
Roles of Infection in Psoriasis

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Bacterial Infection
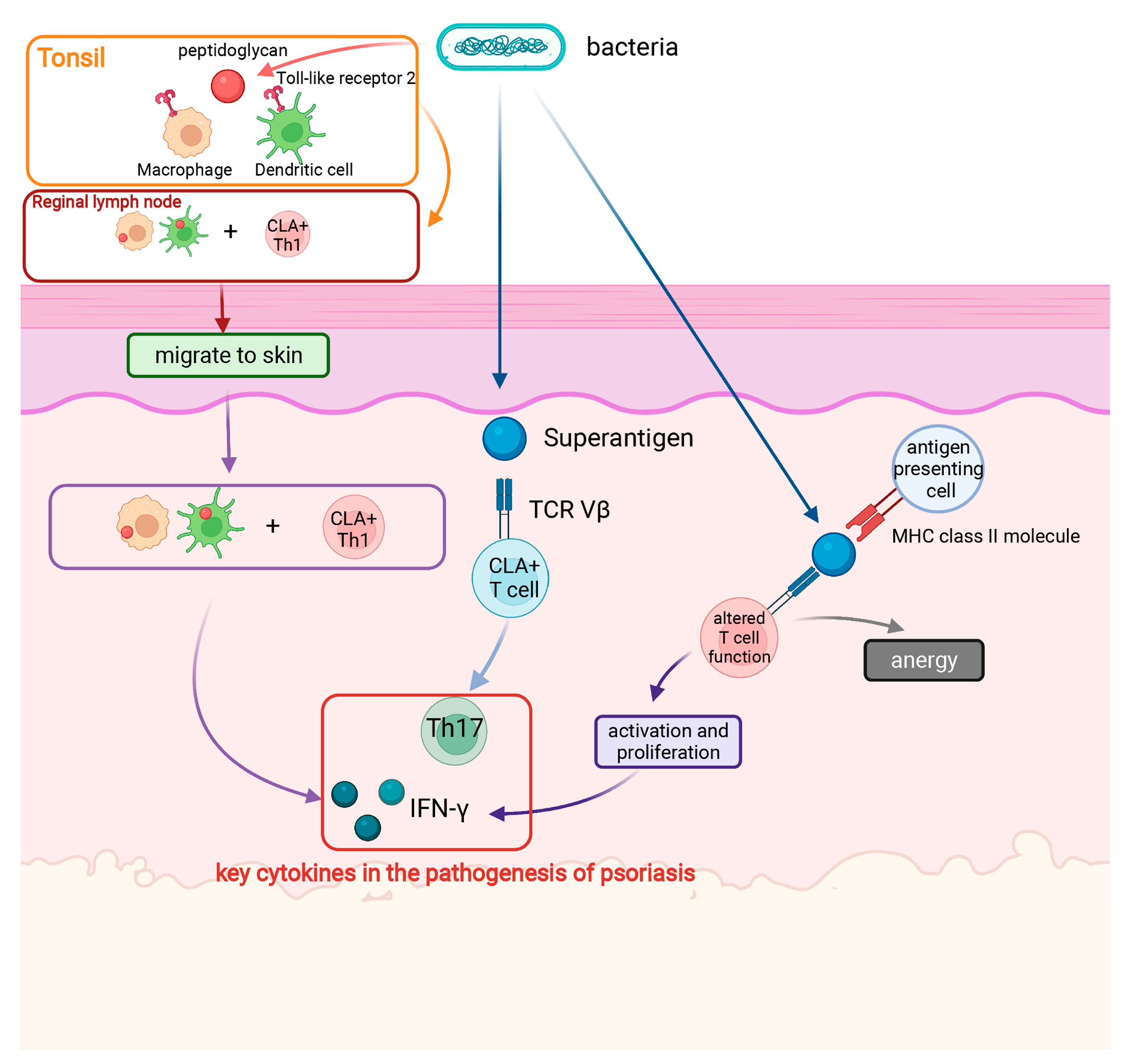
2.1. Streptococcal Infection
2.1.1. Correlation between Streptococcal Infection and Psoriasis
2.1.2. Mechanism of Streptococcal Infection Triggering Psoriasis
2.1.3. Effect of Interventions for Streptococcal Infections (Antibiotics and Tonsillectomy) on the Development of Psoriasis
2.2. Staphylococcus Aureus
2.3. Helicobacter Pylori
2.4. Other Bacteria
3. Viral Infection
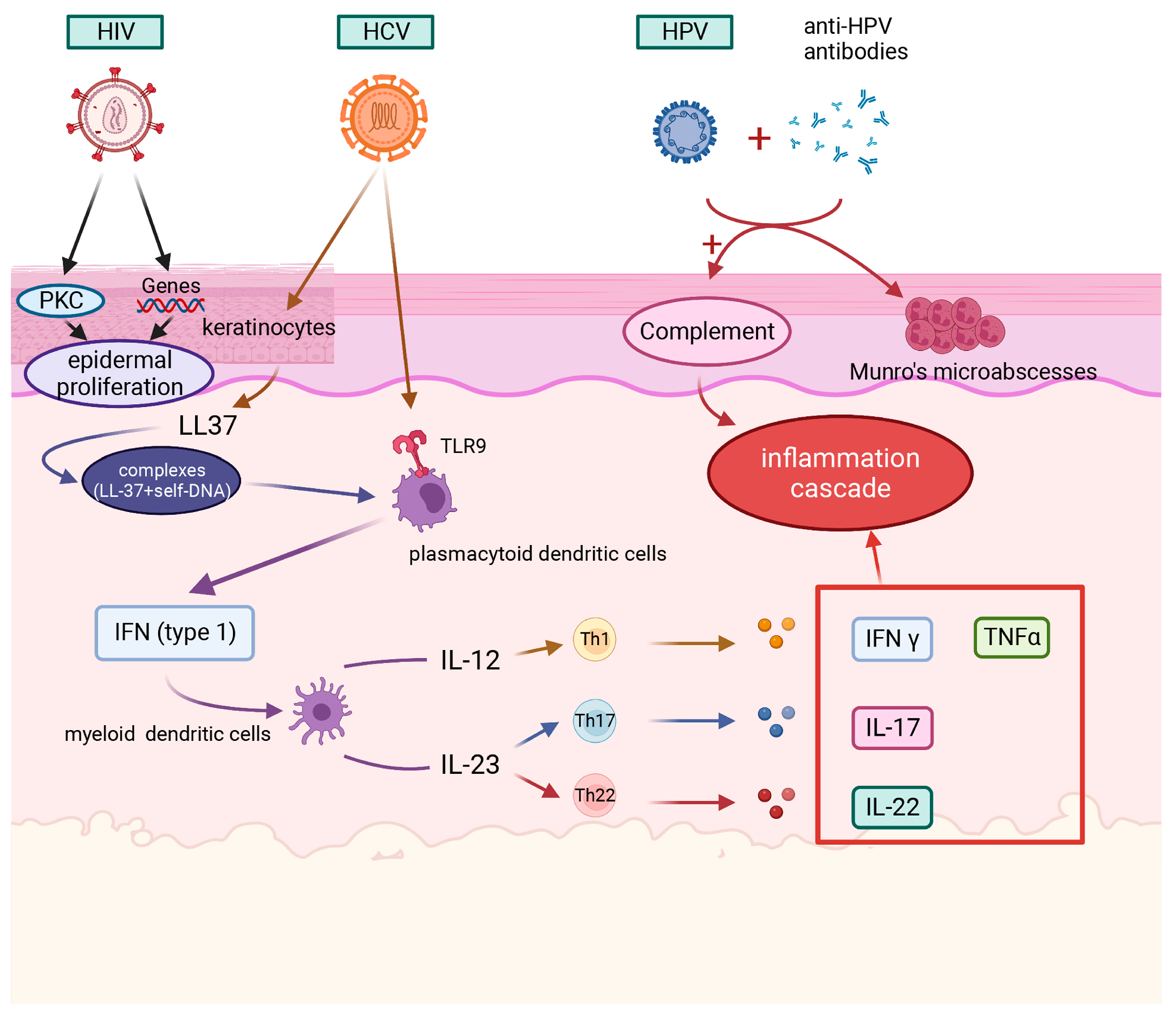
3.1. HIV
3.2. Hepatitis B Virus (HBV)/Hepatitis C Virus (HCV)
3.3. HPV
3.4. CMV
3.5. ZIKV
3.6. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)
4. Fungal Infection
4.1. Malassezia
4.2. Candida
5. Discussion and Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roostaeyan, O.; Kivelevitch, D.; Menter, A. A review article on brodalumab in the treatment of moderate-to-severe plaque psoriasis. Immunotherapy 2017, 9, 963–978. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, J.; Grewal, S.; Langan, S.M.; Mehta, N.N.; Ogdie, A.; Van Voorhees, A.S.; Gelfand, J.M. Psoriasis and comorbid diseases: Epidemiology. J. Am. Acad. Dermatol. 2017, 76, 377–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, D.; Gudjonsson, J.E.; Le, S.; Maverakis, E.; Plazyo, O.; Ritchlin, C.; Scher, J.U.; Singh, R.; Ward, N.L.; Bell, S.; et al. New Frontiers in Psoriatic Disease Research, Part I: Genetics, Environmental Triggers, Immunology, Pathophysiology, and Precision Medicine. J. Investig. Dermatol. 2021, 141, 2112–2122.e3. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Xie, W.; Tao, X.; Liu, N.; Yu, Y.; Huang, Y.; Xu, D.; Fan, Y. Infection-provoked psoriasis: Induced or aggravated (Review). Exp. Ther. Med. 2021, 21, 567. [Google Scholar] [CrossRef]
- Leung, D.Y.; Travers, J.B.; Giorno, R.; Norris, D.A.; Skinner, R.; Aelion, J.; Kazemi, L.V.; Kim, M.H.; Trumble, A.E.; Kotb, M.; et al. Evidence for a streptococcal superantigen-driven process in acute guttate psoriasis. J. Clin. Investig. 1995, 96, 2106–2112. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Lou, F.; Yin, Q.; Gao, Y.; Sun, Y.; Bai, J.; Xu, Z.; Liu, Z.; Cai, W.; Ke, F.; et al. RIG-I antiviral signaling drives interleukin-23 production and psoriasis-like skin disease. EMBO Mol. Med. 2017, 9, 589–604. [Google Scholar] [CrossRef]
- Gao, Z.; Tseng, C.H.; Strober, B.E.; Pei, Z.; Blaser, M.J. Substantial alterations of the cutaneous bacterial biota in psoriatic lesions. PLoS ONE 2008, 3, e2719. [Google Scholar] [CrossRef]
- Okada, K.; Matsushima, Y.; Mizutani, K.; Yamanaka, K. The Role of Gut Microbiome in Psoriasis: Oral Administration of Staphylococcus aureus and Streptococcus danieliae Exacerbates Skin Inflammation of Imiquimod-Induced Psoriasis-Like Dermatitis. Int. J. Mol. Sci. 2020, 21, 3303. [Google Scholar] [CrossRef]
- Saxena, V.N.; Dogra, J. Long-term use of penicillin for the treatment of chronic plaque psoriasis. Eur. J. Dermatol. 2005, 15, 359–362. [Google Scholar]
- Rachakonda, T.D.; Dhillon, J.S.; Florek, A.G.; Armstrong, A.W. Effect of tonsillectomy on psoriasis: A systematic review. J. Am. Acad. Dermatol. 2015, 72, 261–275. [Google Scholar] [CrossRef]
- Navarro-López, V.; Martínez-Andrés, A.; Ramírez-Boscá, A.; Ruzafa-Costas, B.; Núñez-Delegido, E.; Carrión-Gutiérrez, M.A.; Prieto-Merino, D.; Codoñer-Cortés, F.; Ramón-Vidal, D.; Genovés-Martínez, S.; et al. Efficacy and Safety of Oral Administration of a Mixture of Probiotic Strains in Patients with Psoriasis: A Randomized Controlled Clinical Trial. Acta Derm. Venereol. 2019, 99, 1078–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsoi, L.C.; Spain, S.L.; Knight, J.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Ding, J.; Li, Y.; Tejasvi, T.; Gudjonsson, J.E.; et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 2012, 44, 1341–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, R.P.; Duffin, K.C.; Helms, C.; Ding, J.; Stuart, P.E.; Goldgar, D.; Gudjonsson, J.E.; Li, Y.; Tejasvi, T.; Feng, B.J.; et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet. 2009, 41, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, P.E.; Nair, R.P.; Ellinghaus, E.; Ding, J.; Tejasvi, T.; Gudjonsson, J.E.; Li, Y.; Weidinger, S.; Eberlein, B.; Gieger, C.; et al. Genome-wide association analysis identifies three psoriasis susceptibility loci. Nat. Genet. 2010, 42, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Boehncke, W.H.; Schön, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Griffiths, C.E.M.; Armstrong, A.W.; Gudjonsson, J.E.; Barker, J. Psoriasis. Lancet 2021, 397, 1301–1315. [Google Scholar] [CrossRef]
