Regulation of S100As Expression by Inflammatory Cytokines in Chronic Lymphocytic Leukemia

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
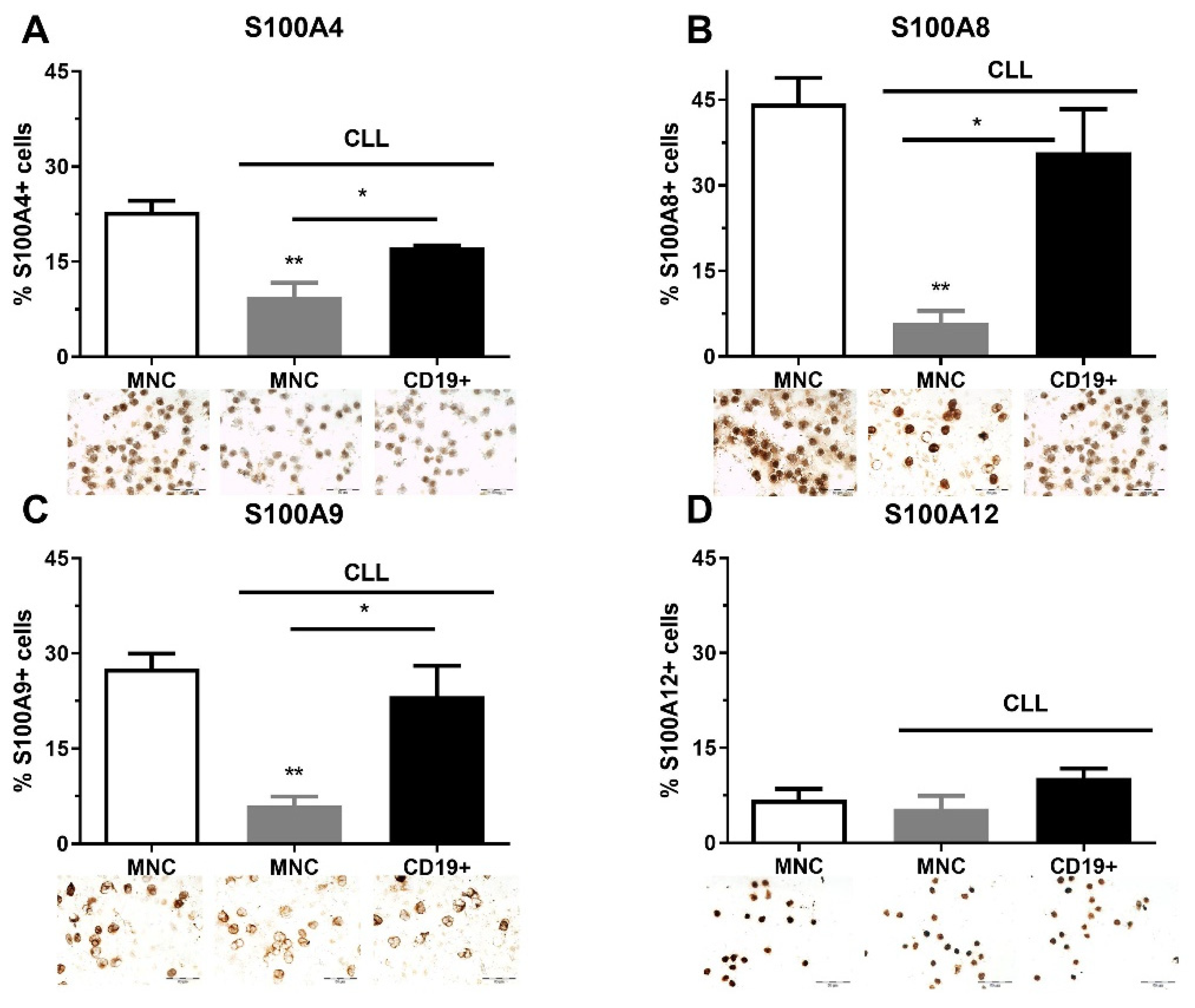
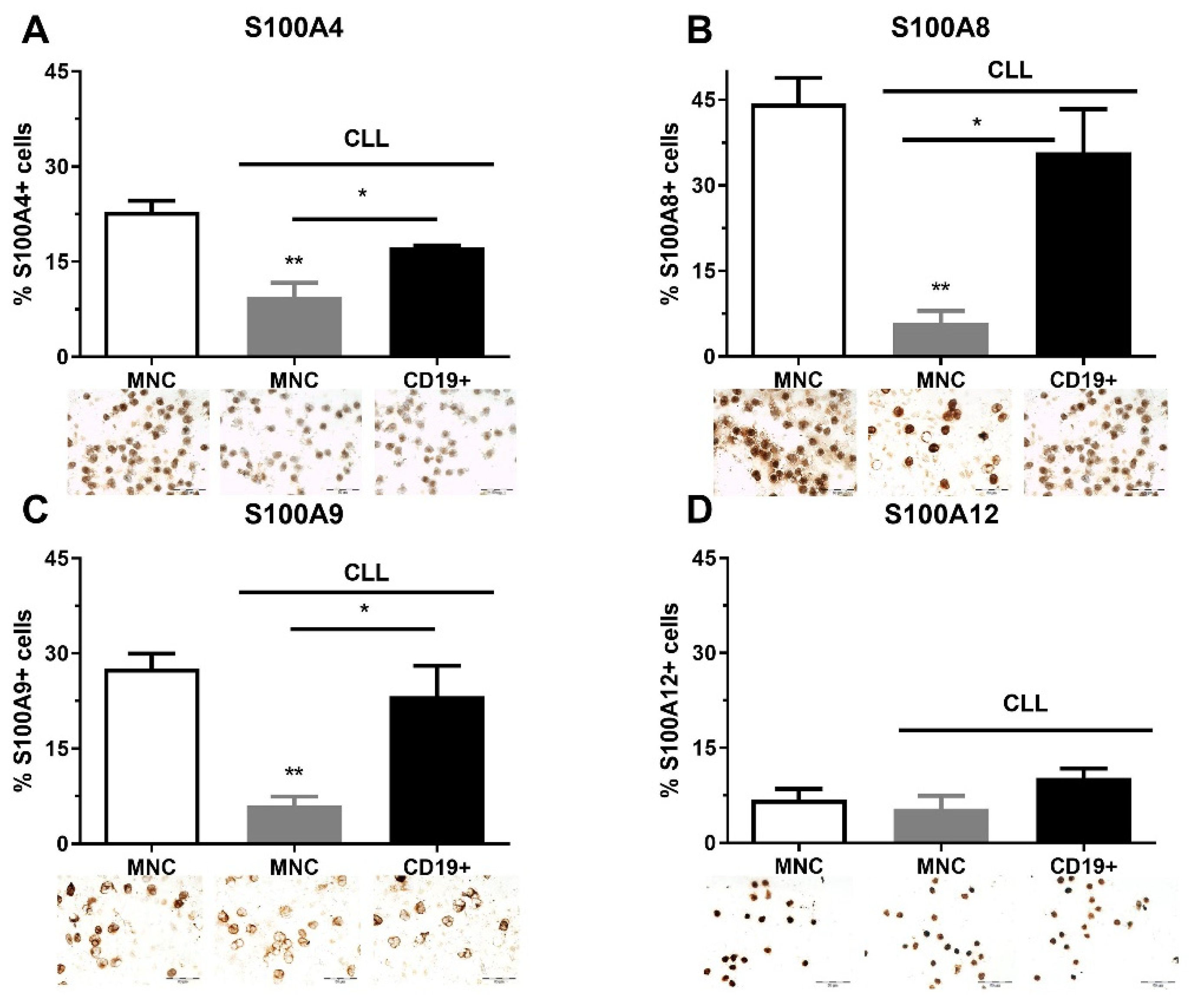
2.1. The Quantity of S100As Immunopositive MNCs and CD19+ Cells in CLL
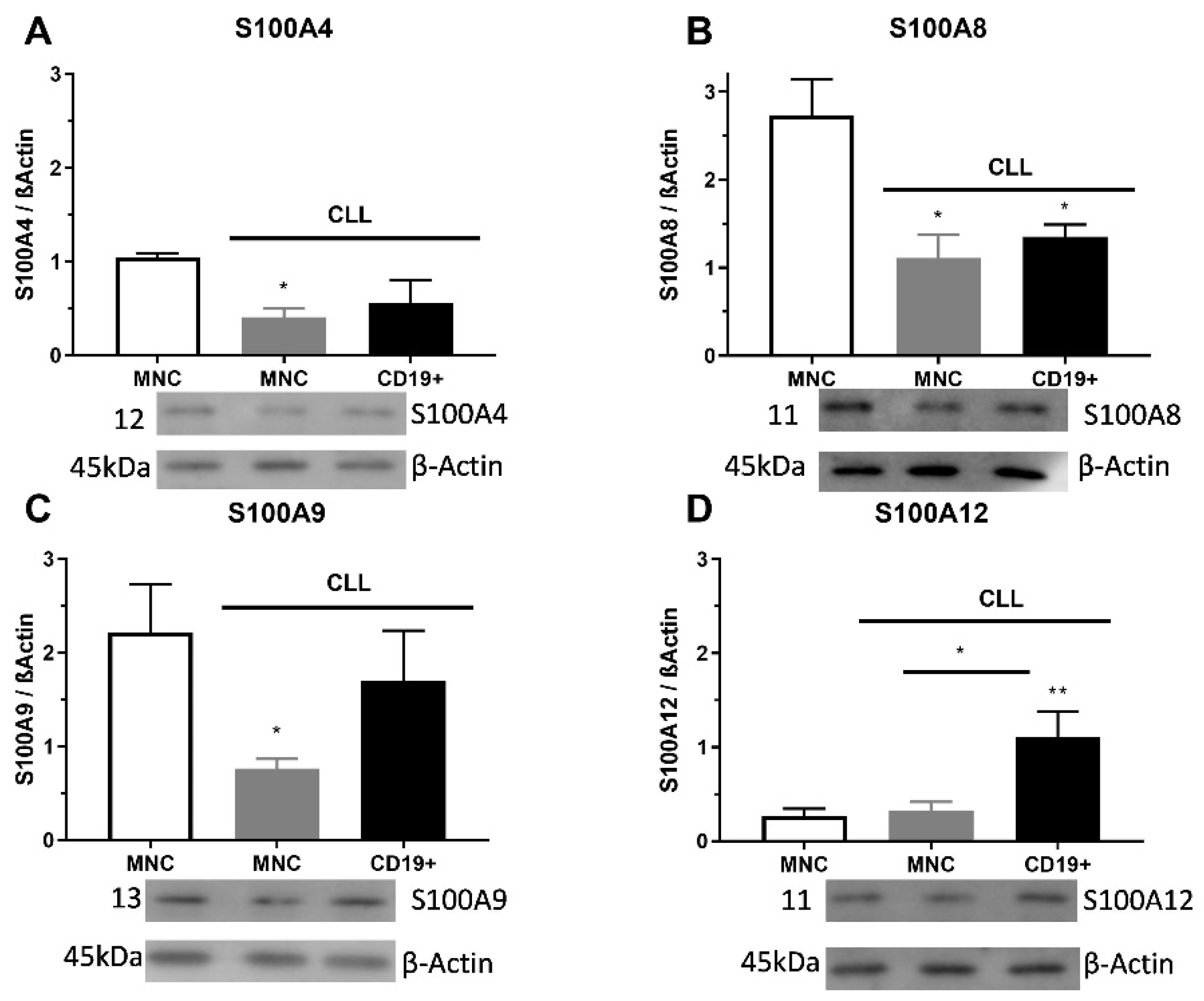
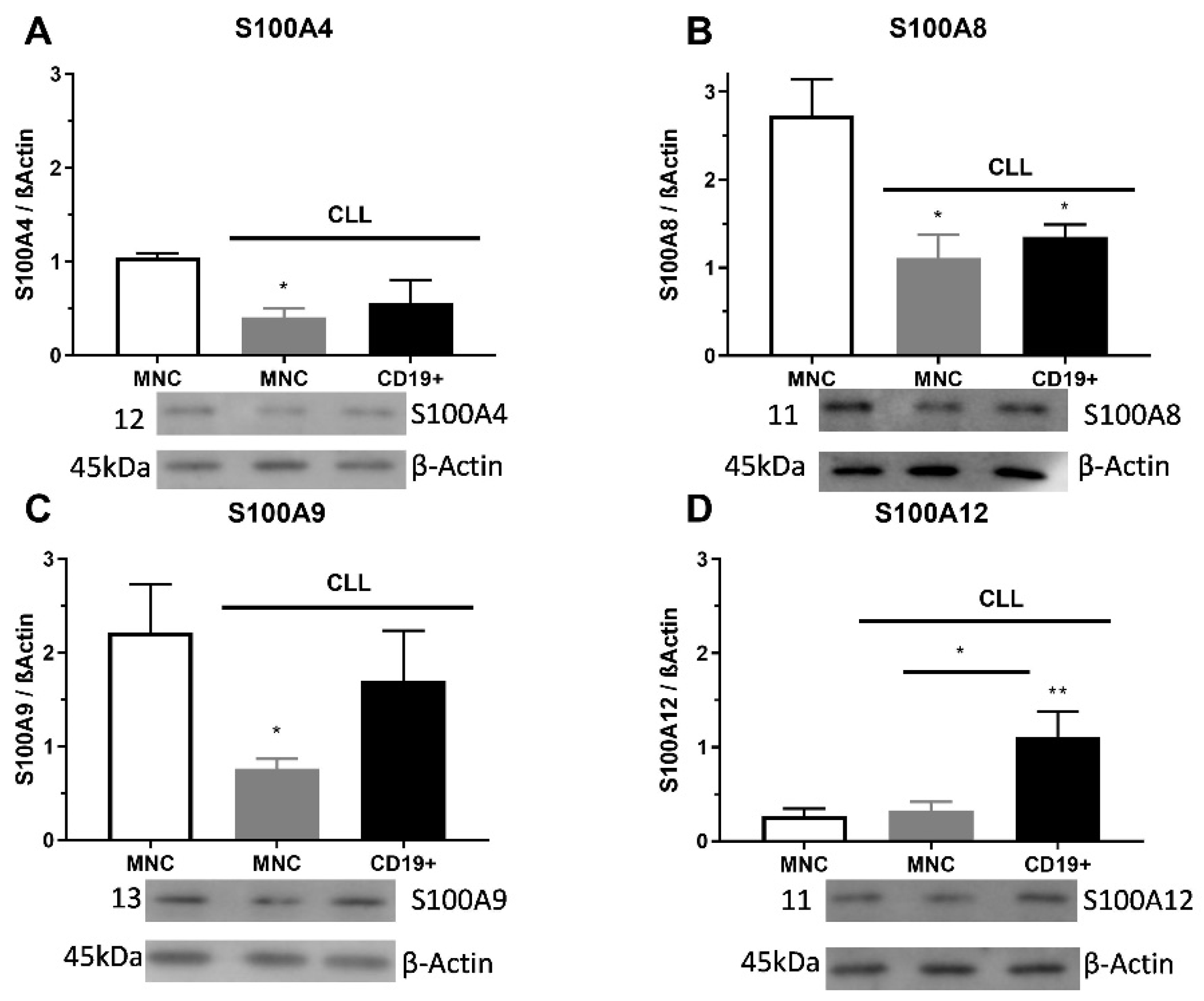
2.2. S100As Protein Expression in MNCs and CD19+ Cells of CLL Patients
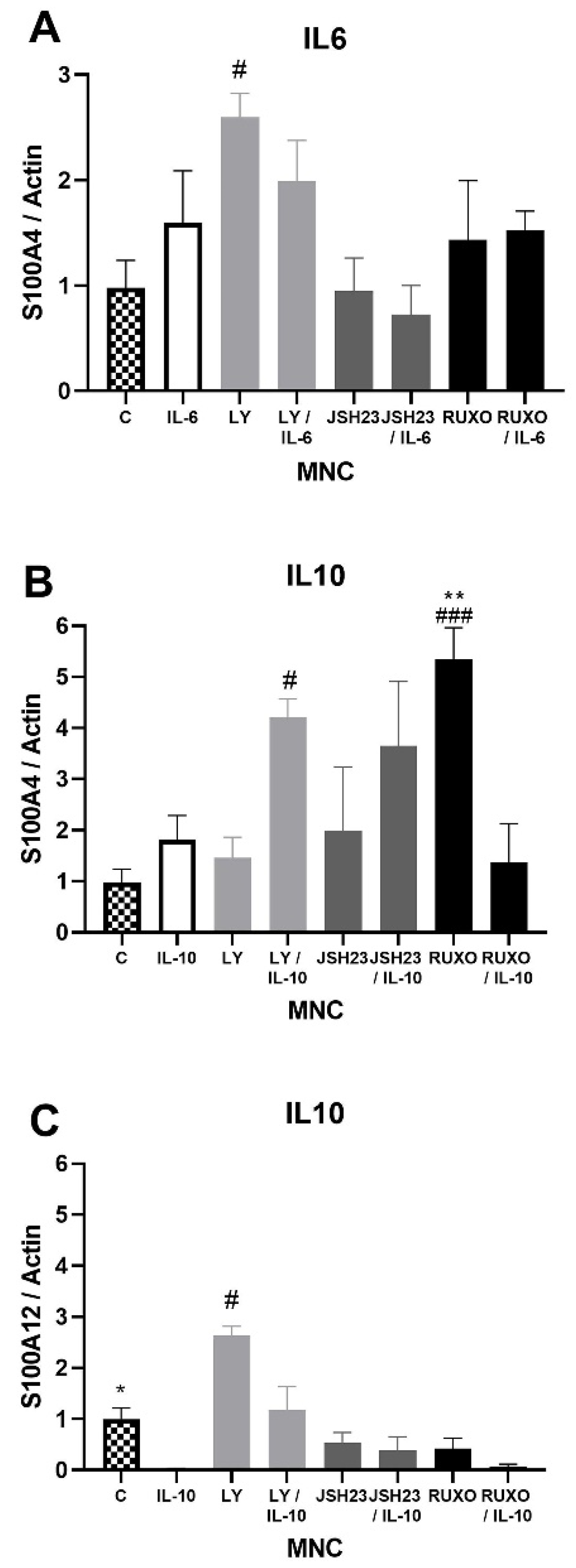
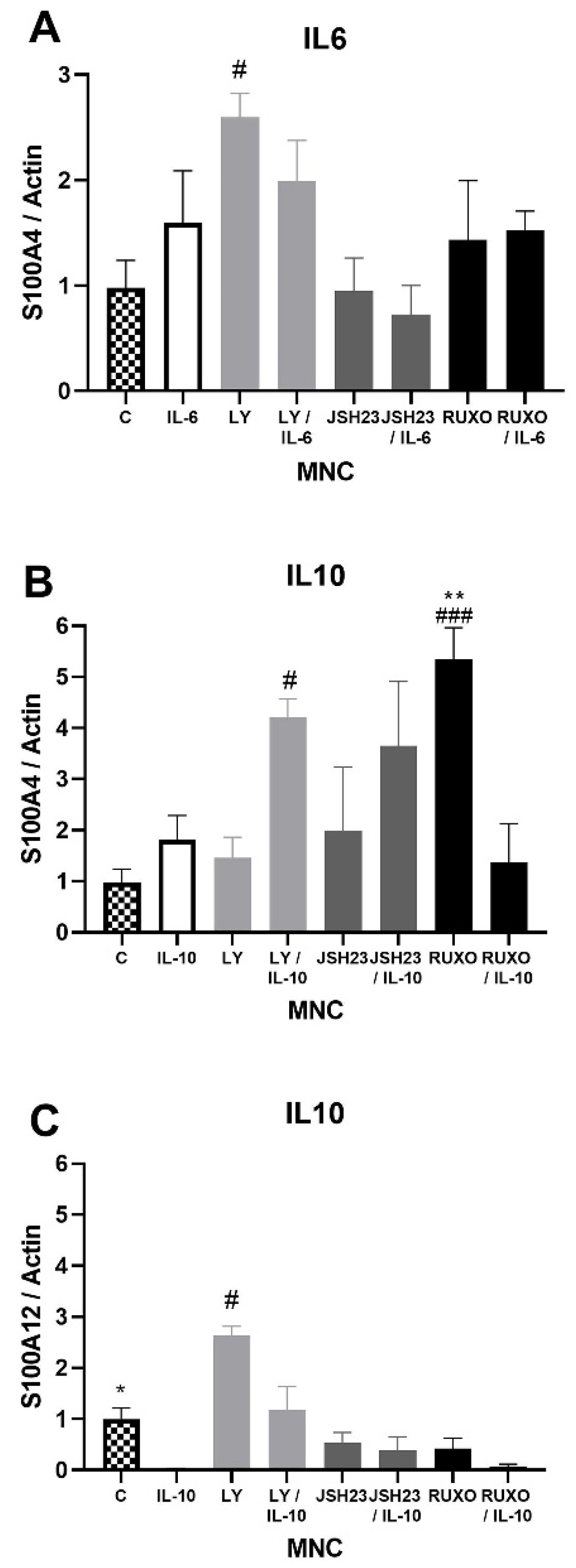
2.3. S100As Gene Expression in MNCs of CLL after IL-6 and IL-10 Treatment
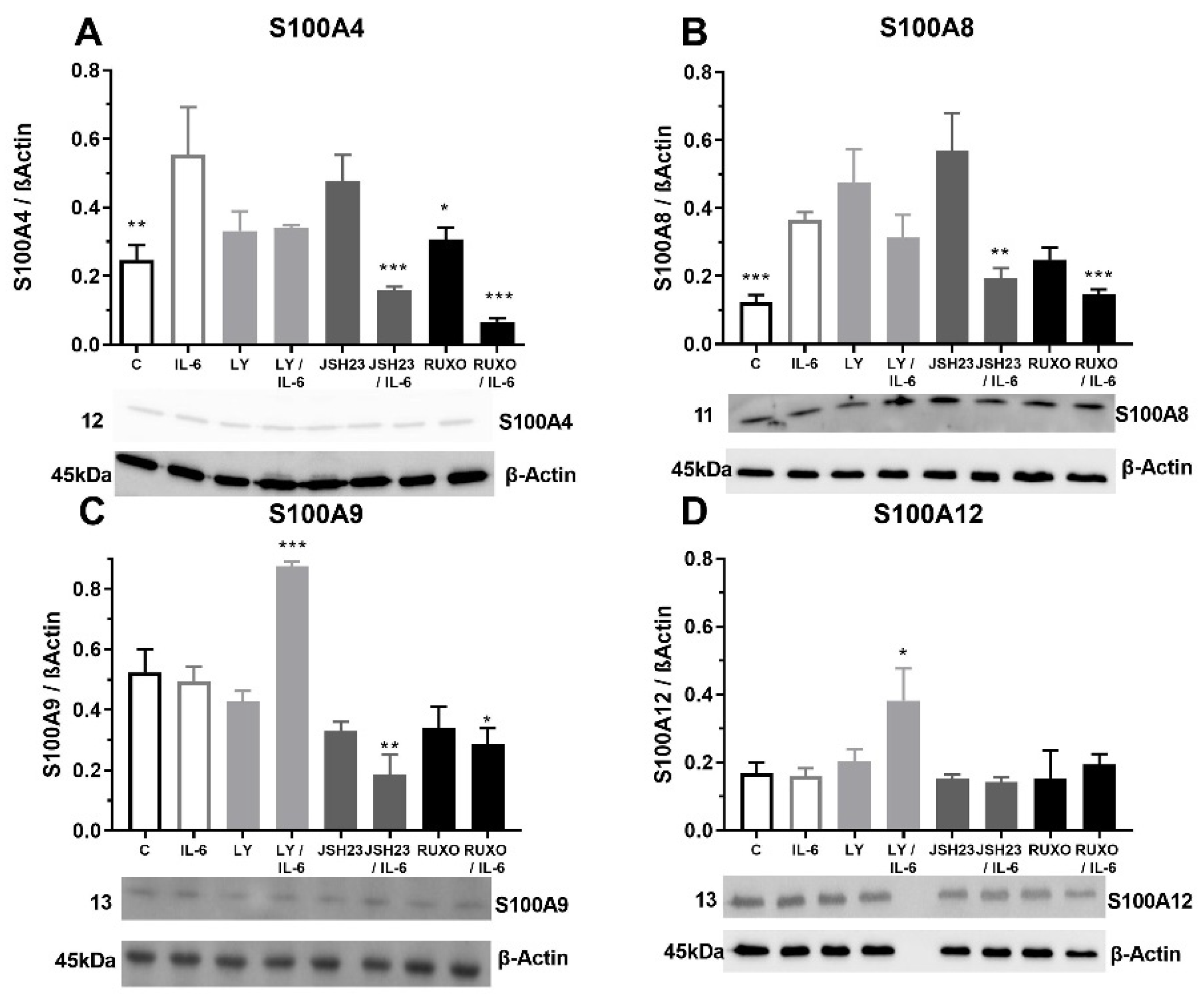
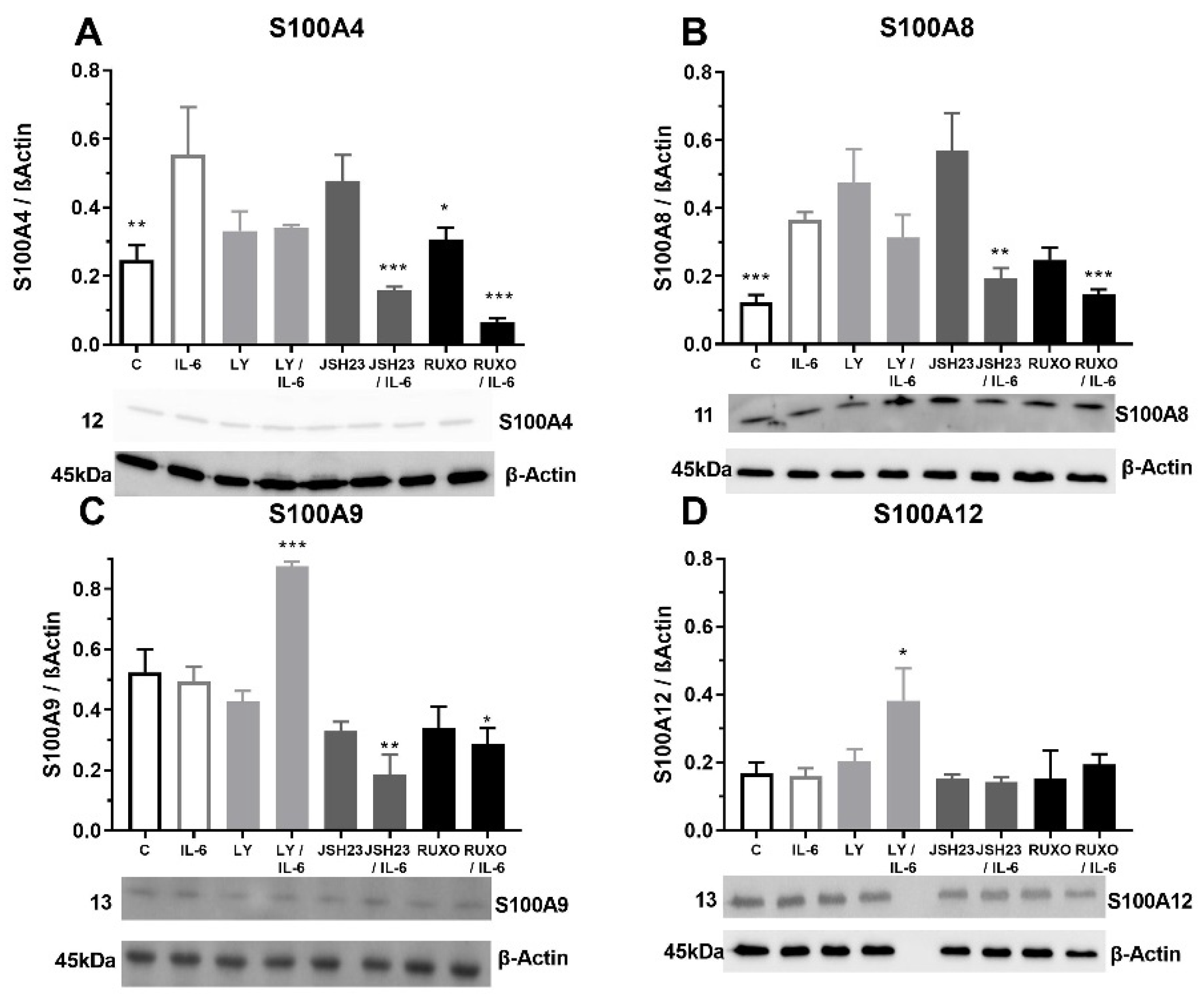
2.4. S100As Protein Expression after Treatment of MNCs with the Proinflammatory Cytokine IL-6
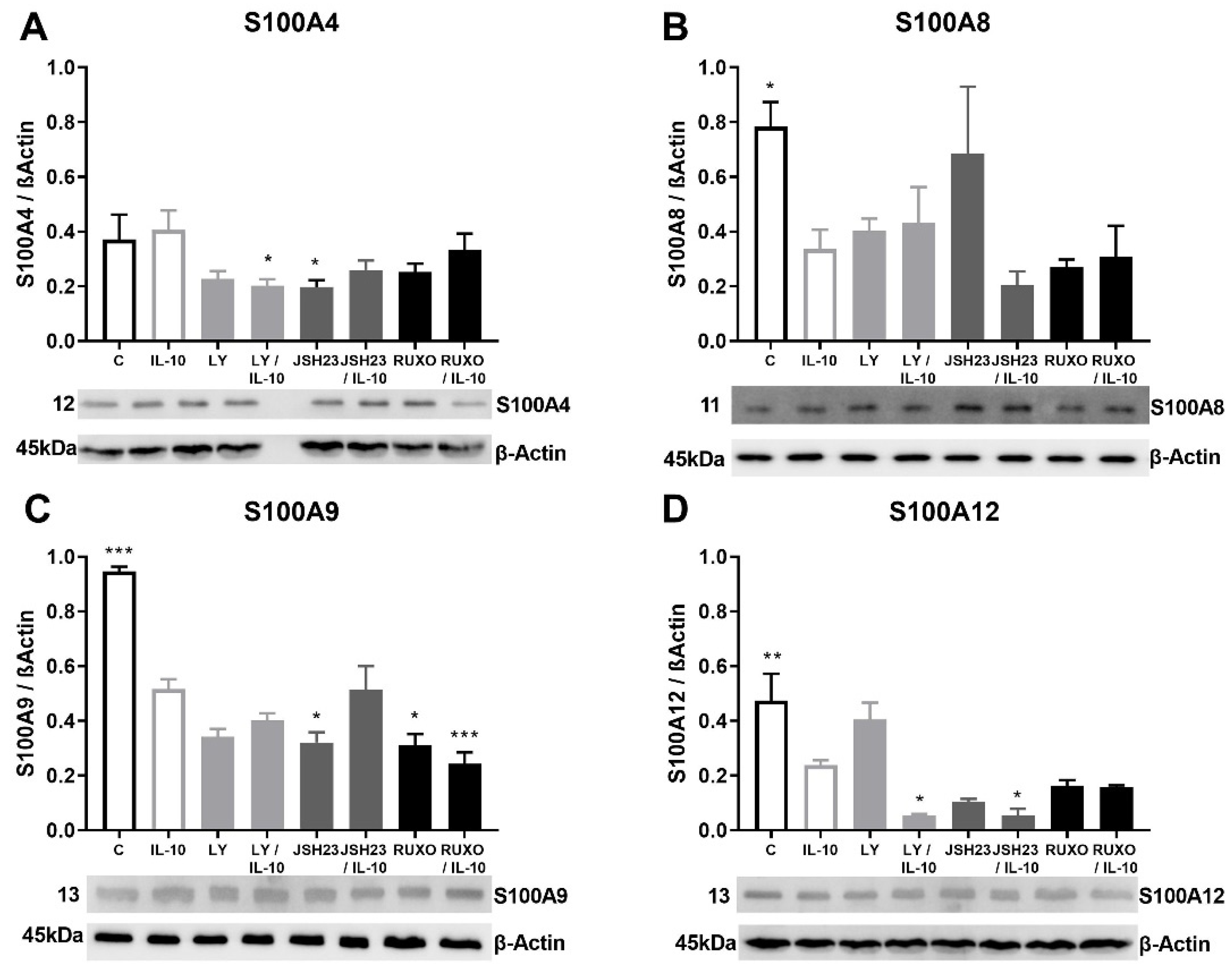
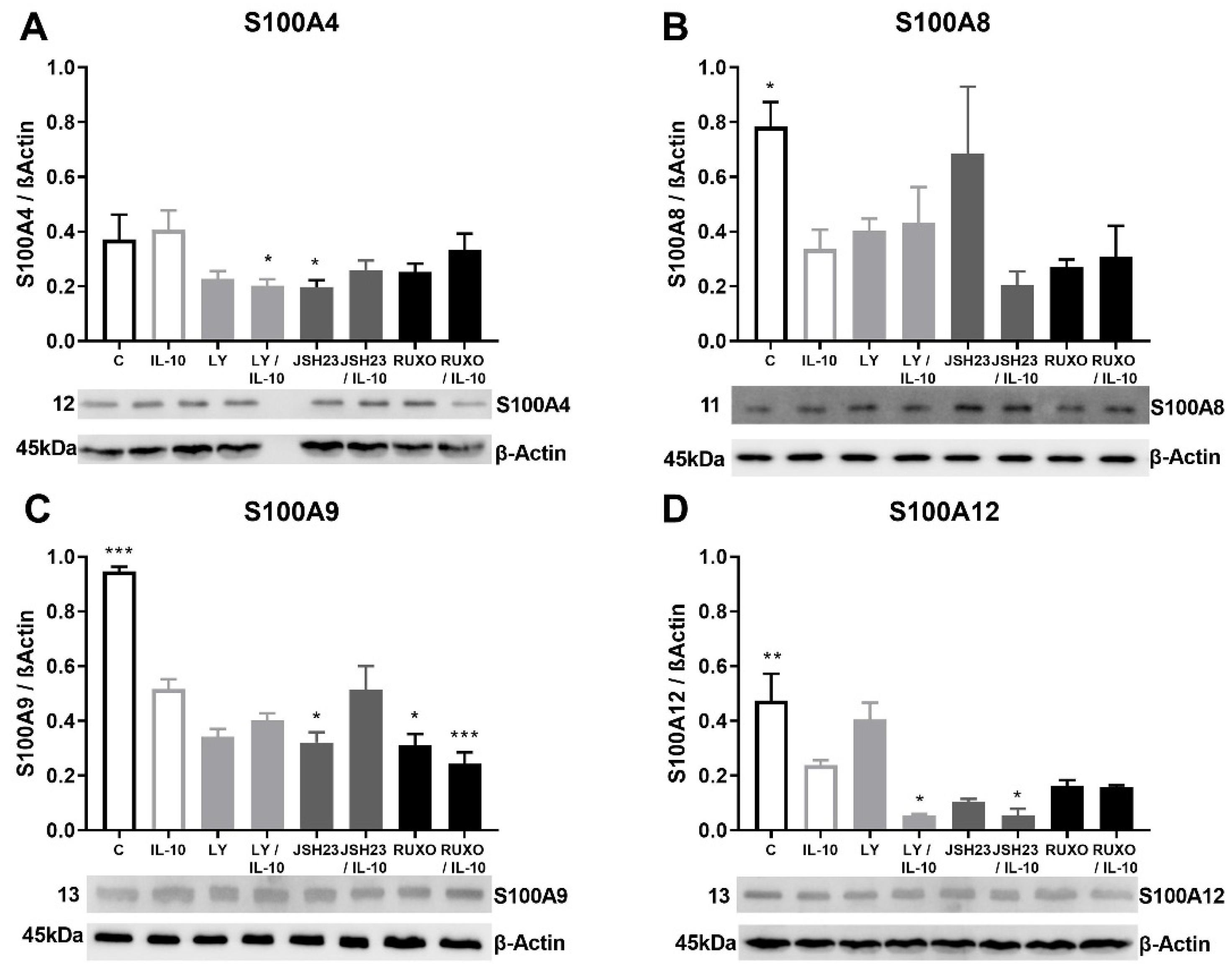
2.5. S100As Protein Expression after Treatment of MNCs with Anti-Inflammatory Cytokine IL-10
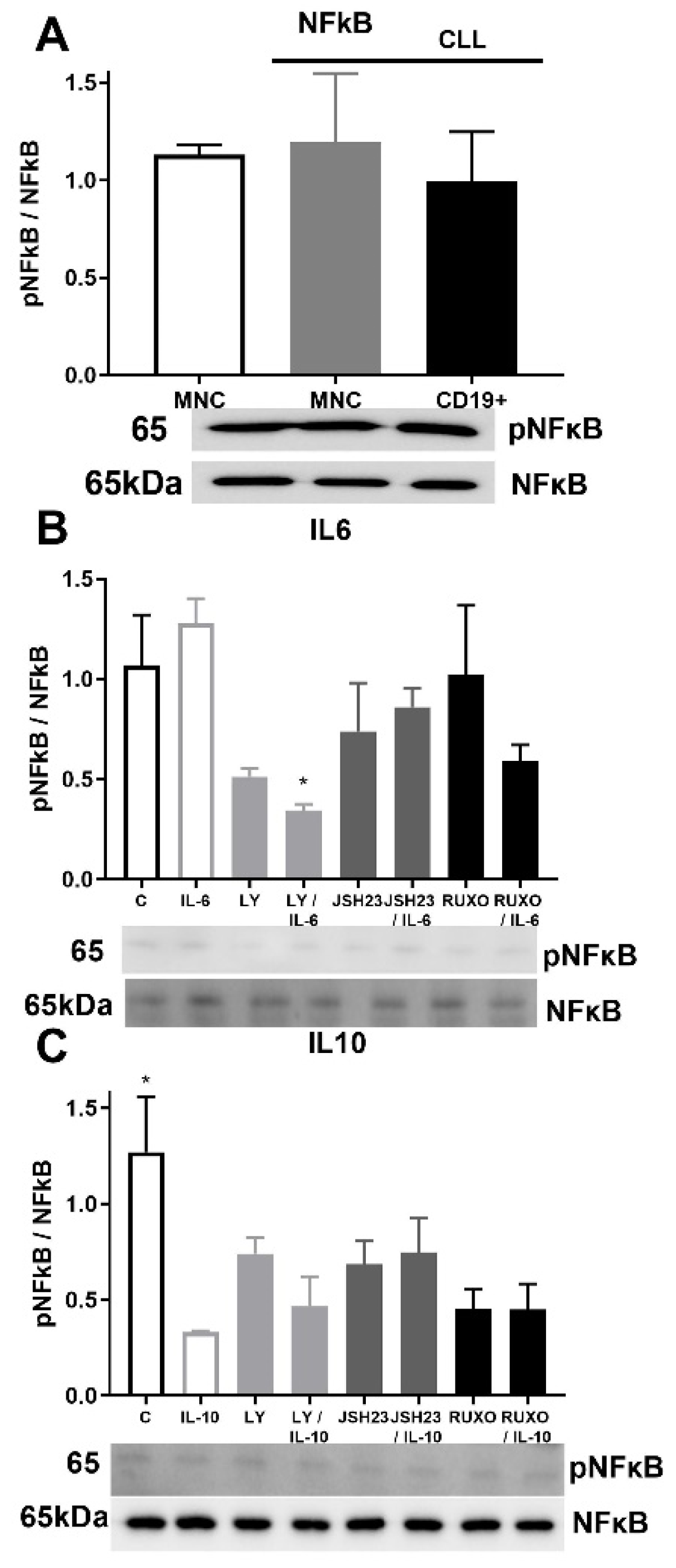
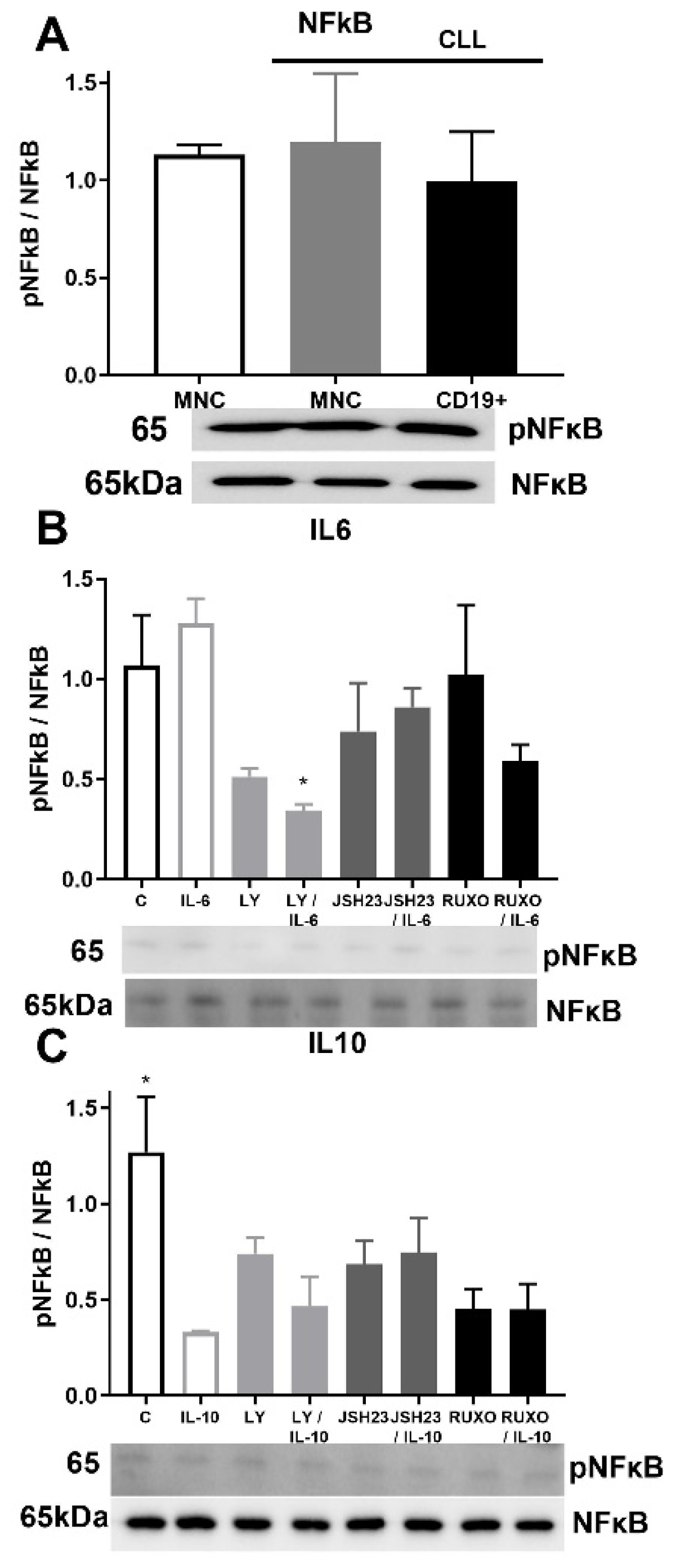
2.6. pNF-κB/NF-κB Signaling in CD19+ and MNCs of CLL after Treatment with IL-6 and IL-10
3. Discussion
4. Materials and Methods
4.1. Western Blot Analysis
4.2. Immunocytochemistry Analysis
4.3. Isolation of RNA and RT-PCR
4.4. RT-q PCR Analysis
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, F.-T.; Jia, L.; Wang, P.; Wang, H.; Farren, T.W.; Agrawal, S.G. STAT3 and NF-kappaB cooperatively control in vitro spontaneous apoptosis and poor chemo-responsiveness in patients with chronic lymphocytic leukemia. Oncotarget 2016, 7, 32031–32045. [Google Scholar]
