
1,3-Benzodioxole Derivatives Improve the Anti-Tumor Efficiency of Arsenicals



Abstract
:1. Introduction
2. Results
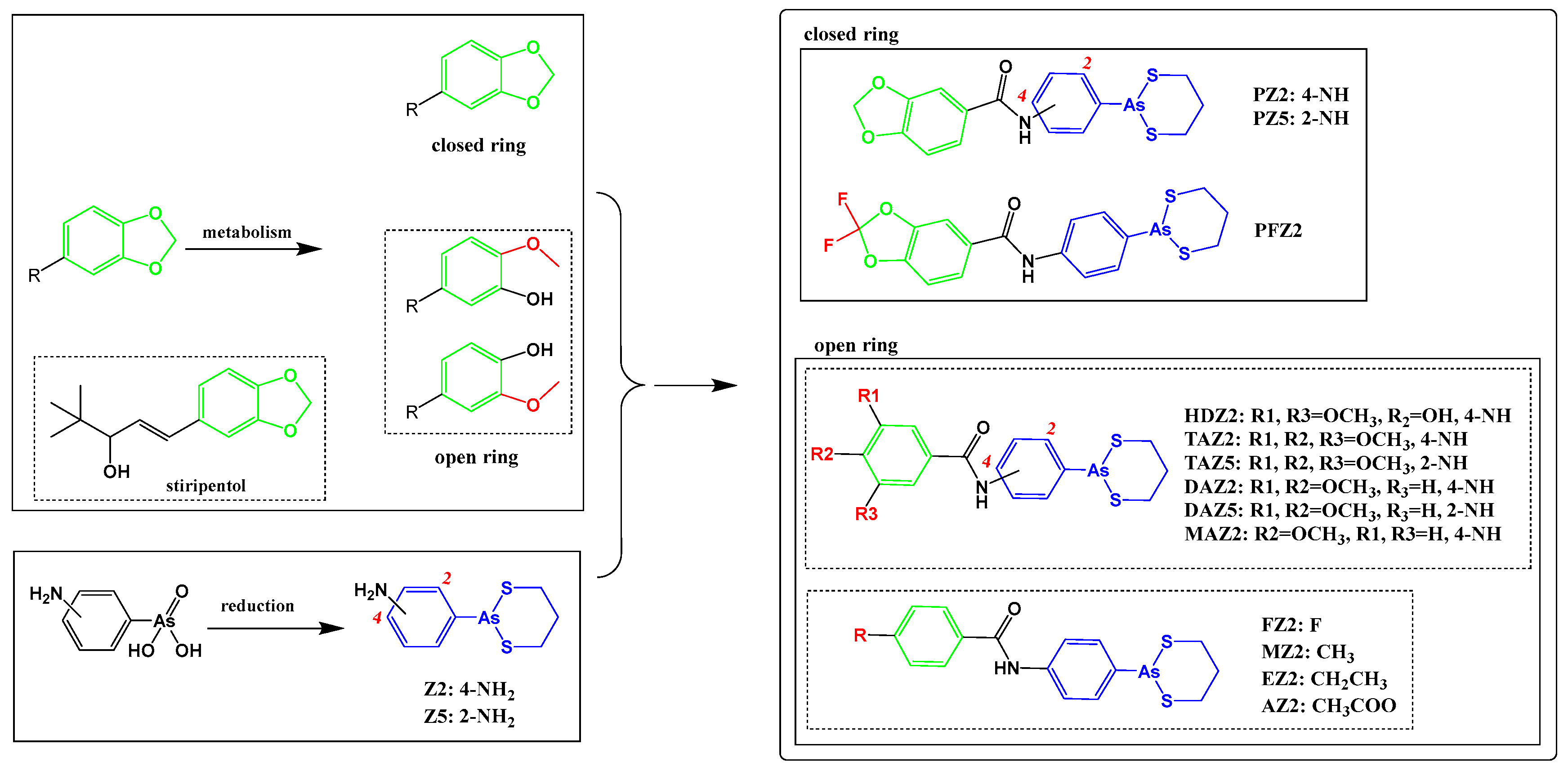
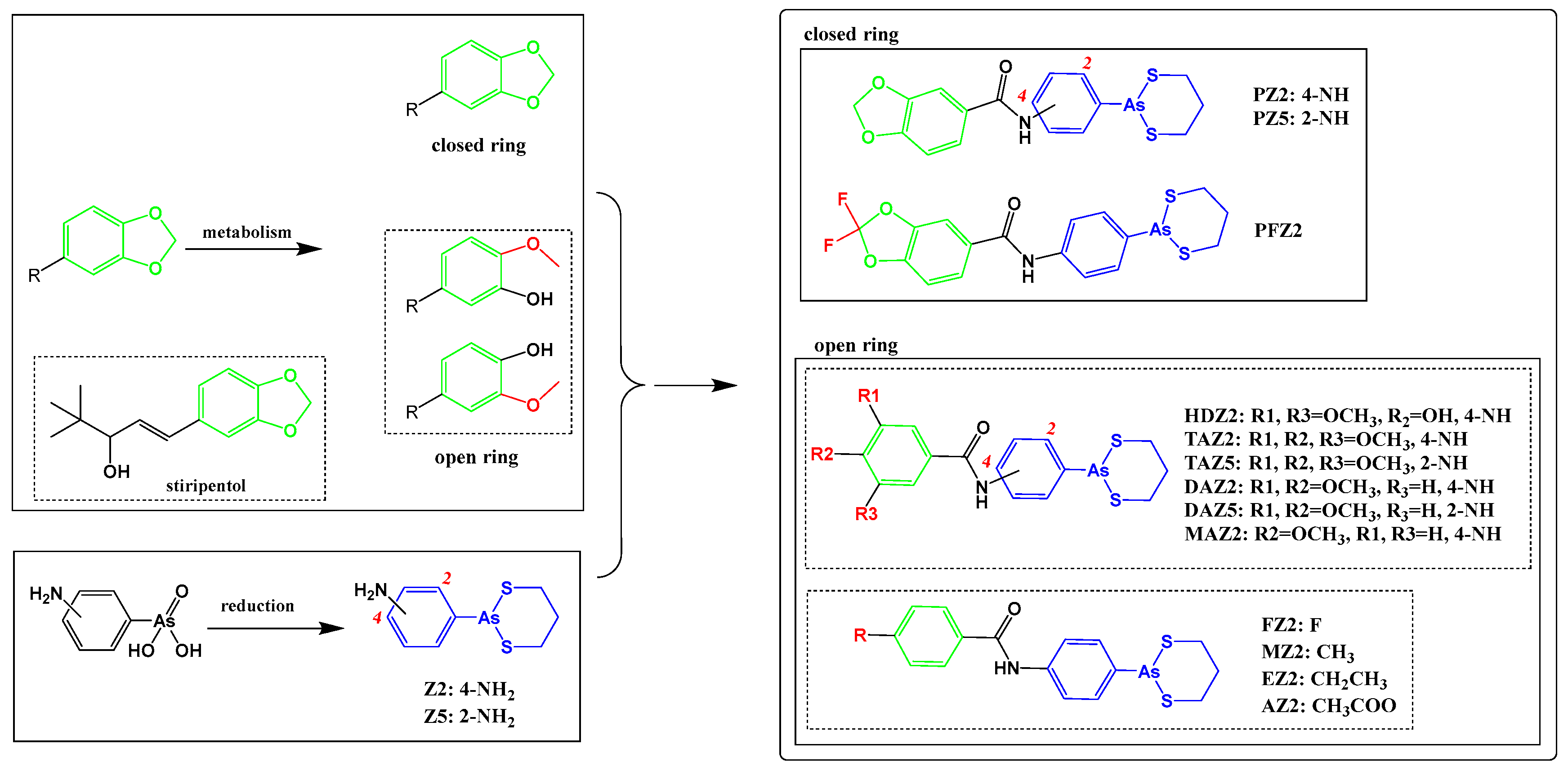
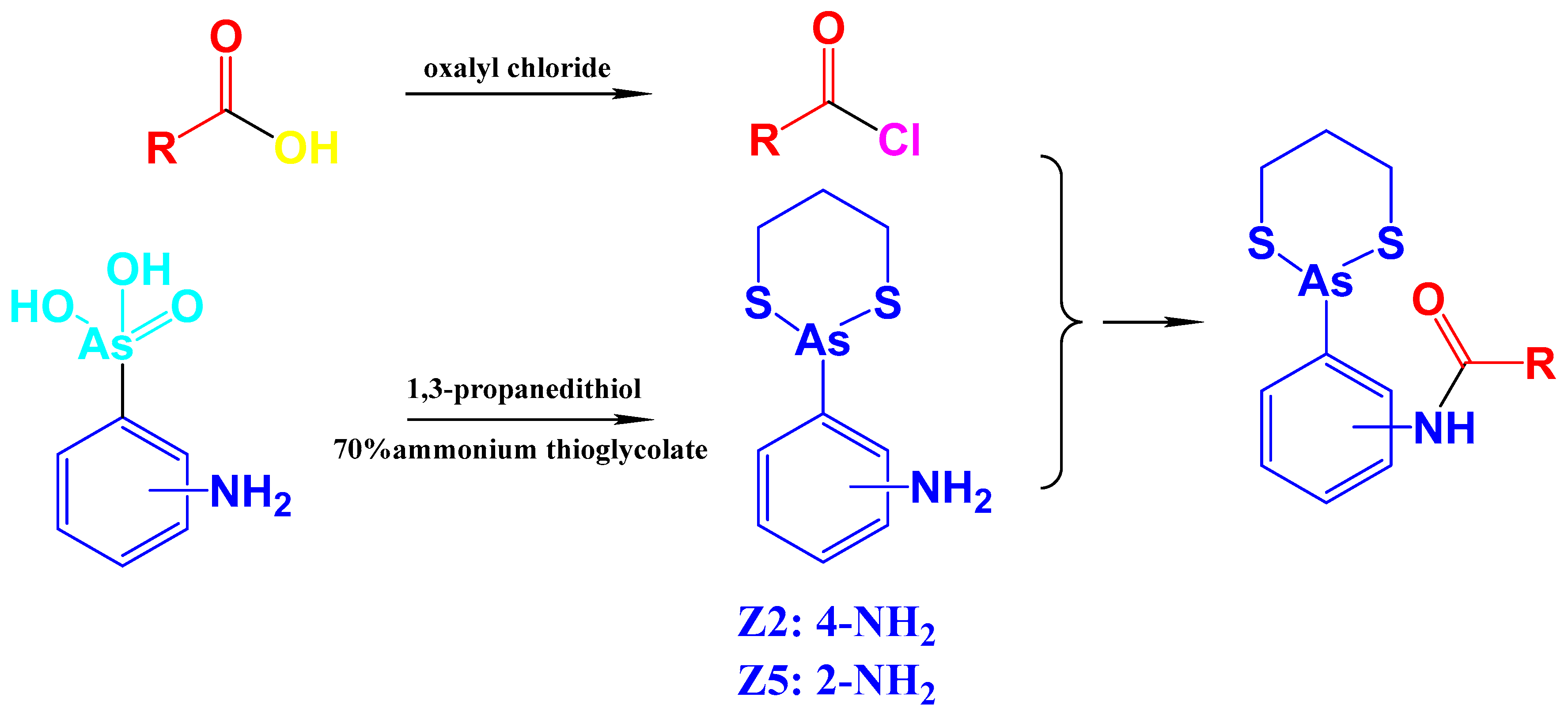
