang, W.; Li, Y. Genetic Analysis and Clinicopathological Features of ALK-rearranged Renal Cell Carcinoma in a Large Series of Resected Chinese Renal Cell Carcinoma Patients and Literature Review. Histopathology 2017, 71, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Wangsiricharoen, S.; Zhong, M.; Ranganathan, S.; Matoso, A.; Argani, P. ALK-Rearranged Renal Cell Carcinoma (RCC): A Report of 2 Cases and Review of the Literature Emphasizing the Distinction between VCL-ALK and Non-VCL-ALK RCC. Int. J. Surg. Pathol. 2021, 29, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Hou, G.; Jiang, Y.; Wang, J.; Li, F.; Cheng, W. Renal Medullary Carcinoma on Dual-Time Point FDG PET/CT Imaging. Clin. Nucl. 2020, 45, 446–447. [Google Scholar] [CrossRef]
- Msaouel, P.; Hong, A.L.; Mullen, E.A.; Atkins, M.B.; Walker, C.L.; Lee, C.-H.; Carden, M.A.; Genovese, G.; Linehan, W.M.; Rao, P. Updated Recommendations on the Diagnosis, Management, and Clinical Trial Eligibility Criteria for Patients with Renal Medullary Carcinoma. Clin. Genitourin. Cancer 2019, 17, 1–6. [Google Scholar] [CrossRef]
- Shi, Z.; Zhuang, Q.; You, R.; Li, Y.; Li, J.; Cao, D. Clinical and computed tomography imaging features of renal medullary carcinoma: A report of six cases. Oncol. Lett. 2016, 11, 261–266. [Google Scholar] [CrossRef]
- Huang, Z.-M.; Wang, H.; Ji, Z.-G. Renal medullary carcinoma masquerading as renal infection: A case report. BMC Nephrol. 2020, 21, 1–4. [Google Scholar] [CrossRef]
- Wei, D.; Yang, Y.; Ricketts, C.J.; Vocke, C.D.; Ball, M.W.; Sourbier, C.; Wangsa, D.; Wangsa, D.; Guha, R.; Zhang, X. Novel Renal Medullary Carcinoma Cell Lines, UOK353 and UOK360, Provide Preclinical Tools to Identify New Therapeutic Treatments. Genes Chromosom. Cancer 2020, 59, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Q.; Ijiri, M.; Rodriguez, R.; Gandour-Edwards, R.; Lee, J.; Tepper, C.G.; Li, Y.; Beckett, L.; Lam, K.; Goodwin, N. Novel Patient Metastatic Pleural Effusion-Derived Xenograft Model of Renal Medullary Carcinoma Demonstrates Therapeutic Efficacy of Sunitinib. Front. Oncol. 2021, 11, 928. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Billis, A.; Shah, R.B.; Moch, H.; Osunkoya, A.O.; Jochum, W.; Hes, O.; Bacchi, C.E.; De Castro, M.G.; Hansel, D.E. Carcinoma of the Collecting Ducts of Bellini and Renal Medullary Carcinoma: Clinicopathologic Analysis of 52 Cases of Rare Aggressive Subtypes of Renal Cell Carcinoma with a Focus on Their Interrelationship. Am. J. Surg. Pathol. 2012, 36, 1265–1278. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, D.D.; Soeung, M.; Perelli, L.; Dondossola, E.; Surasi, D.S.; Tripathi, D.N.; Bertocchio, J.-P.; Carbone, F.; Starbuck, M.W.; Van Alstine, M.L. Association of High-Intensity Exercise with Renal Medullary Carcinoma in Individuals with Sickle Cell Trait: Clinical Observations and Experimental Animal Studies. Cancers 2021, 13, 6022. [Google Scholar] [CrossRef]
- Ezekian, B.; Englum, B.; Gilmore, B.F.; Nag, U.P.; Kim, J.; Leraas, H.J.; Routh, J.C.; Rice, H.E.; Tracy, E.T. Renal Medullary Carcinoma: A National Analysis of 159 Patients. Pediatr. Blood Cancer 2017, 64, e26609. [Google Scholar] [CrossRef]
- Baniak, N.; Tsai, H.; Hirsch, M.S. The Differential Diagnosis of Medullary-Based Renal Masses. Arch. Pathol. Lab. Med. 2021, 145, 1148–1170. [Google Scholar] [CrossRef]