- Liu, Z.; Hazan-Halevy, I.; Harris, D.M.; Li, P.; Ferrajoli, A.; Faderl, S.; Keating, M.-J.; Estrov, Z. STAT-3 Activates NF-kB in Chronic Lymphocytic Leukemia Cells. Mol. Cancer Res. 2011, 9, 507–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balakrishnan, K.; Burger, J.-A.; Wierda, W.-G.; Gandhi, V. AT-101 induces apoptosis in CLL B cells and overcomes stromal cell-mediated Mcl-1 induction and drug resistance. Blood 2010, 113, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Clear, A.; Liu, F.T.; Matthews, J.; Uddin, N.; McCarthy, A.; Hoxha, E.; Durance, C.; Iqbal, S.; Gribben, J.-G. Extracellular HMGB1 promotes differentiation of nurse-like cells in chronic lymphocytic leukemia. Blood 2014, 123, 1709–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar-Hernandez, M.-M.; Blunt, M.D.; Dobson, R.; Yeomans, A.; Thirdborough, S.; Larrayoz, M.; Smith, L.-D.; Linley, A.; Strefford, J.-C.; Davies, A.; et al. IL-4 enhances expression and function of surface IgM in CLL cells. Blood 2016, 127, 3015–3025. [Google Scholar] [CrossRef] [Green Version]
- Drennan, S.; D’Avola, A.; Gao, Y.; Weigel, C.; Chrysostomou, E.; Steele, A.-J.; Zenz, T.; Plass, C.; Johnson, P.-W.; Williams, A.-P.; et al. IL-10 production by CLL cells is enhanced in the anergic IGHV mutated subset and associates with reduced DNA methylation of the IL10 locus. Leukemia 2017, 31, 1686–1694. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.-A.; Ecsédi, P.; Kovács, G.-M.; Póti, Á.-L.; Reményi, A.; Kardos, J.; Gógl, G.; Nyitray, L. High-throughput competitive fluorescence polarization assay reveals functional redundancy in the S100 protein family. FEBS J. 2020, 287, 2834–2846. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [Green Version]