- Ishigame, H.; Kakuta, S.; Nagai, T.; Kadoki, M.; Nambu, A.; Komiyama, Y.; Fujikado, N.; Tanahashi, Y.; Akitsu, A.; Kotaki, H.; et al. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 2009, 30, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Na, L.; Fidel, P.L.; Schwarzenberger, P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J. Infect. Dis. 2004, 190, 624–631. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Meng, S.; Hong, S.; Lin, X.; Jin, W.; Dong, C. IL-17C is required for lethal inflammation during systemic fungal infection. Cell. Mol. Immunol. 2016, 13, 474–483. [Google Scholar] [CrossRef] [Green Version]
- Conti, H.R.; Whibley, N.; Coleman, B.M.; Garg, A.V.; Jaycox, J.R.; Gaffen, S.L. Signaling through IL-17C/IL-17RE is dispensable for immunity to systemic, oral and cutaneous candidiasis. PLoS ONE 2015, 10, e0122807. [Google Scholar] [CrossRef] [Green Version]
- Hansakon, A.; Jeerawattanawart, S.; Pattanapanyasat, K.; Angkasekwinai, P. IL-25 Receptor Signaling Modulates Host Defense against Cryptococcus neoformans Infection. J. Immunol. 2020, 205, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Spanò, F.; Contatore, M.; Guastalla, A.; Penza, E.; Magnani, O.; Puppo, F. Infection risk associated with anti-TNF-α agents: A review. Expert Opin. Drug Saf. 2015, 14, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Hamel, R.; Neyret, A.; Ekchariyawat, P.; Molès, J.P.; Simmons, G.; Chazal, N.; Desprès, P.; Missé, D.; Briant, L. Human keratinocytes restrict chikungunya virus replication at a post-fusion step. Virology 2015, 476, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Boxel-Dezaire, A.H.; Zula, J.A.; Xu, Y.; Ransohoff, R.M.; Jacobberger, J.W.; Stark, G.R. Major differences in the responses of primary human leukocyte subsets to IFN-beta. J. Immunol. 2010, 185, 5888–5899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, B.E.; Martires, K.J.; Ho, R.S. Psoriasis and the Risk of Depression in the US Population: National Health and Nutrition Examination Survey 2009–2012. JAMA Dermatol. 2016, 152, 73–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurd, S.K.; Troxel, A.B.; Crits-Christoph, P.; Gelfand, J.M. The risk of depression, anxiety, and suicidality in patients with psoriasis: A population-based cohort study. Arch. Dermatol. 2010, 146, 891–895. [Google Scholar]
- Nadeem, A.; Ahmad, S.F.; Al-Harbi, N.O.; Fardan, A.S.; El-Sherbeeny, A.M.; Ibrahim, K.E.; Attia, S.M. IL-17A causes depression-like symptoms via NFκB and p38MAPK signaling pathways in mice: Implications for psoriasis associated depression. Cytokine 2017, 97, 14–24. [Google Scholar] [CrossRef]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Marek-Jozefowicz, L.; Czajkowski, R.; Borkowska, A.; Nedoszytko, B.; Żmijewski, M.A.; Cubała, W.J.; Slominski, A.T. The Brain-Skin Axis in Psoriasis-Psychological, Psychiatric, Hormonal, and Dermatological Aspects. Int. J. Mol. Sci. 2022, 23, 669. [Google Scholar] [CrossRef]
- Li, J.; Pu, F.; Peng, C.; Wang, Y.; Zhang, Y.; Wu, S.; Wang, S.; Shen, X.; Li, Y.; Cheng, R.; et al. Antibiotic cocktail-induced gut microbiota depletion in different stages could cause host cognitive impairment and emotional disorders in adulthood in different manners. Neurobiol. Dis. 2022, 170, 105757. [Google Scholar] [CrossRef]