2.1. Arsenicals Were Conjugated with 1,3-Benzodioxole Derivatives
2.2. Conjugated Arsenicals Possessed Good Anti-Proliferation Activity
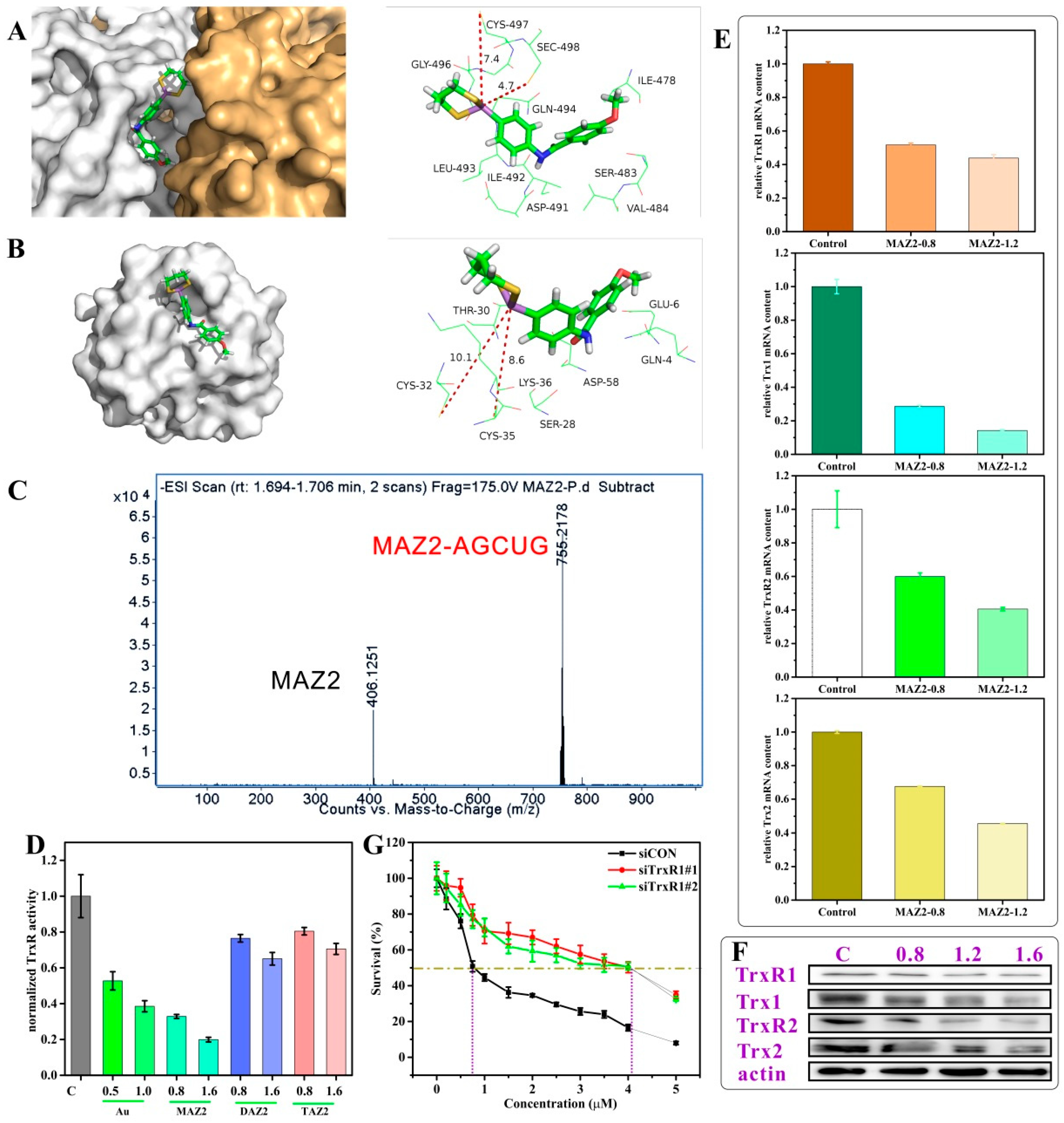
2.3. MAZ2 Inhibited TrxR Activity by Binding to the C-Terminal Sec/Cys Pair