- Hatoum, D.; Yagoub, D.; Ahadi, A.; McGoman, E. Annexin/S100A protein family regulation through p14ARF-p53 activation: A role in cell survival and predicting treatment outcomes in breast cancer. PLoS ONE 2017, 12, e0169925. [Google Scholar] [CrossRef] [Green Version]
- Prieto, D.; Sotelo, N.; Seija, N.; Sernbo, S.; Abreu, C.; Durán, R.; Gil, M.; Sicco, E.; Irigoin, V.; Oliver, C.; et al. S100-A9 protein in exosomes from chronic lymphocytic leukemia cells promotes NF-kappaB activity during disease progression. Blood 2017, 130, 777–788. [Google Scholar] [CrossRef] [Green Version]
- Kovačić, M.; Mitrović-Ajtić, O.; Beleslin-Čokić, B.; Djikić, D.; Subotički, T.; Diklić, M.; Leković, D.; Gotić, M.; Mossuz, P.; Čokić, V.P. TLR4 and RAGE conversely mediate pro-inflammatory S100A8/9-mediated inhibition of proliferation-linked signaling in myeloproliferative neoplasms. Cell. Oncol. 2018, 41, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Barcellini, W.; Imperiali, F.-G.; Zaninoni, A.; Reda, G.; Consonni, D.; Fattizzo, B.; Lonati, S.; Nobili, L.; Zanella, A.; Cortelezzi, A. Toll-like receptor 4 and 9 expressions in B-chronic lymphocytic leukemia: Relationship with infections, autoimmunity, and disease progression. Leuk. Lymphoma 2014, 55, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Skorka, K.; Wlasiuk, P.; Karczmarczyk, A.; Giannopoulos, K. Aberrant Expression of TLR2, TLR7, TLR9, Splicing Variants of TLR4 and MYD88 in Chronic Lymphocytic Leukemia Patients. J. Clin. Med. 2021, 10, 867. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Sun, P.; Zhang, J.-C.; Zhang, Q.; Shang -Long, Y. Proinflammatory effects of S100A8/A9 via TLR4 and RAGE signaling pathways in BV-2 microglial cells. Int. J. Mol. Med. 2017, 40, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Fayad, L.; Keating, M.