- Needham, B.D.; Funabashi, M.; Adame, M.D.; Wang, Z.; Boktor, J.C.; Haney, J.; Wu, W.L.; Rabut, C.; Ladinsky, M.S.; Hwang, S.J.; et al. A gut-derived metabolite alters brain activity and anxiety behaviour in mice. Nature 2022, 602, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; O’Riordan, K.J.; Cowan, C.S.M.; Sandhu, K.V.; Bastiaanssen, T.F.S.; Boehme, M.; Codagnone, M.G.; Cussotto, S.; Fulling, C.; Golubeva, A.V.; et al. The Microbiota-Gut-Brain Axis. Physiol. Rev. 2019, 99, 1877–2013. [Google Scholar] [CrossRef] [PubMed]
- Rademaker, M.; Agnew, K.; Anagnostou, N.; Andrews, M.; Armour, K.; Baker, C.; Foley, P.; Gebauer, K.; Gupta, M.; Marshman, G.; et al. Psoriasis and infection. A clinical practice narrative. Australas. J. Dermatol. 2019, 60, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Weyrich, L.S.; Dixit, S.; Farrer, A.G.; Cooper, A.J.; Cooper, A.J. The skin microbiome: Associations between altered microbial communities and disease. Australas. J. Dermatol. 2015, 56, 268–274. [Google Scholar] [CrossRef]
- Alekseyenko, A.V.; Perez-Perez, G.I.; De Souza, A.; Strober, B.; Gao, Z.; Bihan, M.; Li, K.; Methé, B.A.; Blaser, M.J. Community differentiation of the cutaneous microbiota in psoriasis. Microbiome 2013, 1, 31. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Issa, N.; Afifi, L.; Jeon, C.; Chang, H.W.; Liao, W. The Role of the Skin and Gut Microbiome in Psoriatic Disease. Curr. Dermatol. Rep. 2017, 6, 94–103. [Google Scholar] [CrossRef]
- Hedin, C.R.H.; Sonkoly, E.; Eberhardson, M.; Ståhle, M. Inflammatory bowel disease and psoriasis: Modernizing the multidisciplinary approach. J. Intern. Med. 2021, 290, 257–278. [Google Scholar] [CrossRef]
- Shelley, W.B.; Wood, M.G.; Beerman, H. Pustular psoriasis elicited by streptococcal antigen and localized to the sweat pore. J. Investig. Dermatol. 1975, 65, 466–471. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.M. The relationship of streptococcus fecalis to psoriasis. J. Investig. Dermatol. 1953, 20, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Ishikawa, H. Unspecific increase of antistreptolysin-O titer in acute guttate psoriasis. J. Dermatol. 1977, 4, 187–191. [Google Scholar] [CrossRef]
- Mallbris, L.; Wolk, K.; Sánchez, F.; Ståhle, M. HLA-Cw*0602 associates with a twofold higher prevalence of positive streptococcal throat swab at the onset of psoriasis: A case control study. BMC Dermatol. 2009, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norrlind, R. The significance of infections in the origination of psoriasis. Acta Rheumatol. Scand. 1955, 1, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.B.; Jadeja, S.; Allawh, R.M.; Goyal, K. Psoriasis, chronic tonsillitis, and biofilms: Tonsillar pathologic findings supporting a microbial hypothesis. Ear Nose Throat J. 2018, 97, 79–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.L.; Ku, Y.H.; Yip, H.T.; Wei, J.C. Tonsillectomy and the subsequent risk of psoriasis: A nationwide population-based cohort study. J. Am. Acad. Dermatol. 2021, 85, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Osterlund, A.; Popa, R.; Nikkilä, T.; Scheynius, A.; Engstrand, L. Intracellular reservoir of Streptococcus pyogenes in vivo: A possible explanation for recurrent pharyngotonsillitis. Laryngoscope 1997, 107, 640–647. [Google Scholar] [CrossRef]
- Molinari, G.; Chhatwal, G.S. Streptococcal invasion. Curr. Opin. Microbiol. 1999, 2, 56–61. [Google Scholar] [CrossRef]
- Diluvio, L.; Vollmer, S.; Besgen, P.; Ellwart, J.W.; Chimenti, S.; Prinz, J.C. Identical TCR beta-chain rearrangements in streptococcal angina and skin lesions of patients with psoriasis vulgaris. J. Immunol. 2006, 176, 7104–7111. [Google Scholar] [CrossRef]
- Baker, B.S.; Laman, J.D.; Powles, A.; van der Fits, L.; Voerman, J.S.; Melief, M.J.; Fry, L. Peptidoglycan and peptidoglycan-specific Th1 cells in psoriatic skin lesions. J. Pathol. 2006, 209, 174–181. [Google Scholar] [CrossRef]
- Capon, F.; Semprini, S.; Chimenti, S.; Fabrizi, G.; Zambruno, G.; Murgia, S.; Carcassi, C.; Fazio, M.; Mingarelli, R.; Dallapiccola, B.; et al. Fine mapping of the PSORS4 psoriasis susceptibility region on chromosome 1q21. J. Investig. Dermatol. 2001, 116, 728–730. [Google Scholar] [CrossRef] [Green Version]
