
A New Bistable Switch Model of Alzheimer’s Disease Pathogenesis

 ,
,  and
and {kind=link}
Abstract
:1. Introduction
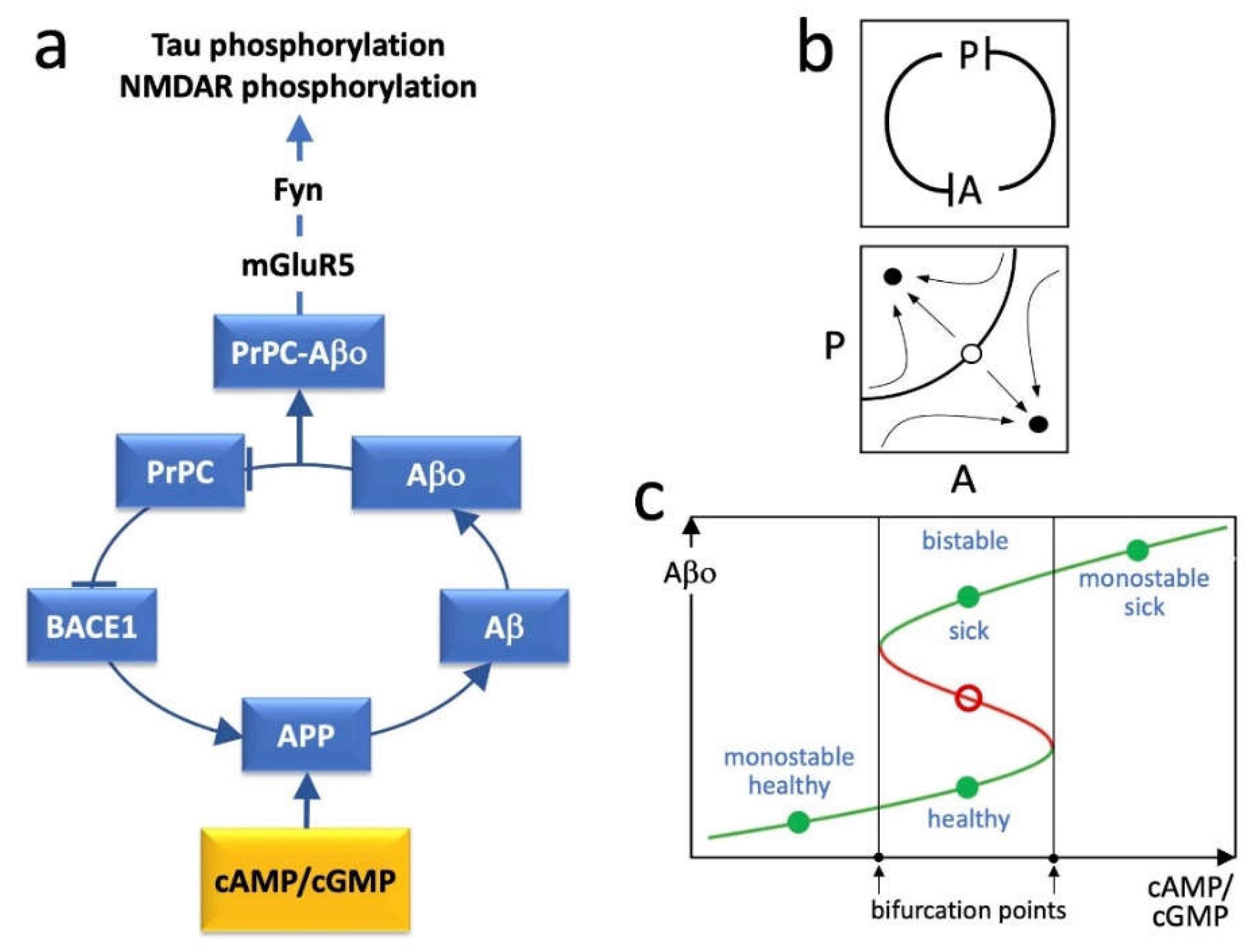
2. BACE1, PrPC and the Positive Feedback Loop
3. Loop Dynamics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sheppard, O.; Coleman, M. Alzheimer’s Disease: Etiology, Neuropathology and Pathogenesis. In Alzheimer’s Disease: Drug Discovery; Huang, X., Ed.; Exon Publications: Brisbane, Australia, 2020. [Google Scholar]
- Burlando, B. Loopomics: A new functional approach to life. J. Appl. Physiol. 2017, 123, 1011–1013. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolkenhauer, O.; Mesarovic, M. Feedback dynamics and cell function: Why systems biology is called Systems Biology. Mol. Biosyst. 2005, 1, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. Microbe sensing, positive feedback loops, and the pathogenesis of inflammatory diseases. Immunol. Rev. 2009, 227, 248–263. [Google Scholar] [CrossRef] [PubMed]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid beta Oligomers (AbetaOs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, H.H.; Whitehouse, I.J.; Baybutt, H.; Brown, D.; Kellett, K.A.; Jackson, C.D.; Turner, A.J.; Piccardo, P.; Manson, J.C.; Hooper, N.M. Prion protein interacts with BACE1 protein and differentially regulates its activity toward wild type and Swedish mutant amyloid precursor protein. J. Biol. Chem. 2011, 286, 33489–33500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchini, F.; Franco, E.; Giordano, G. A structural classification of candidate oscillatory and multistationary biochemical systems. Bull. Math. Biol. 2014, 76, 2542–2569. [Google Scholar] [CrossRef] [PubMed]
- Canepa, E.; Domenicotti, C.; Marengo, B.; Passalacqua, M.; Marinari, U.M.; Pronzato, M.A.; Fedele, E.; Ricciarelli, R. Cyclic adenosine monophosphate as an endogenous modulator of the amyloid-beta precursor protein metabolism. IUBMB Life 2013, 65, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Ricciarelli, R.; Puzzo, D.; Bruno, O.; Canepa, E.; Gardella, E.; Rivera, D.; Privitera, L.; Domenicotti, C.; Marengo, B.; Marinari, U.M.; et al. A novel mechanism for cyclic adenosine monophosphate-mediated memory formation: Role of amyloid beta. Ann. Neurol. 2014, 75, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, A.; Ricciarelli, R.; Gulisano, W.; Rivera, D.; Rebosio, C.; Calcagno, E.; Tropea, M.R.; Conti, S.; Das, U.; Roy, S.; et al. Amyloid-beta Peptide Is Needed for cGMP-Induced Long-Term Potentiation and Memory. J. Neurosci. 2017, 37, 6926–6937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H.; Kerrisk, M.E.; Vortmeyer, A.; Wisniewski, T.; Koleske, A.J.; et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 2013, 79, 887–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygaard, H.B. Targeting Fyn Kinase in Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Blanchini, F.; Cuba Samaniego, C.; Franco, E.; Giordano, G. Aggregates of Monotonic Step Response Systems: A Structural Classification. IEEE Trans. Control. Netw. Syst. 2018, 5, 782–792. [Google Scholar] [CrossRef]