-J.; Reuben, J.-M.; O’Brien, S.; Lee, B.-N.; Lerner, S.; Kurzrock, R. Interleukin-6 and interleukin-10 levels in chronic lymphocytic leukemia: Correlation with phenotypic characteristics and outcome. Blood 2001, 97, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Heikkilä, K.; Ebrahim, S.; Lawlor, D.-A. Systematic review of the association between circulating interleukin-6 (IL-6) and cancer. Eur. J. Cancer 2008, 44, 937–945. [Google Scholar] [CrossRef]
- Alhakeem, S.-S.; McKenna, M.-K.; Oben, K.-Z.; Noothi, S.-K.; Rivas, J.-R.; Hildebrandt, G.-C.; Fleischman, R.-A.; Rangnekar, V.-M.; Muthusamy, N.; Bondada, S. Chronic Lymphocytic Leukemia-Derived IL-10 Suppresses Antitumor Immunity. J. Immunol. 2018, 200, 4180–4189. [Google Scholar] [CrossRef]
- Kurt, A.-N.-C.; Aygun, A.D.; Godekmerdan, A.; Kurt, A.; Dogan, Y.; Yilmaz, E. Serum IL-1beta, IL-6, IL-8, and TNF-alpha levels in early diagnosis and management of neonatal sepsis. Mediat. Inflamm. 2007, 2007, 31397. [Google Scholar] [CrossRef]
- Rozovski, U.; Wu, J.Y.; Harris, D.M.; Liu, Z.; Li, P.; Hazan-Halevy, I.; Ferrajoli, A.; Burger, J.A.; O’Brien, S.; Jain, N.; et al. Stimulation of the B-cell receptor activates the JAK2/ STAT3 signaling pathway in chronic lymphocytic leukemia cells. Blood 2014, 123, 3797–3802. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, P.-C.; Behrmann, I.; Haan, S.; Hermanns, H.-M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signaling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Rozovski, U.; Grgurevic, S.; Bueso-Ramos, C.; Harris, D.-M.; Li, P.; Liu, Z.; Wu, J.