- Philpott, D.J.; Girardin, S.E. The role of Toll-like receptors and Nod proteins in bacterial infection. Mol. Immunol. 2004, 41, 1099–1108. [Google Scholar] [CrossRef]
- Spaulding, A.R.; Salgado-Pabón, W.; Kohler, P.L.; Horswill, A.R.; Leung, D.Y.; Schlievert, P.M. Staphylococcal and streptococcal superantigen exotoxins. Clin. Microbiol. Rev. 2013, 26, 422–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdimarsson, H.; Sigmundsdóttir, H.; Jónsdóttir, I. Is psoriasis induced by streptococcal superantigens and maintained by M-protein-specific T cells that cross-react with keratin? Clin. Exp. Immunol. 1997, 107 (Suppl S1), 21–24. [Google Scholar] [PubMed]
- Tokura, Y.; Seo, N.; Ohshima, A.; Wakita, H.; Yokote, R.; Furukawa, F.; Takigawa, M. Hyporesponsiveness of peripheral blood lymphocytes to streptococcal superantigens in patients with guttate psoriasis: Evidence for systemic stimulation of T cells with superantigens released from focally infecting Streptococcus pyogenes. Arch. Dermatol. Res. 1999, 291, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y.; Gately, M.; Trumble, A.; Ferguson-Darnell, B.; Schlievert, P.M.; Picker, L.J. Bacterial superantigens induce T cell expression of the skin-selective homing receptor, the cutaneous lymphocyte-associated antigen, via stimulation of interleukin 12 production. J. Exp. Med. 1995, 181, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Carrión, F.; Fernandez, M.; Iruretagoyena, M.; Coelho Andrade, L.E.; Odete-Hilário, M.; Figueroa, F. Selective depletion of Vbeta2+CD8+ T cells in peripheral blood from rheumatic heart disease patients. J. Autoimmun. 2003, 20, 183–190. [Google Scholar] [CrossRef]
- Baker, B.S.; Powles, A.; Fry, L. Peptidoglycan: A major aetiological factor for psoriasis? Trends Immunol. 2006, 27, 545–551. [Google Scholar] [CrossRef]
- Ghazizadeh, R.; Shimizu, H.; Tosa, M.; Ghazizadeh, M. Pathogenic mechanisms shared between psoriasis and cardiovascular disease. Int. J. Med. Sci. 2010, 7, 284–289. [Google Scholar] [CrossRef] [Green Version]
- Ferran, M.; Galván, A.B.; Rincón, C.; Romeu, E.R.; Sacrista, M.; Barboza, E.; Giménez-Arnau, A.; Celada, A.; Pujol, R.M.; Santamaria-Babí, L.F. Streptococcus induces circulating CLA(+) memory T-cell-dependent epidermal cell activation in psoriasis. J. Investig. Dermatol. 2013, 133, 999–1007. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Romeu, E.; Ferran, M.; Sagristà, M.; Gómez, J.; Giménez-Arnau, A.; Herszenyi, K.; Hóllo, P.; Celada, A.; Pujol, R.; Santamaria-Babí, L.F. Streptococcus pyogenes-induced cutaneous lymphocyte antigen-positive T cell-dependent epidermal cell activation triggers TH17 responses in patients with guttate psoriasis. J. Allergy Clin. Immunol. 2016, 138, 491–499.e6. [Google Scholar] [CrossRef] [Green Version]
- Valdimarsson, H.; Thorleifsdottir, R.H.; Sigurdardottir, S.L.; Gudjonsson, J.E.; Johnston, A. Psoriasis-as an autoimmune disease caused by molecular mimicry. Trends Immunol. 2009, 30, 494–501. [Google Scholar] [CrossRef]
- Pérez-Lorenzo, R.; Zambrano-Zaragoza, J.F.; Saul, A.; Jiménez-Zamudio, L.; Reyes-Maldonado, E.; García-Latorre, E. Autoantibodies to autologous skin in guttate and plaque forms of psoriasis and cross-reaction of skin antigens with streptococcal antigens. Int. J. Dermatol. 1998, 37, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Besgen, P.; Trommler, P.; Vollmer, S.; Prinz, J.C. Ezrin, maspin, peroxiredoxin 2, and heat shock protein 27: Potential targets of a streptococcal-induced autoimmune response in psoriasis. J. Immunol. 2010, 184, 5392–5402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshita, J.; Shin, D.B.; Ogdie, A.; Gelfand, J.M. Risk of Serious Infection, Opportunistic Infection, and Herpes Zoster among Patients with Psoriasis in the United Kingdom. J. Investig. Dermatol. 2018, 138, 1726–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupire, G.; Droitcourt, C.; Hughes, C.; Le Cleach, L. Antistreptococcal interventions for guttate and chronic plaque psoriasis. Cochrane Database Syst. Rev. 2019, 3, Cd011571. [Google Scholar] [CrossRef]
- Dogan, B.; Karabudak, O.; Harmanyeri, Y. Antistreptococcal treatment of guttate psoriasis: A controlled study. Int. J. Dermatol. 2008, 47, 950–952. [Google Scholar] [CrossRef]