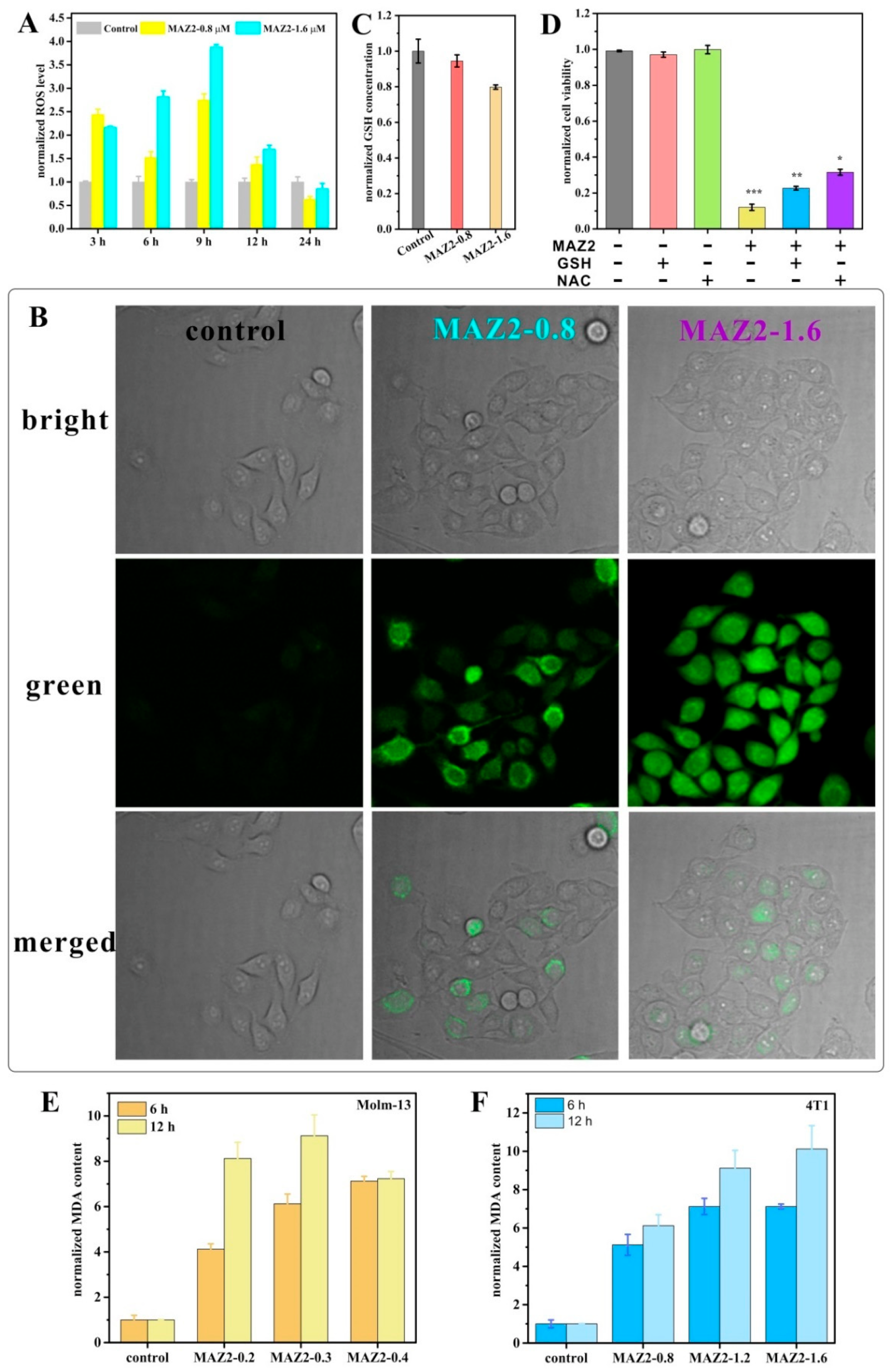
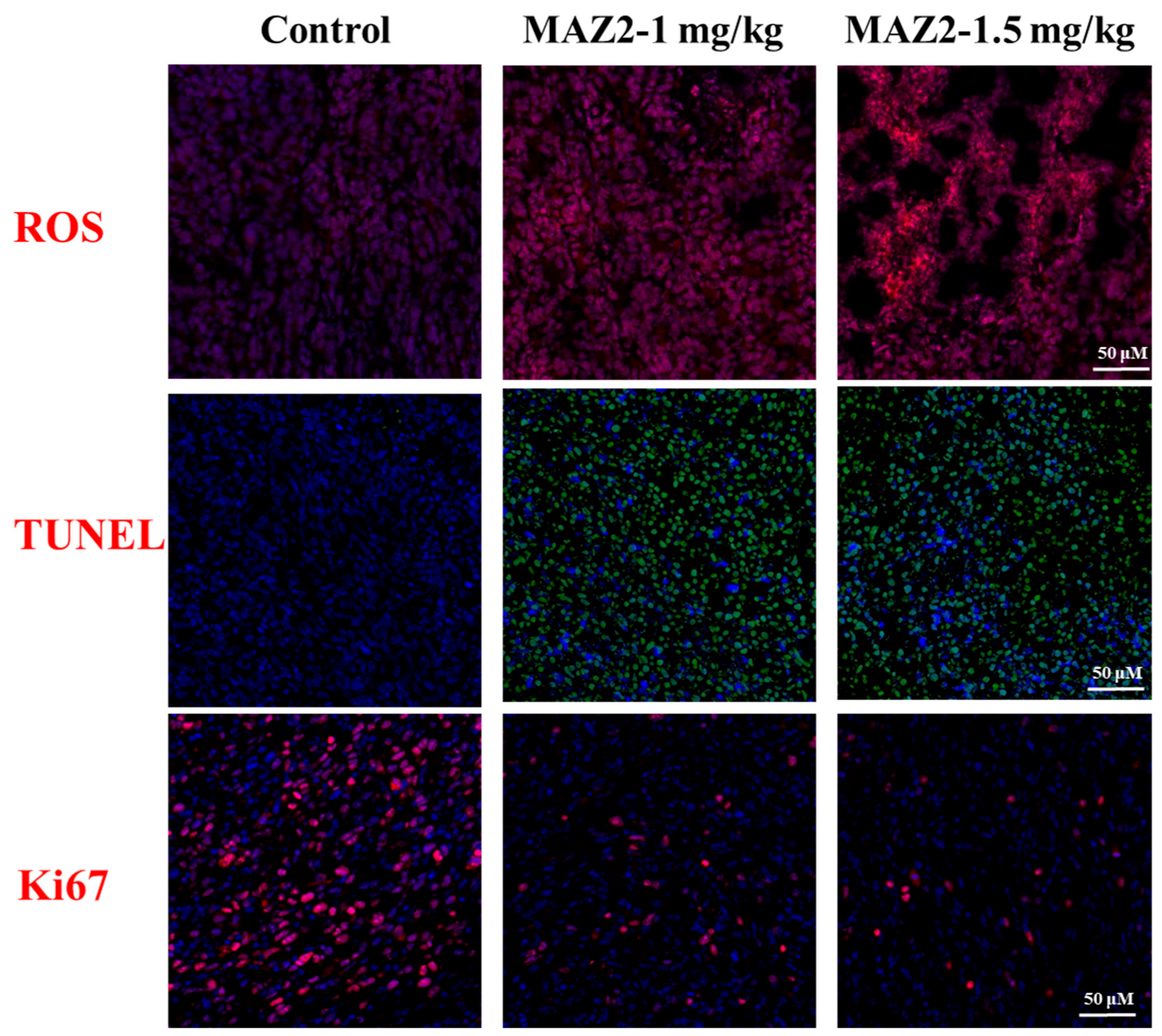
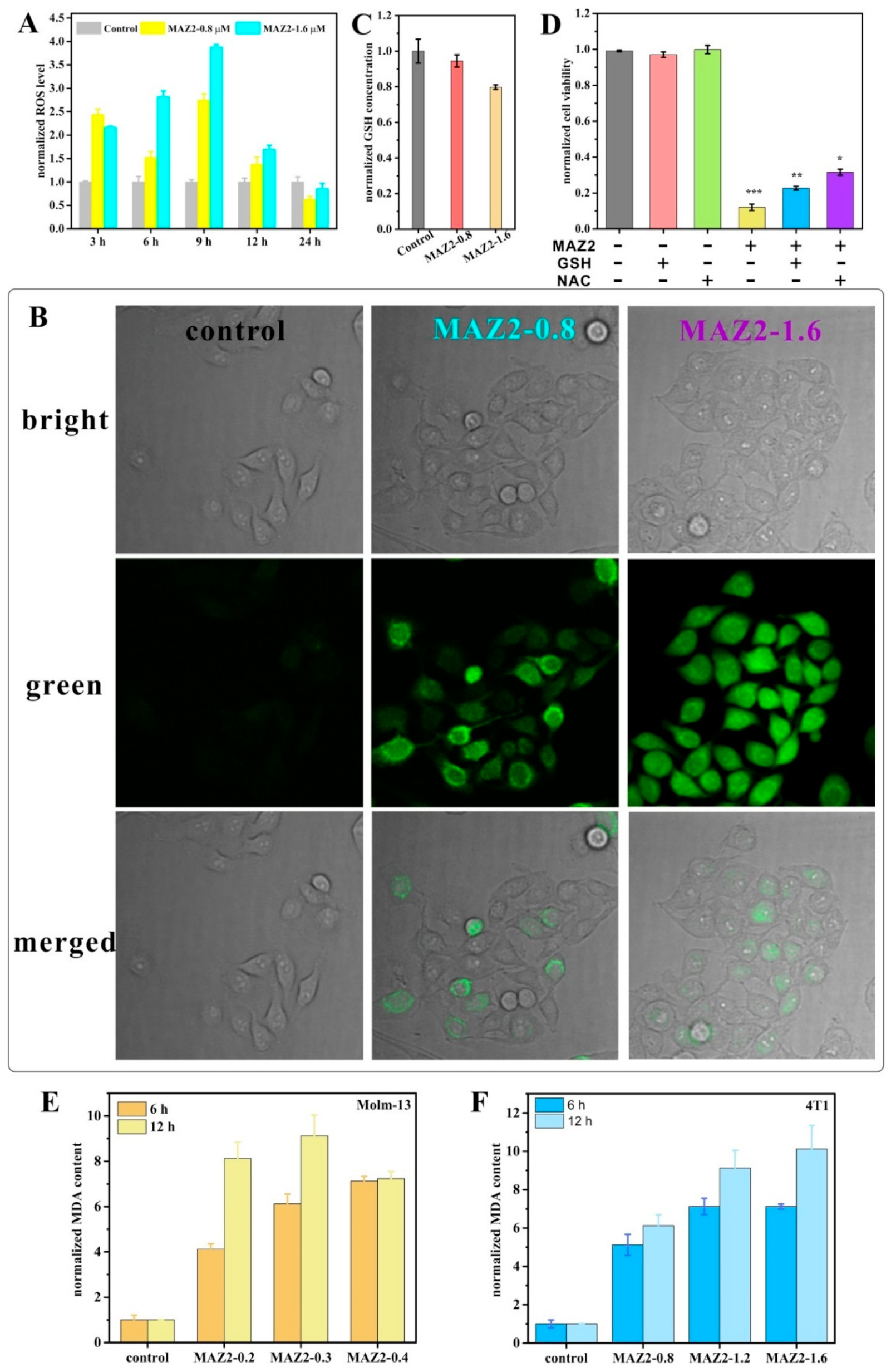
2.4. ROS Bursting after TrxR Inhibition by Arsenicals
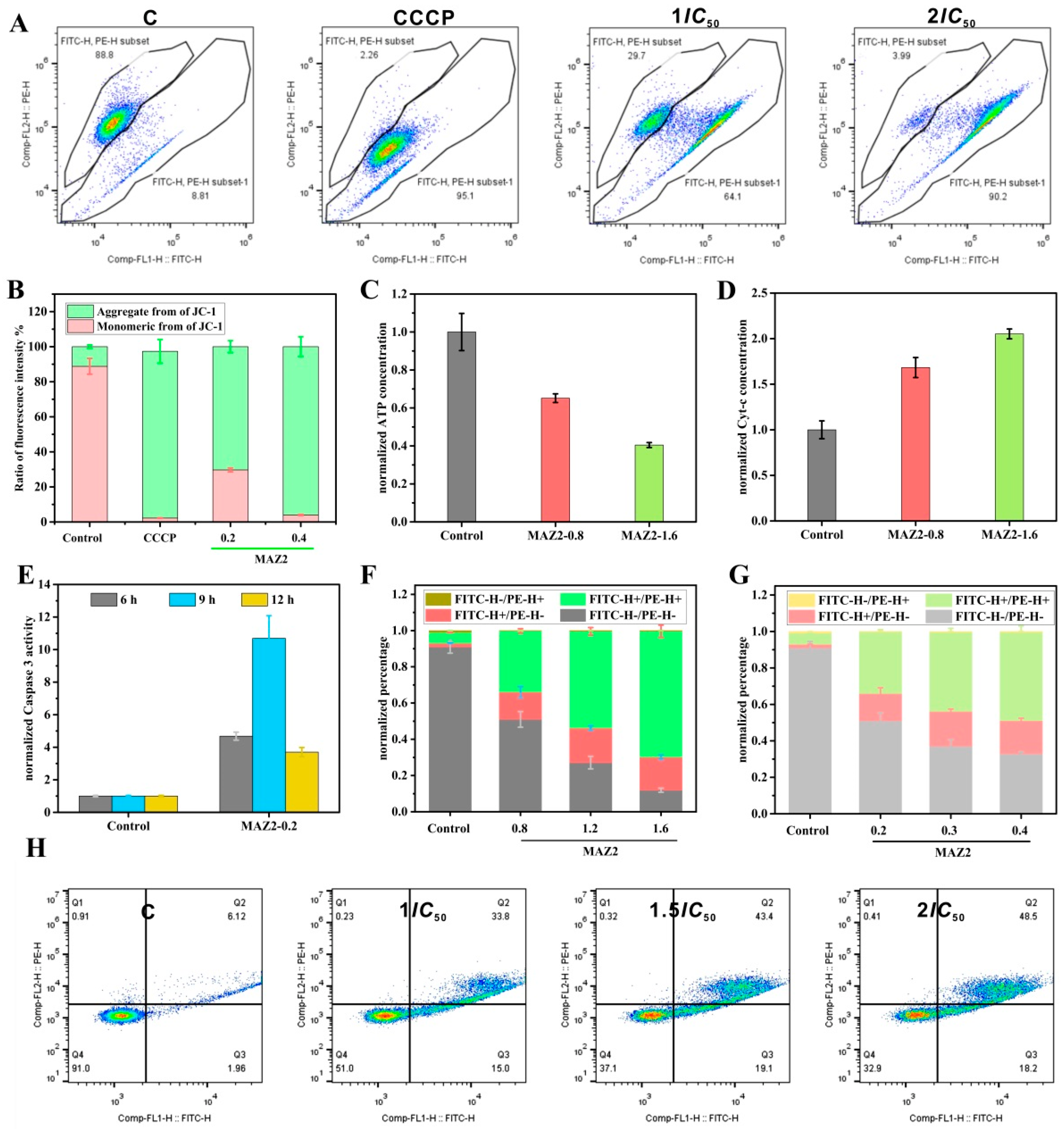
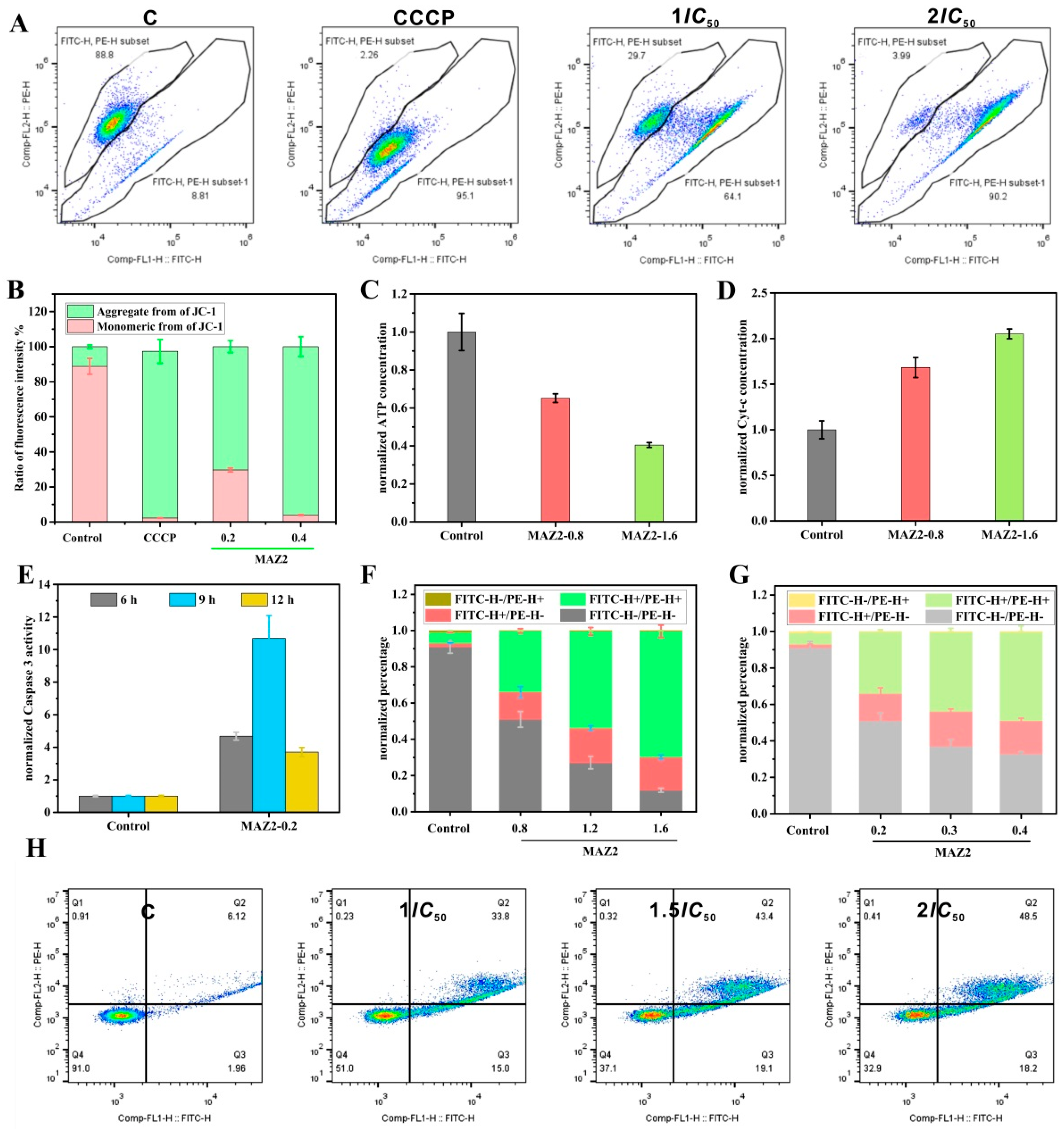