-Y.; Jain, P.; Wierda, W.; Burger, J.; et al. Aberrant LPL expression, driven by STAT3, mediates free fatty acid metabolism in CLL cells. Mol. Cancer Res. 2015, 13, 944–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliopoulos, D.; Hirsch, H.-A.; Struhl, K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesina, M.; Kurkowski, M.-U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foell, D.; Frosch, M.; Sorg, C.; Roth, J. Phagocyte-specific calcium-binding S100 proteins as clinical laboratory markers of inflammation. Clin. Chim. Acta 2004, 344, 37–51. [Google Scholar] [CrossRef]
- Fei, F.; Qu, J.; Zhang, M.; Li, Y.; Zhang, S. S100A4 in cancer progression and metastasis: A systematic review. Oncotarget 2017, 8, 73219–73239. [Google Scholar] [CrossRef] [Green Version]
- Bresnick, A.-R.; Weber, D.-J.; Zimmer, D.B. S100 proteins in cancer. Nat. Rev. Cancer 2015, 15, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Tao, T.; Raftery, M.-J.; Youssef, P.; Di Girolamo, N.; Geczy, C.L. Proinflammatory properties of the human S100 protein S100A12. J. Leukoc. Biol. 2001, 69, 986–994. [Google Scholar]
- Hasegawa, T.; Kosaki, A.; Kimura, T.; Matsubara, H.; Mori, Y.; Okigaki, M.; Masaki, H.; Toyoda, N.; Inoue-Shibata, M.; Kimura, Y.; et al. The regulation of EN-RAGE (S100A12) gene expression in human THP-1 macrophages. Atherosclerosis 2003, 171, 211–218. [Google Scholar] [CrossRef]
- Böttcher, M.; Panagiotidis, K.; Bruns, H.; Stumpf, M.; Völkl, S.; Geyh, S.; Dietel, B.; Schroeder, T.; Mackensen, A.; Mougiakakos, D. Bone marrow stroma cells promote induction of a chemoresistant and prognostic unfavorable S100A8/A9high AML cell subset. Blood Adv. 2022; in press. [Google Scholar] [CrossRef]