- Simões, J.F.; Ribeiro, J.; Ferreira, B.R.; Paiva, S. The role of tonsillectomy in psoriasis treatment. BMJ Case Rep. 2015, 2015, bcr2014206899. [Google Scholar] [CrossRef] [Green Version]
- Thorleifsdottir, R.H.; Sigurdardottir, S.L.; Sigurgeirsson, B.; Olafsson, J.H.; Sigurdsson, M.I.; Petersen, H.; Arnadottir, S.; Gudjonsson, J.E.; Johnston, A.; Valdimarsson, H. Improvement of psoriasis after tonsillectomy is associated with a decrease in the frequency of circulating T cells that recognize streptococcal determinants and homologous skin determinants. J. Immunol. 2012, 188, 5160–5165. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.B.; Odum, N.; Gerwien, J.; Svejgaard, A.; Bendtzen, K.; Bregentholt, S.; Röpke, C.; Geisler, C.; Dohlsten, M.; Kaltoft, K. Staphylococcal enterotoxin-A directly stimulates signal transduction and interferon-gamma production in psoriatic T-cell lines. Tissue Antigens 1998, 52, 530–538. [Google Scholar] [CrossRef]
- Han, M.H.; Jang, K.A.; Sung, K.J.; Moon, K.C.; Koh, J.K.; Choi, J.H. A case of guttate psoriasis following Kawasaki disease. Br. J. Dermatol. 2000, 142, 548–550. [Google Scholar] [CrossRef]
- Halasz, C.L. Helicobacter pylori antibodies in patients with psoriasis. Arch. Dermatol. 1996, 132, 95–96. [Google Scholar] [CrossRef]
- Azizzadeh, M.; Nejad, Z.V.; Ghorbani, R.; Pahlevan, D. Relationship between Helicobacter pylori infection and psoriasis. Ann. Saudi Med. 2014, 34, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Whitehead, M. Clearance of chronic psoriasis after eradication therapy for Helicobacter pylori infection. J. Eur. Acad. Dermatol. Venereol. 2008, 22, 753–754. [Google Scholar] [CrossRef] [PubMed]
- Martin Hübner, A.; Tenbaum, S.P. Complete remission of palmoplantar psoriasis through Helicobacter pylori eradication: A case report. Clin. Exp. Dermatol. 2008, 33, 339–340. [Google Scholar] [CrossRef] [PubMed]
- Yen, Y.F.; Jen, I.A.; Chen, M.; Lan, Y.C.; Lee, C.Y.; Chuang, P.H.; Lee, Y.; Arthur Chen, Y.M. HIV Infection Increases the Risk of Incident Psoriasis: A Nationwide Population-Based Cohort Study in Taiwan. J. Acquir. Immune Defic. Syndr. 2017, 75, 493–499. [Google Scholar] [CrossRef]
- Mallon, E.; Young, D.; Bunce, M.; Gotch, F.M.; Easterbrook, P.J.; Newson, R.; Bunker, C.B. HLA-Cw*0602 and HIV-associated psoriasis. Br. J. Dermatol. 1998, 139, 527–533. [Google Scholar] [CrossRef]
- Kim, C.M.; Vogel, J.; Jay, G.; Rhim, J.S. The HIV tat gene transforms human keratinocytes. Oncogene 1992, 7, 1525–1529. [Google Scholar] [PubMed]
- Vogel, J.; Cepeda, M.; Enk, A.; Ngo, L.; Jay, G. The hiv tat gene is a promoter of epidermal skin tumors. Int. J. Oncol. 1995, 7, 727–733. [Google Scholar] [CrossRef]
- Mahoney, S.E.; Duvic, M.; Nickoloff, B.J.; Minshall, M.; Smith, L.C.; Griffiths, C.E.; Paddock, S.W.; Lewis, D.E. Human immunodeficiency virus (HIV) transcripts identified in HIV-related psoriasis and Kaposi’s sarcoma lesions. J. Clin. Investig. 1991, 88, 174–185. [Google Scholar] [CrossRef] [Green Version]
- Fife, D.J.; Waller, J.M.; Jeffes, E.W.; Koo, J.Y. Unraveling the paradoxes of HIV-associated psoriasis: A review of T-cell subsets and cytokine profiles. Dermatol. Online J. 2007, 13, 4. [Google Scholar] [CrossRef]
- Austin, L.M.; Ozawa, M.; Kikuchi, T.; Walters, I.B.; Krueger, J.G. The majority of epidermal T cells in Psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: A type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J. Investig. Dermatol. 1999, 113, 752–759. [Google Scholar]
- Parker, S.R. The skin and HIV: No superficial matter. Top. Antivir. Med. 2014, 22, 680–684. [Google Scholar] [PubMed]