2.5. Apoptosis was Induced by MAZ2 Incubation
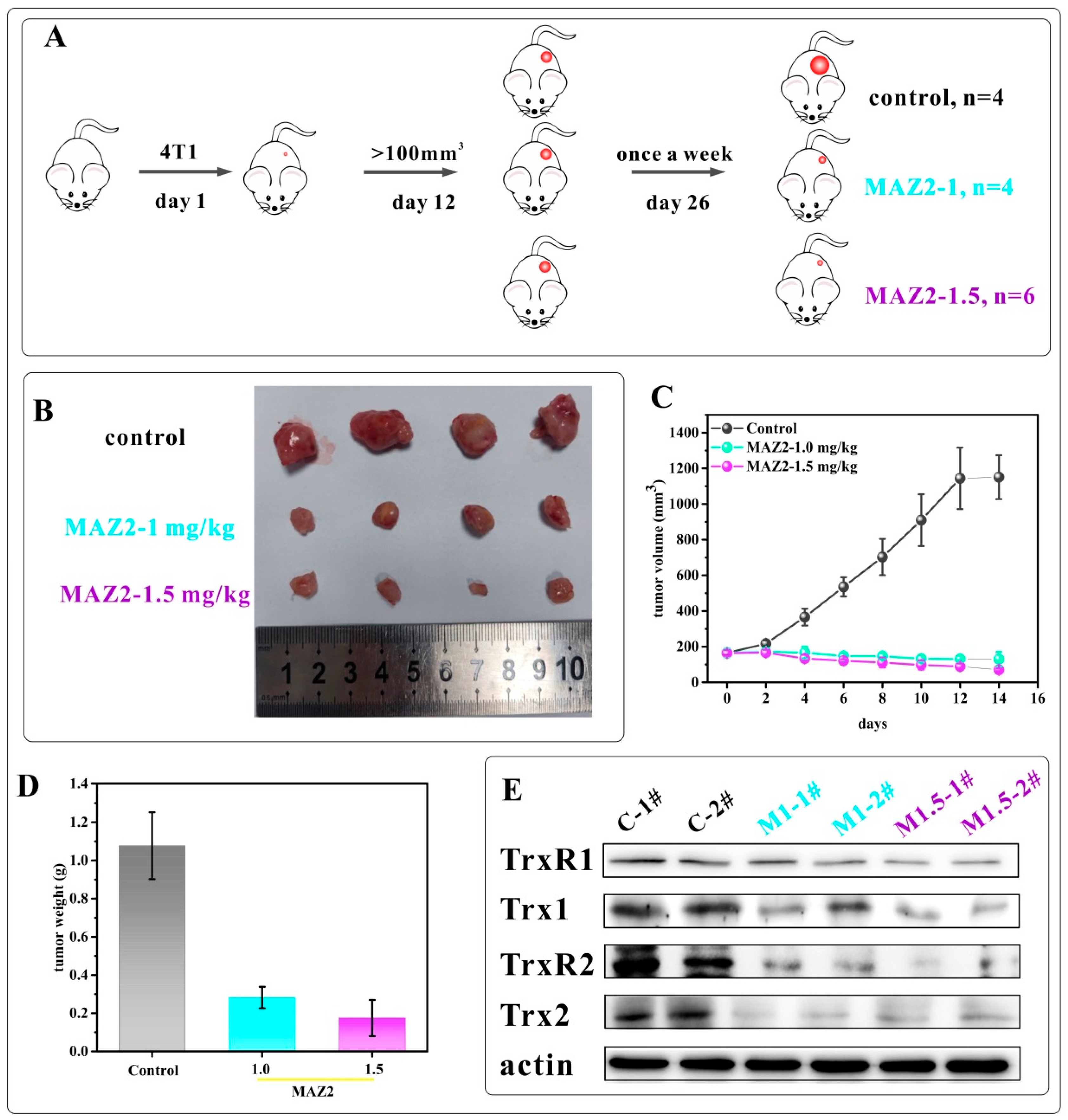
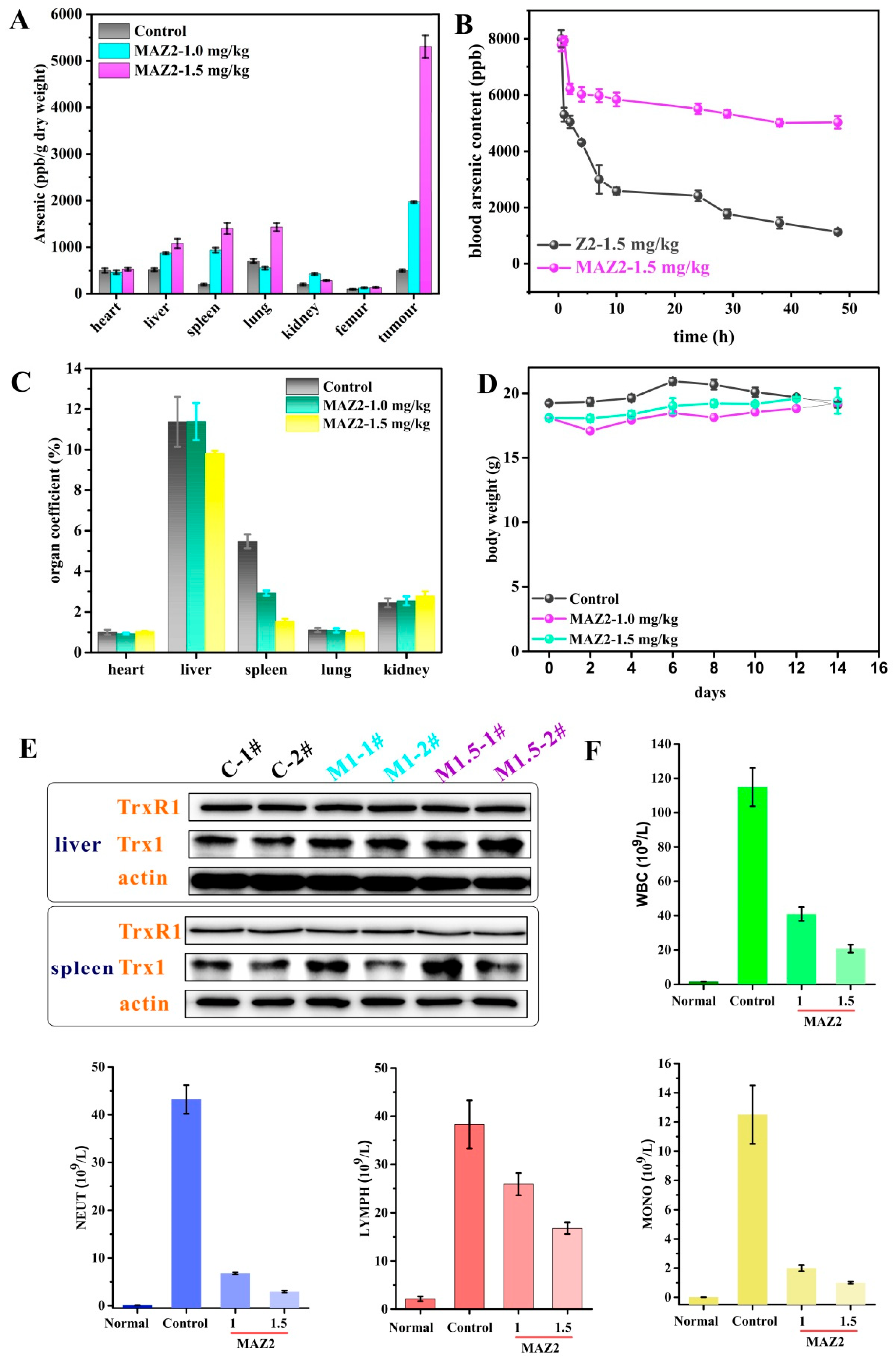
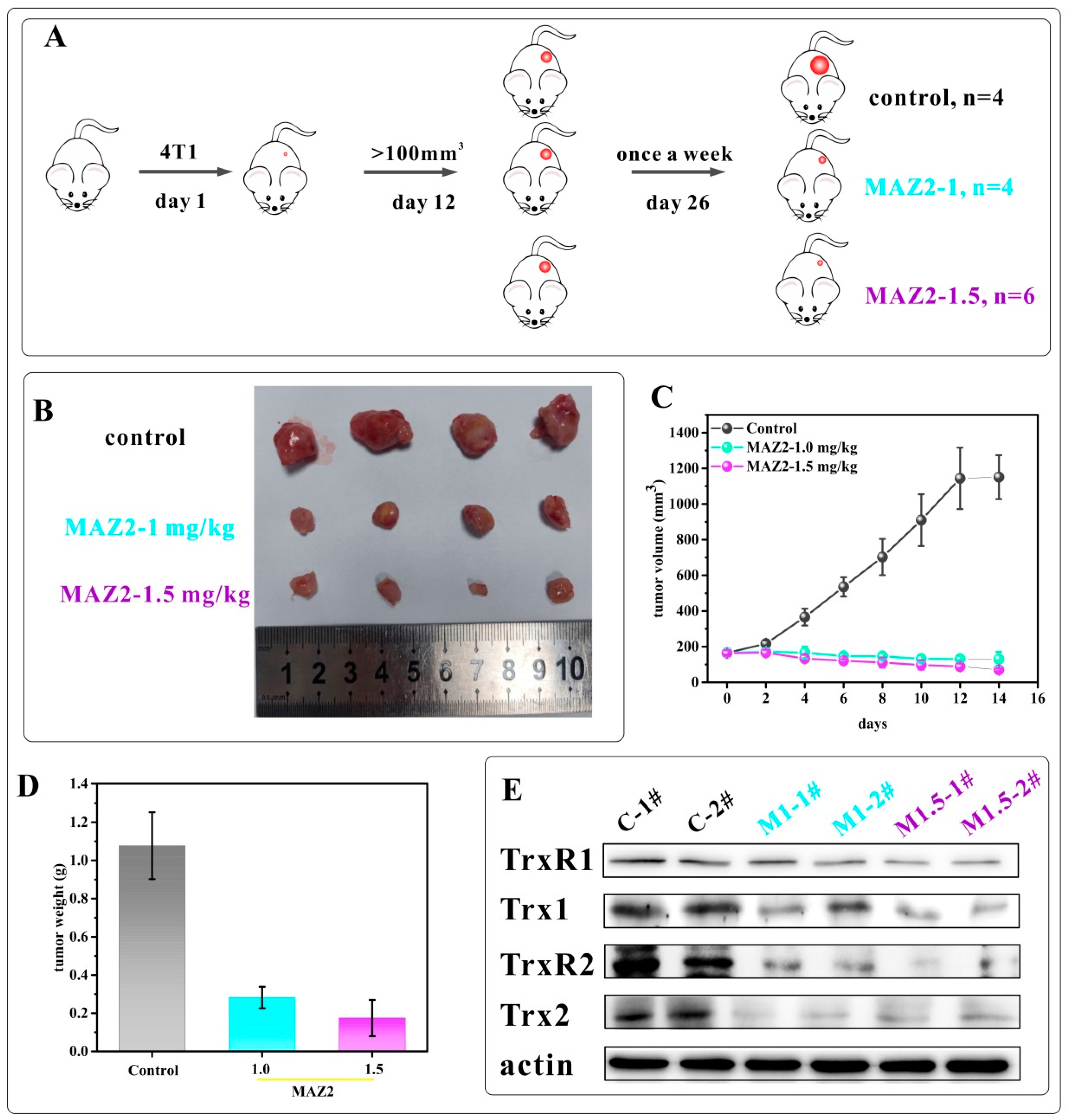
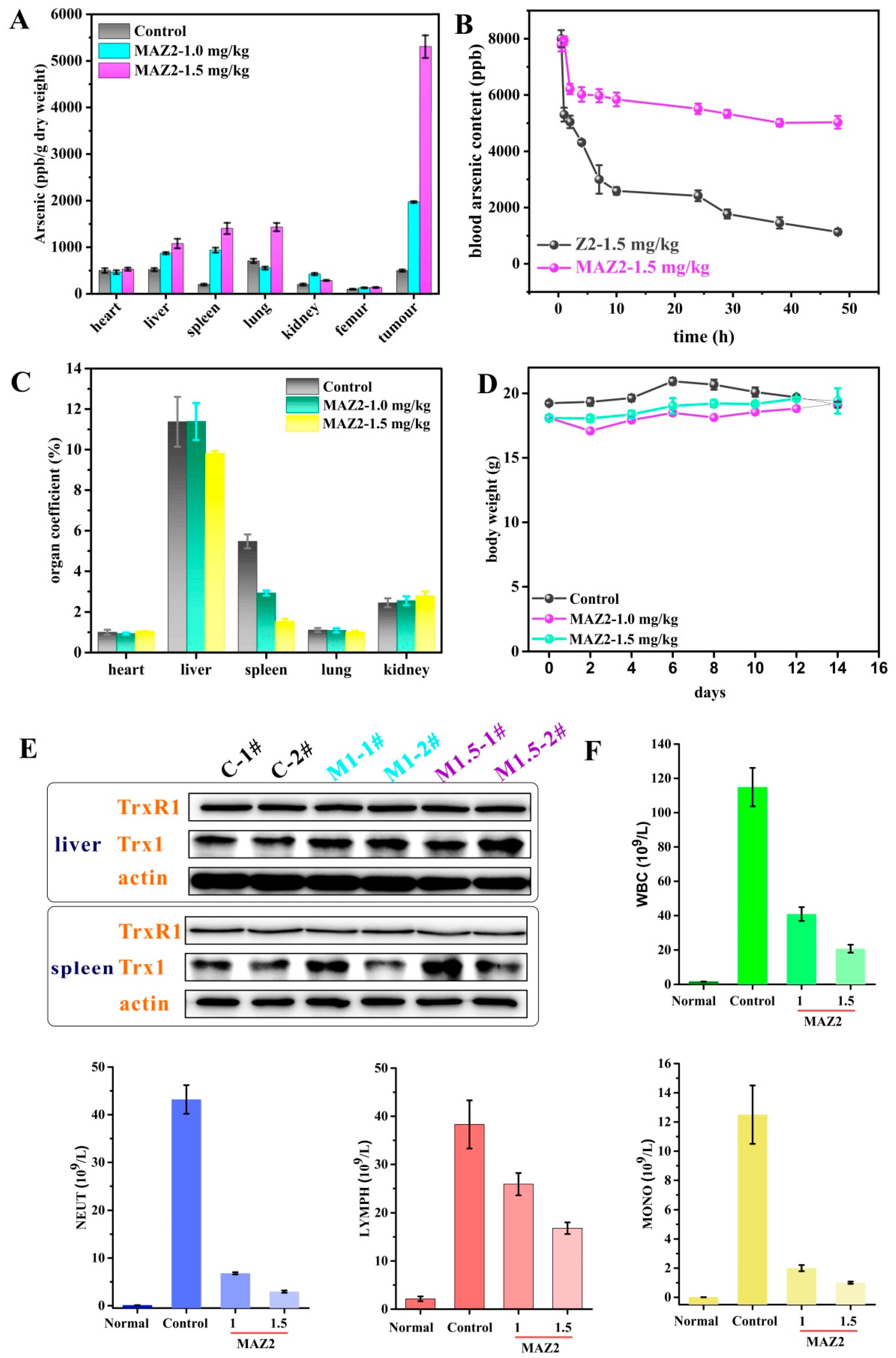
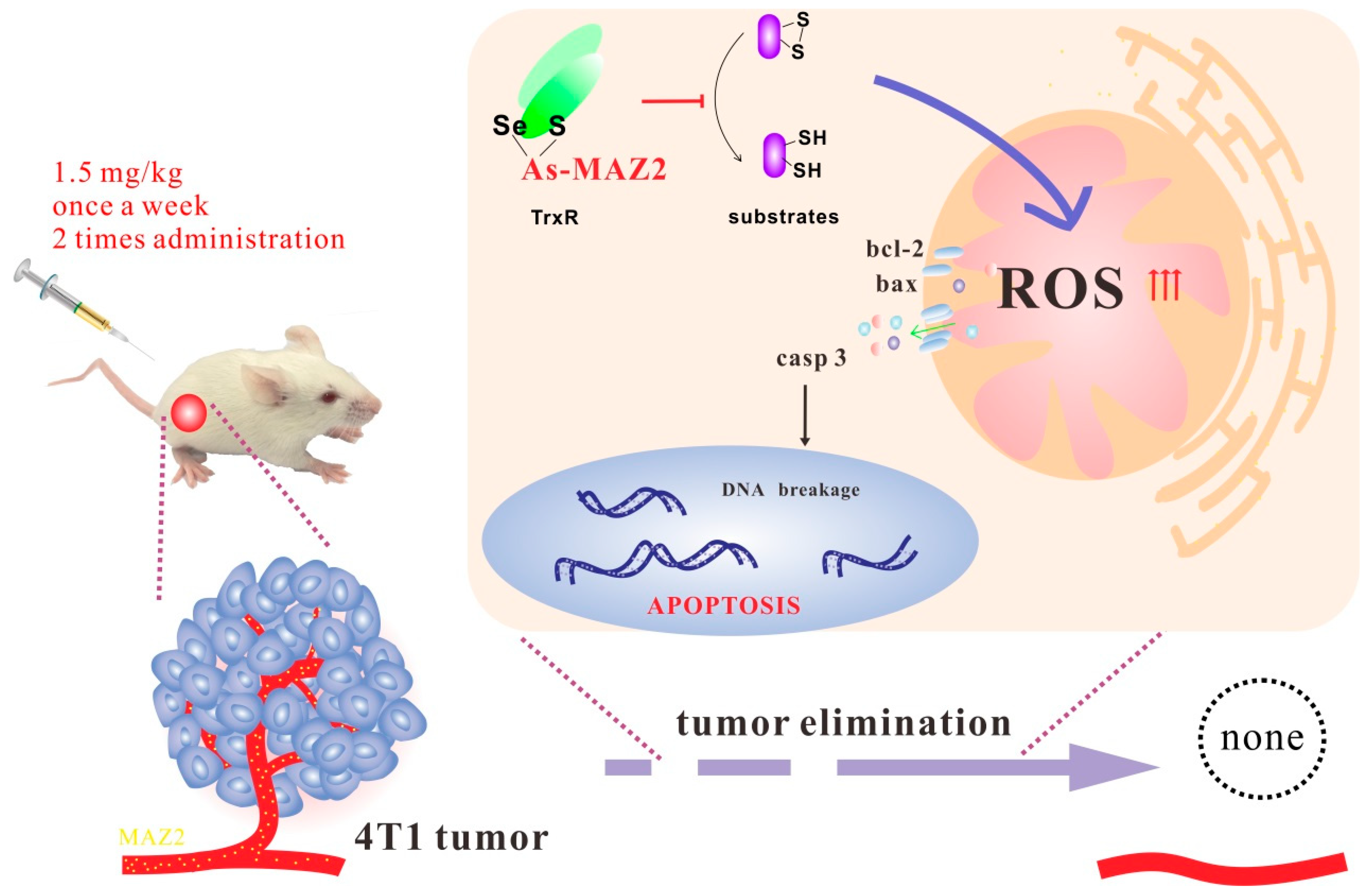