- Karjalainen, R.; Pemovska, T.; Popa, M.; Liu, M.; Javarappa, K.; Majumder, M.; Yadav, B.; Tamborero, D.; Tang, J.; Bychkov, D.; et al. JAK1/2 and BCL2 inhibitors synergize to counteract bone marrow stromal cell-induced protection of AML. Blood 2017, 130, 789–802. [Google Scholar] [CrossRef]
- Roberts, A.-W.; Davids, M.-S.; Pagel, M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef]
- Moia, R.; Diop, F.; Favini, C.; Kodipad, A.A.; Gaidano, G. Potential of BCL2 as a target for chronic lymphocytic leukemia treatment. Expert Rev. Hematol. 2018, 11, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Gu, A.; Lee, J.S.; Yang, E.J.; Kashif, A.; Hong, H.; Kim, G.; Park, B.S.; Lee, S.J.; Kim, I.S. Suppressive effects of S100A8 and S100A9 on neutrophil apoptosis by cytokine release of human bronchial epithelial cells in asthma. Int. J. Med. Sci. 2020, 17, 498–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Guerra, M.; Colomer, D. NF-kappaB as a therapeutic target in chronic lymphocytic leukemia. Expert Opin. Ther. Targets 2010, 14, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Hertlein, E.; Wagner, A.-J.; Jones, J.; Lin, T.-S.; Maddocks, K.J.; Towns, W.H.; Goettl, V.-M.; Zhang, X.; Jarjoura, D.; Raymond, C.-A.; et al. 17-DMAG targets the NF-{kappa}B family of proteins to induce apoptosis in CLL: Clinical implications of HSP90 inhibition. Blood 2010, 116, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Hazan-Halevy, I.; Harris, D.; Liu, Z.; Liu, J.; Li, P.; Chen, X.; Shanker, S.; Ferrajoli, A.; Keating, M.-J.; Estrov, Z. STAT-3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 2010, 115, 2852–2863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohlhaas, V.; Blakemore, S.