- Lew, W.; Bowcock, A.M.; Krueger, J.G. Psoriasis vulgaris: Cutaneous lymphoid tissue supports T-cell activation and “Type ” inflammatory gene expression. Trends Immunol. 2004, 25, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Schlaak, J.F.; Buslau, M.; Jochum, W.; Hermann, E.; Girndt, M.; Gallati, H.; Meyer zum Büschenfelde, K.H.; Fleischer, B. T cells involved in psoriasis vulgaris belong to the Th1 subset. J. Investig. Dermatol. 1994, 102, 145–149. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.A.; Dobmeyer, J.M.; Dobmeyer, T.S.; Pape, M.; Ottmann, O.G.; Helm, E.B.; Hoelzer, D.; Rossol, R. Demonstration of the Th1 to Th2 cytokine shift during the course of HIV-1 infection using cytoplasmic cytokine detection on single cell level by flow cytometry. AIDS 1997, 11, 1111–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roederer, M.; Dubs, J.G.; Anderson, M.T.; Raju, P.A.; Herzenberg, L.A.; Herzenberg, L.A. CD8 naive T cell counts decrease progressively in HIV-infected adults. J. Clin. Investig. 1995, 95, 2061–2066. [Google Scholar] [CrossRef] [PubMed]
- Morar, N.; Willis-Owen, S.A.; Maurer, T.; Bunker, C.B. HIV-associated psoriasis: Pathogenesis, clinical features, and management. Lancet. Infect. Dis. 2010, 10, 470–478. [Google Scholar] [CrossRef]
- Torres, B.A.; Johnson, H.M. Identification of an HIV-1 Nef peptide that binds to HLA class II antigens. Biochem. Biophys. Res. Commun. 1994, 200, 1059–1065. [Google Scholar] [CrossRef]
- Townsley-Fuchs, J.; Neshat, M.S.; Margolin, D.H.; Braun, J.; Goodglick, L. HIV-1 gp120: A novel viral B cell superantigen. Int. Rev. Immunol. 1997, 14, 325–338. [Google Scholar] [CrossRef]
- Fielder, L.M.; Harvey, V.M.; Kishor, S.I. Necrolytic acral erythema: Case report and review of the literature. Cutis 2008, 81, 355–360. [Google Scholar]
- Yamamoto, T.; Katayama, I.; Nishioka, K. Psoriasis and hepatitis C virus. Acta Derm. Venereol. 1995, 75, 482–483. [Google Scholar]
- Cohen, A.D.; Weitzman, D.; Birkenfeld, S.; Dreiher, J. Psoriasis associated with hepatitis C but not with hepatitis B. Dermatology 2010, 220, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Afshar, M.; Martinez, A.D.; Gallo, R.L.; Hata, T.R. Induction and exacerbation of psoriasis with Interferon-alpha therapy for hepatitis C: A review and analysis of 36 cases. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 771–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, A.W.; Read, C. Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review. Jama 2020, 323, 1945–1960. [Google Scholar] [CrossRef] [PubMed]
- Spearman, C.W.; Dusheiko, G.M.; Hellard, M.; Sonderup, M. Hepatitis C. Lancet 2019, 394, 1451–1466. [Google Scholar]
- Imafuku, S.; Naito, R.; Nakayama, J. Possible association of hepatitis C virus infection with late-onset psoriasis: A hospital-based observational study. J. Dermatol. 2013, 40, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Chun, K.; Afshar, M.; Audish, D.; Kabigting, F.; Paik, A.; Gallo, R.; Hata, T. Hepatitis C may enhance key amplifiers of psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Claudy, A.L.; Touraine, J.L.; Mitanne, D. Epidermodysplasia verruciformis induced by a new human papillomavirus (HPV-8). Report of a case without immune dysfunction. Effect of treatment with an aromatic retinoid. Arch. Dermatol. Res. 1982, 274, 213–219. [Google Scholar] [CrossRef]
- Chen, M.L.; Kao, W.M.; Huang, J.Y.; Hung, Y.M.; Wei, J.C. Human papillomavirus infection associated with increased risk of new-onset psoriasis: A nationwide population-based cohort study. Int. J. Epidemiol. 2020, 49, 786–797. [Google Scholar] [CrossRef]
- Cronin, J.G.; Mesher, D.; Purdie, K.; Evans, H.; Breuer, J.; Harwood, C.A.; McGregor, J.M.; Proby, C.M. Beta-papillomaviruses and psoriasis: An intra-patient comparison of human papillomavirus carriage in skin and hair. Br. J. Dermatol. 2008, 159, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Simeone, P.; Teson, M.; Latini, A.; Carducci, M.; Venuti, A. Human papillomavirus type 5 in primary keratinocytes from psoriatic skin. Exp. Dermatol. 2005, 14, 824–829. [Google Scholar] [CrossRef]
- de Villiers, E.M.; Ruhland, A. Do specific human papillomavirus types cause psoriasis? Arch. Dermatol. 2001, 137, 384. [Google Scholar]
- Lee, L.A.; Huang, C.G.; Tsao, K.C.; Liao, C.T.; Kang, C.J.; Chang, K.P.; Huang, S.F.; Chen, I.H.; Fang, T.J.; Li, H.Y.; et al. Human Papillomavirus Infections are Common and Predict Mortality in a Retrospective Cohort Study of Taiwanese Patients With Oral Cavity Cancer. Medicine 2015, 94, e2069. [Google Scholar] [CrossRef] [PubMed]