2.6. 4T1 Tumors Were Eliminated by Two Administrations of MAZ2
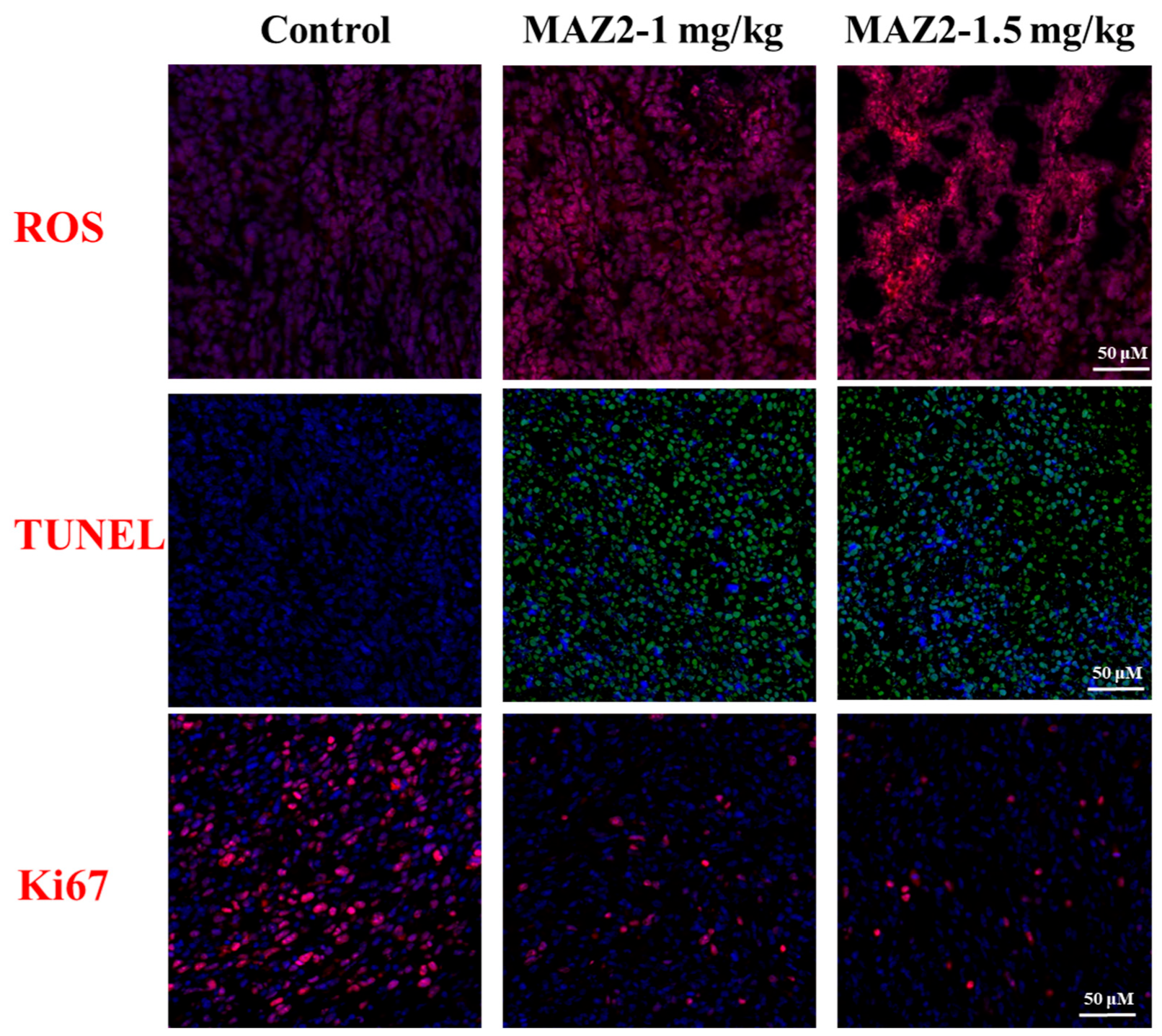
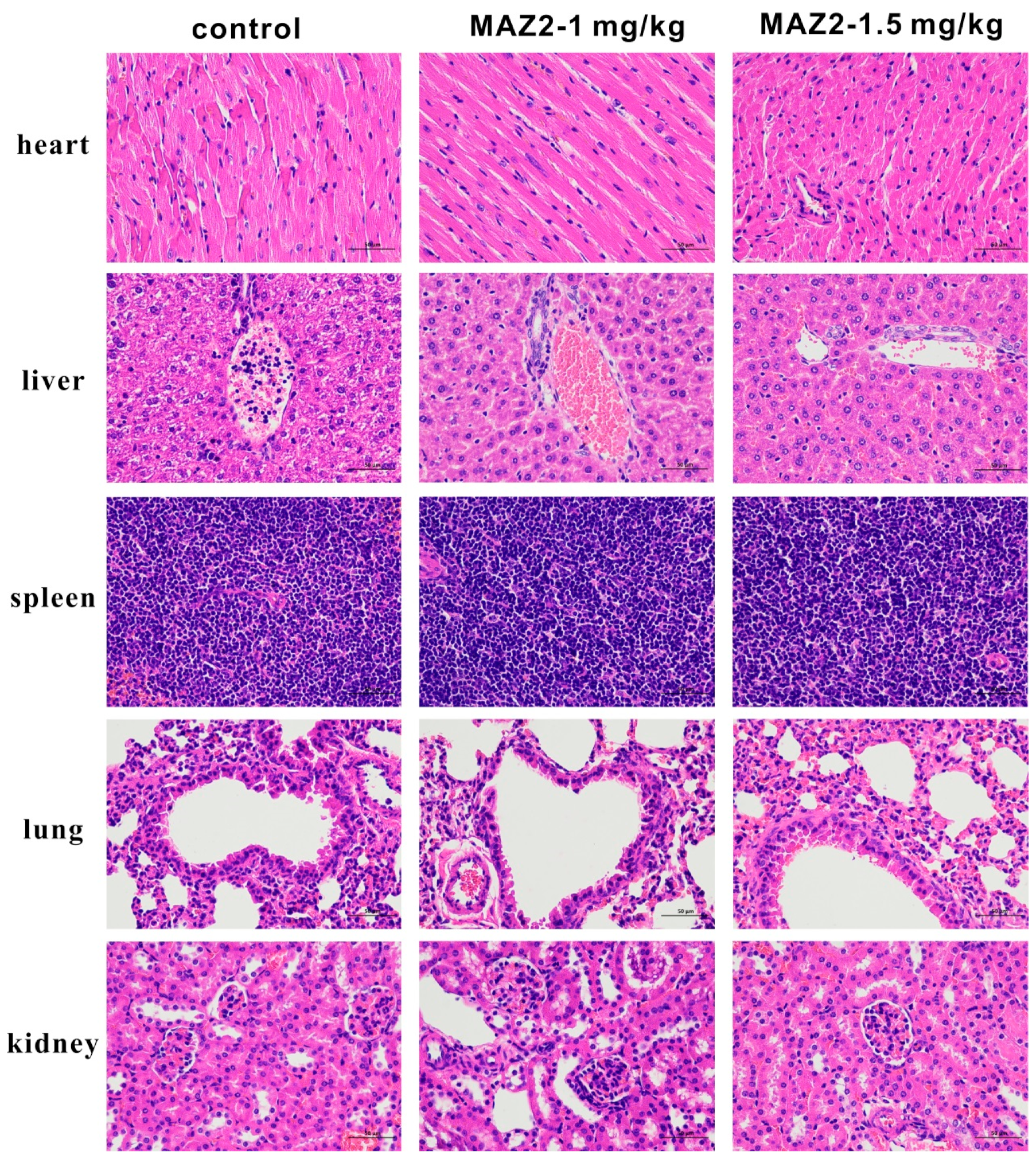
2.7. MAZ2 Administration Had No Obvious Side Effects
3. Discussion
4. Materials and Methods