-J.; Al-Maarri, M.; Nickel, N.; Pal, M.; Roth, A.; Hövelmeyer, N.; Schäfer, S.-C.; Knittel, G.; Lohneis, P.; et al. Active Akt signalling triggers CLL toward Richter transformation via overactivation of Notch1. Blood 2021, 137, 646–660. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, C.-J.; Martin, B.-N.; Bulek, K.; Kang, Z.; Zhao, J.; Bian, G.; Carman, J.-A.; Gao, J.; Dongre, A.; et al. IL-17 induced NOTCH1 activation in oligodendrocyte progenitor cells enhances proliferation and inflammatory gene expression. Nat. Commun. 2017, 8, 15508. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.; Mangaonkar, A. Idelalisib. A Novel PI3Kδ Inhibitor for Chronic Lymphocytic Leukemia. Ann. Pharmacother. 2015, 49, 1162–1170. [Google Scholar] [CrossRef]
- Sharman, J.-P.; Coutre, S.-E.; Furman, R.-R.; Cheson, B.-D.; Pagel, J.-M.; Hillmen, P.; Barrientos, J.-C.; Zelenetz, A.-D.; Kipps, T.-J.; Flinn, I.-W.; et al. Final Results of a Randomized, Phase III Study of Rituximab with or Without Idelalisib Followed by Open-Label Idelalisib in Patients with Relapsed Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2019, 37, 1391–1402. [Google Scholar] [CrossRef]
- Herman, S.-E.; Gordon, A.-L.; Wagner, A.J.; Heerema, N.-A.; Zhao, W.; Flynn, J.-M.; Jones, J.; Andritsos, L.; Puri, K.-D.; Lannutti, B.-J.; et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood 2010, 116, 2078–2088. [Google Scholar] [CrossRef] [Green Version]