- Favre, M.; Orth, G.; Majewski, S.; Baloul, S.; Pura, A.; Jablonska, S. Psoriasis: A possible reservoir for human papillomavirus type 5, the virus associated with skin carcinomas of epidermodysplasia verruciformis. J. Investig. Dermatol. 1998, 110, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahé, E.; Bodemer, C.; Descamps, V.; Mahé, I.; Crickx, B.; De Prost, Y.; Favre, M. High frequency of detection of human papillomaviruses associated with epidermodysplasia verruciformis in children with psoriasis. Br. J. Dermatol. 2003, 149, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Majewski, S.; Jablonska, S. Possible involvement of epidermodysplasia verruciformis human papillomaviruses in the immunopathogenesis of psoriasis: A proposed hypothesis. Exp. Dermatol. 2003, 12, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Majewski, S.; Jablonska, S. Do epidermodysplasia verruciformis human papillomaviruses contribute to malignant and benign epidermal proliferations? Arch. Dermatol. 2002, 138, 649–654. [Google Scholar] [CrossRef]
- Griffiths, P.; Reeves, M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat. Reviews. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef]
- Philipp, S.; Wolk, K.; Kreutzer, S.; Wallace, E.; Ludwig, N.; Roewert, J.; Höflich, C.; Volk, H.D.; Sterry, W.; Sabat, R. The evaluation of psoriasis therapy with biologics leads to a revision of the current view of the pathogenesis of this disorder. Expert Opin. Ther. Targets 2006, 10, 817–831. [Google Scholar] [CrossRef]
- Yoneda, K.; Matsuoka-Shirahige, Y.; Demitsu, T.; Kubota, Y. Pustular psoriasis precipitated by cytomegalovirus infection. Br. J. Dermatol. 2012, 167, 1186–1189. [Google Scholar] [CrossRef]
- Salem, S.A.; Zuel-Fakkar, N.M.; Fathi, G.; Abd El-Reheem, S.M.; Abd El-monem El-Tabakh, A.; Ragab, D.M. Comparative study of human papilloma virus in untreated and ultraviolet-treated psoriatic patients. Photodermatol. Photoimmunol. Photomed. 2010, 26, 78–82. [Google Scholar] [CrossRef]
- Paniz Mondolfi, A.E.; Hernandez Perez, M.; Blohm, G.; Marquez, M.; Mogollon Mendoza, A.; Hernandez-Pereira, C.E.; Escalona, M.A.; Lodeiro Colatosti, A.; Rothe DeArocha, J.; Rodriguez Morales, A.J. Generalized pustular psoriasis triggered by Zika virus infection. Clin. Exp. Dermatol. 2018, 43, 171–174. [Google Scholar] [CrossRef]
- Joob, B.; Wiwanitkit, V. Psoriasis triggered by Zika virus infection. Clin. Exp. Dermatol. 2018, 43, 937. [Google Scholar] [CrossRef] [PubMed]
- Gananandan, K.; Sacks, B.; Ewing, I. Guttate psoriasis secondary to COVID-19. BMJ Case Rep. 2020, 13, e237367. [Google Scholar] [CrossRef] [PubMed]
- Rouai, M.; Rabhi, F.; Mansouri, N.; Jaber, K.; Dhaoui, R. New-onset guttate psoriasis secondary to COVID-19. Clin. Case Rep. 2021, 9, e04542. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, R.J.; Cobb, C.B.C.; Telang, G.H.; Firoz, E.F. New-onset pustular psoriasis in the setting of severe acute respiratory syndrome coronavirus 2 infection causing coronavirus disease 2019. JAAD Case Rep. 2020, 6, 1360–1362. [Google Scholar] [CrossRef] [PubMed]
- Sbidian, E.; Madrange, M.; Viguier, M.; Salmona, M.; Duchatelet, S.; Hovnanian, A.; Smahi, A.; Le Goff, J.; Bachelez, H. Respiratory virus infection triggers acute psoriasis flares across different clinical subtypes and genetic backgrounds. Br. J. Dermatol. 2019, 181, 1304–1306. [Google Scholar] [CrossRef]
- Marrakchi, S.; Guigue, P.; Renshaw, B.R.; Puel, A.; Pei, X.Y.; Fraitag, S.; Zribi, J.; Bal, E.; Cluzeau, C.; Chrabieh, M.; et al. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 2011, 365, 620–628. [Google Scholar] [CrossRef]
- Hewson, C.A.; Jardine, A.; Edwards, M.R.; Laza-Stanca, V.; Johnston, S.L. Toll-like receptor 3 is induced by and mediates antiviral activity against rhinovirus infection of human bronchial epithelial cells. J. Virol. 2005, 79, 12273–12279. [Google Scholar] [CrossRef] [Green Version]