4.1. Chemicals and Instruments
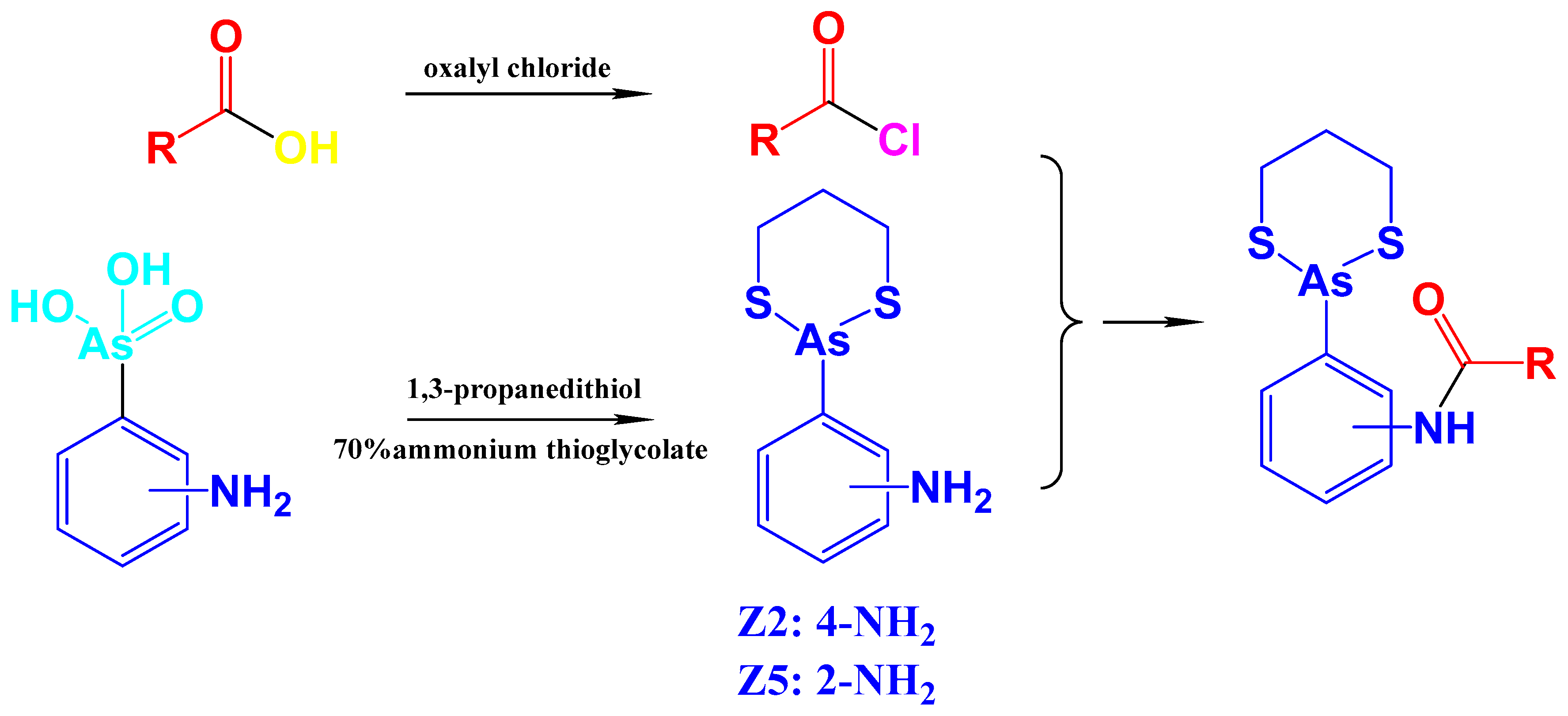
4.2. Synthesis and Characterization of Z2 and Z5
- Z2: 4-(1,3,2-dithiarsinan-2-yl)aniline. Yield: 2.62 g (52.1%). 1H NMR (400 MHz, CDCl3) δ: 7.66–7.64 (d, J = 8.0 Hz, 2H), 6.79–6.77 (d, J = 8.0 Hz, 2H), 3.86 (s, NH2, 2H), 2.93–2.87 (m, 2H), 2.75–2.69 (m, 2H), 2.19–2.10 (m, 1H), 1.97–1.89 (m, 1H). 13C NMR (400 MHz, CDCl3): 147.54, 133.81, 125.73, 115.65, 28.72, 26.44. ESI-MS: calculated for C9H12NS2As: 272.9627, found 273.9708[M + H]+.