- Beckermann, K.E.; Sharma, D.; Chaturvedi, S.; Msaouel, P.; Abboud, M.R.; Allory, Y.; Bourdeaut, F.; Calderaro, J.; De Cubas, A.A.; Derebail, V.K. Renal Medullary Carcinoma: Establishing Standards in Practice. J. Oncol. 2017, 13, 414–421. [Google Scholar] [CrossRef]
- Lee, R.S.; Stewart, C.; Carter, S.L.; Ambrogio, L.; Cibulskis, K.; Sougnez, C.; Lawrence, M.S.; Auclair, D.; Mora, J.; Golub, T.R. A Remarkably Simple Genome Underlies Highly Malignant Pediatric Rhabdoid Cancers. J. Clin. Investig. 2012, 122, 2983–2988. [Google Scholar] [CrossRef]
- Hohmann, A.F.; Vakoc, C.R. A Rationale to Target the SWI/SNF Complex for Cancer Therapy. Trends Genet. 2014, 30, 356–363. [Google Scholar] [CrossRef]
- Geller, J.I.; Roth, J.J.; Biegel, J.A. Biology and Treatment of Rhabdoid Tumor. Crit. Rev. Oncog. 2015, 20, 3–4. [Google Scholar] [CrossRef]
- Thomas, C.; Oehl-Huber, K.; Bens, S.; Soschinski, P.; Koch, A.; Nemes, K.; Oyen, F.; Kordes, U.; Kool, M.; Frühwald, M.C. Transposable element insertion as a mechanism of SMARCB1 inactivation in atypical teratoid/rhabdoid tumor. Genes Chromosom. Cancer 2021, 60, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Bookhout, C.; Bouldin, T.W.; Ellison, D.W. Atypical Teratoid/Rhabdoid Tumor with Retained INI 1 (SMARCB1) Expression and Loss of BRG 1 (SMARCA4). Neuropathology 2018, 38, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Carlo, M.I.; Chaim, J.; Patil, S.; Kemel, Y.; Schram, A.M.; Woo, K.; Coskey, D.; Nanjangud, G.J.; Voss, M.H.; Feldman, D.R. Genomic Characterization of Renal Medullary Carcinoma and Treatment Outcomes. Clin. Genitourin. Cancer 2017, 15, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, E.M.; Guzzo, T.J.; Furge, K.A.; Netto, G.; Westphal, M.; Dykema, K.; Yang, X.; Zhou, M.; Teh, B.T.; Pavlovich, C.P. Renal Medullary Carcinoma: Molecular, Pathological and Clinical Evidence for Treatment with Topoisomerase-inhibiting Therapy. Br. J. Urol. 2010, 106, 62–65. [Google Scholar] [CrossRef]
- Albadine, R.; Wang, W.; Brownlee, N.A.; Toubaji, A.; Billis, A.; Argani, P.; Epstein, J.I.; Garvin, A.J.; Cousi, R.; Schaeffer, E.M. Topoisomerase II α Status in Renal Medullary Carcinoma: Immuno-Expression and Gene Copy Alterations of a Potential Target of Therapy. J. Urol. 2009, 182, 735–740. [Google Scholar] [CrossRef][Green Version]
- Imtiaz, S.; Zekri, J. Metastatic Renal Medullary Carcinoma: Response to Chemotherapy and Unusual Long Survival. J. Unexplored Med. Data 2017, 2, 105–109. [Google Scholar] [CrossRef]
- Tran, J.; Ornstein, M.C. Clinical review on the management of metastatic renal cell carcinoma. J. Oncol. Pract. 2022, 18, 187–196. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, Y.; Hong, A.L. Recent Advances in Renal Medullary Carcinoma. Int. J. Mol. Sci. 2022, 23, 7097. https://doi.org/10.3390/ijms23137097
Su Y, Hong AL. Recent Advances in Renal Medullary Carcinoma. International Journal of Molecular Sciences. 2022; 23(13):7097. https://doi.org/10.3390/ijms23137097
Chicago/Turabian StyleSu, Yongdong, and Andrew L. Hong. 2022. "Recent Advances in Renal Medullary Carcinoma" International Journal of Molecular Sciences 23, no. 13: 7097. https://doi.org/10.3390/ijms23137097
APA StyleSu, Y., & Hong, A. L. (2022). Recent Advances in Renal Medullary Carcinoma. International Journal of Molecular Sciences, 23(13), 7097. https://doi.org/10.3390/ijms23137097