- Ozaras, R.; Berk, A.; Ucar, D.H.; Duman, H.; Kaya, F.; Mutlu, H. COVID-19 and exacerbation of psoriasis. Dermatol. Ther. 2020, 33, e13632. [Google Scholar] [CrossRef]
- Aydogan, K.; Tore, O.; Akcaglar, S.; Oral, B.; Ener, B.; Tunalı, S.; Saricaoglu, H. Effects of Malassezia yeasts on serum Th1 and Th2 cytokines in patients with guttate psoriasis. Int. J. Dermatol. 2013, 52, 46–52. [Google Scholar] [CrossRef]
- Gomez-Moyano, E.; Crespo-Erchiga, V.; Martínez-Pilar, L.; Godoy Diaz, D.; Martínez-García, S.; Lova Navarro, M.; Vera Casaño, A. Do Malassezia species play a role in exacerbation of scalp psoriasis? J. De Mycol. Med. 2014, 24, 87–92. [Google Scholar] [CrossRef]
- Bonifaz, A.; Rojas, R.; Tirado-Sánchez, A.; Chávez-López, D.; Mena, C.; Calderón, L.; María, P.O. Superficial Mycoses Associated with Diaper Dermatitis. Mycopathologia 2016, 181, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E.W.; Belew, P.; Bale, G. Effect of topical applications of heavy suspensions of killed Malassezia ovalis on rabbit skin. Mycopathologia 1980, 72, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Skinner, R.B., Jr.; Noah, P.W.; Taylor, R.M.; Zanolli, M.D.; West, S.; Guin, J.D.; Rosenberg, E.W. Double-blind treatment of seborrheic dermatitis with 2% ketoconazole cream. J. Am. Acad. Dermatol. 1985, 12, 852–856. [Google Scholar] [CrossRef]
- Taheri Sarvtin, M.; Shokohi, T.; Hajheydari, Z.; Yazdani, J.; Hedayati, M.T. Evaluation of candidal colonization and specific humoral responses against Candida albicans in patients with psoriasis. Int. J. Dermatol. 2014, 53, e555–e560. [Google Scholar] [CrossRef] [PubMed]
- Vijaya Chandra, S.H.; Srinivas, R.; Dawson, T.L., Jr.; Common, J.E. Cutaneous Malassezia: Commensal, Pathogen, or Protector? Front. Cell. Infect. Microbiol. 2020, 10, 614446. [Google Scholar] [CrossRef]
- Rudramurthy, S.M.; Honnavar, P.; Chakrabarti, A.; Dogra, S.; Singh, P.; Handa, S. Association of Malassezia species with psoriatic lesions. Mycoses 2014, 57, 483–488. [Google Scholar] [CrossRef]
- Lober, C.W.; Belew, P.W.; Rosenberg, E.W.; Bale, G. Patch tests with killed sonicated microflora in patients with psoriasis. Arch. Dermatol. 1982, 118, 322–325. [Google Scholar] [CrossRef]
- Bunse, T.; Mahrle, G. Soluble Pityrosporum-derived chemoattractant for polymorphonuclear leukocytes of psoriatic patients. Acta Derm. Venereol. 1996, 76, 10–12. [Google Scholar]
- Baroni, A.; Paoletti, I.; Ruocco, E.; Agozzino, M.; Tufano, M.A.; Donnarumma, G. Possible role of Malassezia furfur in psoriasis: Modulation of TGF-beta1, integrin, and HSP70 expression in human keratinocytes and in the skin of psoriasis-affected patients. J. Cutan. Pathol. 2004, 31, 35–42. [Google Scholar] [CrossRef]
- Hernández-Santos, N.; Gaffen, S.L. Th17 cells in immunity to Candida albicans. Cell Host Microbe 2012, 11, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Javad, G.; Taheri Sarvtin, M.; Hedayati, M.T.; Hajheydari, Z.; Yazdani, J.; Shokohi, T. Evaluation of Candida Colonization and Specific Humoral Responses against Candida albicans in Patients with Atopic Dermatitis. BioMed Res. Int. 2015, 2015, 849206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Wang, R.; Song, Y.; Wan, Z.; Chen, W.; Li, H.; Li, R. Dysbiosis of nail microbiome in patients with psoriasis. Exp. Dermatol. 2022, 31, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Furue, M.; Furue, K.; Tsuji, G.; Nakahara, T. Interleukin-17A and Keratinocytes in Psoriasis. Int. J. Mol. Sci. 2020, 21, 1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, S.; Yao, Z. Roles of Infection in Psoriasis. Int. J. Mol. Sci. 2022, 23, 6955. https://doi.org/10.3390/ijms23136955
Zhou S, Yao Z. Roles of Infection in Psoriasis. International Journal of Molecular Sciences. 2022; 23(13):6955. https://doi.org/10.3390/ijms23136955
Chicago/Turabian StyleZhou, Shihui, and Zhirong Yao. 2022. "Roles of Infection in Psoriasis" International Journal of Molecular Sciences 23, no. 13: 6955. https://doi.org/10.3390/ijms23136955
APA StyleZhou, S., & Yao, Z. (2022). Roles of Infection in Psoriasis. International Journal of Molecular Sciences, 23(13), 6955. https://doi.org/10.3390/ijms23136955