- Moia, R.; Patriarca, A.; Schipani, M.; Ferri, V.; Favini, C.; Sagiraju, S.; Al Essa, W.; Gaidano, G. Precision Medicine Management of Chronic Lymphocytic Leukemia. Cancers 2020, 12, 642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subotički, T.; Mitrović Ajtić, O.; Beleslin-Čokić, B.-B.; Nienhold, R.; Diklić, M.; Djikić, D.; Leković, D.; Bulat, T.; Marković, D.; Gotić, M.; et al. Angiogenic factors are increased in circulation granulocytes and CD34+ cells of myeloproliferative neoplasm. Mol. Carcinog. 2017, 56, 567–579. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitrović Ajtić, O.; Subotički, T.; Diklić, M.; Đikić, D.; Vukotić, M.; Dragojević, T.; Živković, E.; Antić, D.; Čokić, V. Regulation of S100As Expression by Inflammatory Cytokines in Chronic Lymphocytic Leukemia. Int. J. Mol. Sci. 2022, 23, 6952. https://doi.org/10.3390/ijms23136952
Mitrović Ajtić O, Subotički T, Diklić M, Đikić D, Vukotić M, Dragojević T, Živković E, Antić D, Čokić V. Regulation of S100As Expression by Inflammatory Cytokines in Chronic Lymphocytic Leukemia. International Journal of Molecular Sciences. 2022; 23(13):6952. https://doi.org/10.3390/ijms23136952
Chicago/Turabian StyleMitrović Ajtić, Olivera, Tijana Subotički, Miloš Diklić, Dragoslava Đikić, Milica Vukotić, Teodora Dragojević, Emilija Živković, Darko Antić, and Vladan Čokić. 2022. "Regulation of S100As Expression by Inflammatory Cytokines in Chronic Lymphocytic Leukemia" International Journal of Molecular Sciences 23, no. 13: 6952. https://doi.org/10.3390/ijms23136952
APA StyleMitrović Ajtić, O., Subotički, T., Diklić, M., Đikić, D., Vukotić, M., Dragojević, T., Živković, E., Antić, D., & Čokić, V. (2022). Regulation of S100As Expression by Inflammatory Cytokines in Chronic Lymphocytic Leukemia. International Journal of Molecular Sciences, 23(13), 6952. https://doi.org/10.3390/ijms23136952