- Huang, B.; Lu, M.; Jolly, M.K.; Tsarfaty, I.; Onuchic, J.; Ben-Jacob, E. The three-way switch operation of Rac1/RhoA GTPase-based circuit controlling amoeboid-hybrid-mesenchymal transition. Sci. Rep. 2014, 4, 6449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mucci, V.; Indovina, I.; Browne, C.J.; Blanchini, F.; Giordano, G.; Marinelli, L.; Burlando, B. Mal de Debarquement Syndrome: A Matter of Loops? Front. Neurol. 2020, 11, 576860. [Google Scholar] [CrossRef] [PubMed]
- Burlando, B.; Milanese, M.; Giordano, G.; Bonifacino, T.; Ravera, S.; Blanchini, F.; Bonanno, G. A multistationary loop model of ALS unveils critical molecular interactions involving mitochondria and glucose metabolism. PLoS ONE 2020, 15, e0244234. [Google Scholar] [CrossRef] [PubMed]
- Montalto, G.; Caudano, F.; Sturla, L.; Bruzzone, S.; Salis, A.; Damonte, G.; Prickaerts, J.; Fedele, E.; Ricciarelli, R. Protein kinase G phosphorylates the Alzheimer’s disease-associated tau protein at distinct Ser/Thr sites. Biofactors 2021, 47, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Villa, V.; Montalto, G.; Caudano, F.; Fedele, E.; Ricciarelli, R. Selective inhibition of phosphodiesterase 4D increases tau phosphorylation at Ser214 residue. Biofactors 2022. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Conolly, R.B.; Kaminski, N.E.; Thomas, R.S.; Andersen, M.E.; Zhang, Q. A bistable switch underlying B-cell differentiation and its disruption by the environmental contaminant 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Sci. 2010, 115, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burlando, B.; Losacco, S.; Villa, V.; Fedele, E.; Ricciarelli, R. A New Bistable Switch Model of Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2022, 23, 7061. https://doi.org/10.3390/ijms23137061
Burlando B, Losacco S, Villa V, Fedele E, Ricciarelli R. A New Bistable Switch Model of Alzheimer’s Disease Pathogenesis. International Journal of Molecular Sciences. 2022; 23(13):7061. https://doi.org/10.3390/ijms23137061
Chicago/Turabian StyleBurlando, Bruno, Serena Losacco, Viviana Villa, Ernesto Fedele, and Roberta Ricciarelli. 2022. "A New Bistable Switch Model of Alzheimer’s Disease Pathogenesis" International Journal of Molecular Sciences 23, no. 13: 7061. https://doi.org/10.3390/ijms23137061
APA StyleBurlando, B., Losacco, S., Villa, V., Fedele, E., & Ricciarelli, R. (2022). A New Bistable Switch Model of Alzheimer’s Disease Pathogenesis. International Journal of Molecular Sciences, 23(13), 7061. https://doi.org/10.3390/ijms23137061