- Z5: 2-(1,3,2-dithiarsinan-2-yl)aniline. Yield: 1.94g (40.1%). 1H NMR (400 MHz, DMSO-d6) δ: 7.62–6.60 (d, J = 8 Hz, 2H), 7.19–7.15 (m, 1H), 6.76–6.70 (m, 2H), 5.45 (s, NH2, 2H), 2.94–2.91 (m, 4H), 2.08–2.00 (m, 1H), 1.92–1.83 (m, 1H). 13C NMR (400 MHz, DMSO-d6): 151.63, 134.66, 131.79, 118.42, 117.83, 116.59, 28.95, 27.84. ESI-MS: m/z calculated for C9H12NS2As: 272.9627, found 273.9700[M + H]+.
4.3. Synthesis and Characterization of Conjugates of Arsenical Precursors and 1,3-Benzodioxole Derivatives
- PZ2: N-(4-(1,3,2-dithiarsinan-2-yl)phenyl)benzo[d][1,3]dioxole-5-carboxamide (P: Piperonylic acid). Yield: 644 mg (51%). 1H NMR (400 MHz, DMSO) δ: 10.25 (s, NH, 1H), 7.95–7.93 (m, 2H), 7.80–7.77 (m, 2H), 7.60–7.58 (d, J = 8.0 Hz, 1H), 7.53 (s, 1H), 7.08–7.06 (d, J = 8.0 Hz, 1H), 6.14 (s, 2H), 2.84–2.73 (m, 4H), 2.03–1.89 (m, 2H). 13C NMR (400 MHz, DMSO): 165.19, 150.67, 147.88, 140.86, 133.13, 132.21, 128.96, 123.37, 121.32, 108.45, 108.24, 102.34, 28.36, 26.15. ESI-MS: m/z calculated for C17H16AsNO3S2 [M]: 420.9788; [M – H]−: 419.9715; found 419.9713.
- PZ5: N-(2-(1,3,2-dithiarsinan-2-yl)phenyl)benzo[d][1,3]dioxole-5-carboxamide (P: Piperonylic acid). Yield: 505 mg (40%). 1H NMR (400 MHz, DMSO) δ: 10.46 (s, NH, 1H), 8.10–8.08 (d, J = 8 Hz, 1H), 7.52–7.49 (m, 2H), 7.42–7.35 (m, 2H), 7.08–7.06 (d, J = 8 Hz, 1H), 2.94–2.87 (m, 2H), 2.82–2.76 (m, 2H), 2.05–1.75(m, 2H). 13C NMR (400 MHz, DMSO): 165.67, 150.75, 147.88, 140.64, 143.79,143.17, 130.92, 128.16, 126.79, 126.22, 123.62, 108.51, 108.21, 102.34, 29.17, 28.18. ESI-MS: m/z calculated for C17H16AsNO3S2 [M]: 420.9793; [M + H]+: 421.9860; found 421.9856; [2M + Na]+: 864.9467; found 864.9459.
- MAZ2: 2-(4-(1,3,2-dithiarsinan-2-yl)phenyl)-1-(4-methoxyphenyl)ethan-1-one (MA: 4-methoxybenzoic acid). Yield: 105 mg (35%). 1H NMR (400 MHz, DMSO) δ: 10.29 (s, NH, 1H), 8.00–7.95 (m, 4H), 7.80–7.78 (d, J = 8 Hz, 2H), 7.09–7.07 (d, J = 8 Hz, 2H), 3.85 (s, 3H), 2.85–2.74 (m, 4H), 2.05–1.89 (m, 2H). 13C NMR (400 MHz, DMSO): 135.45, 134.14, 134.04, 130.81, 130.69, 119.40, 118.55, 115.40, 55.0, 18.96, 14.88. ESI-MS: m/z calculated for C17H18AsNO2S2 [M]: 407.0021; [M – H]−: 405.9922; found 405.9975.
- DAZ2: N-(4-(1,3,2-dithiarsinan-2-yl)phenyl)-3,4-dimethoxybenzamide (DA: 3,4-dimethoxybenzoic acid). Yield: 206 mg (40%). 1H NMR (400 MHz, DMSO) δ: 10.27 (s, NH, 1H), 7.97–7.94 (d, J = 8 Hz, 2H), 7.81–7.79 (d, J = 8 Hz, 2H), 7.67–7.64 (s, 1H), 7.56–7.55 (s, 1H), 7.12–7.10 (d, J = 8 Hz, 2H), 3.86 (s, 6H), 2.85–2.74 (m, 4H), 2.03–1.90 (m, 2H). 13C NMR (400 MHz, DMSO): 165.60, 152.26, 148.80, 140.93, 133.12, 132.14, 127.22, 121.42, 111.56, 111.38, 56.17, 56.11, 28.38, 26.18. ESI-MS: m/z calculated for C18H20AsNO3S2 [M]: 437.0018; [M – H]−: 436.0028; found 436.0024.
- DAZ5: N-(2-(1,3,2-dithiarsinan-2-yl)phenyl)-3,4-dimethoxybenzamide (DA: 3,4-dimethoxybenzoic acid). Yield: 123 mg (42%). 1H NMR (400 MHz, DMSO) δ: 10.47 (s, NH, 1H), 8.11–8.09 (d, J = 8 Hz, 1H), 7.64–7.62 (d, J = 8 Hz, 1H), 7.54–7.49 (m, 2H), 7.42–7.37 (m, 2H), 7.12–7.10 (d, J = 8 Hz, 1H), 3.85 (s, 6H), 2.95–2.89 (m, 2H), 2.84–2.78(m, 2H), 2.04–2.00(m, 1H), 1.84–1.75 (m, 2H). 13C NMR (400 MHz, DMSO): 166.07, 152.08, 148.62, 140.77, 134.30, 130.98, 126.20, 121.81, 111.50, 56.18, 56.11, 29.22, 28.29. ESI-MS: m/z calculated for C18H20AsNO3S2 [M]: 437.0018; [M − H]−: 436.0028; found 436.0057.
- TAZ2: N-(4-(1,3,2-dithiarsinan-2-yl)phenyl)-3,4,5-trimethoxybenzamide (TA: 3,4,5-tirmethoxybenzoic acid). Yield: 234 mg (52%). 1H NMR (400 MHz, DMSO) δ: 10.32 (s, NH, 1H), 7.95–7.93 (d, J = 8 Hz, 2H), 7.83–7.81 (d, J = 8 Hz, 2H), 7.30 (s, 2H), 3.89 (s, 6H), 3.75 (s, 3H), 2.84–2.74 (m, 4H), 2.03–1.93 (m, 2H). 13C NMR (400 MHz, DMSO): 165.60, 153.11, 140.90, 140.68, 133.16, 132.48, 130.31, 129.07, 121.57, 105.85, 60.61, 56.60, 28.36, 26.15. ESI-MS: m/z calculated for C19H22AsNO4S2 [M]: 467.0221; [M − H]−: 466.0133; found 466.0178.
- TAZ5: N-(2-(1,3,2-dithiarsinan-2-yl)phenyl)-3,4,5-trimethoxybenzamide (TA: 3,4,5-tirmethoxybenzoic acid). Yield: 131 mg (35%). 1H NMR (400 MHz, DMSO) δ: 10.53 (s, NH, 1H), 8.12–8.10 (d, J = 8 Hz, 1H), 7.55–7.51 (m, 1H), 7.45–7.41 (m, 1H), 7.38–7.36 (d, J = 8 Hz, 1H), 7.31 (s, 2H), 3.89 (s, 6H), 3.76 (s, 3H), 2.96–2.90 (m, 2H), 2.85–2.79 (m, 2H), 2.08–2.00 (m, 1H), 1.84–1.74 (m, 1H). 13C NMR (400 MHz, DMSO): 165.97, 153.14, 141.00, 140.57, 134.92, 134.47, 131.04, 129.36, 127.01, 126.33, 105.86, 60.61, 56.58, 29.19, 28.28. ESI-MS: m/z calculated for C19H22AsNO4S2 [M]: 467.0221; [M − H]−: 466.0133; found 466.0189.
- FZ2: N-(4-(1,3,2-dithiarsinan-2-yl)phenyl)-4-fluorobenzamide (F: 4-fluorobenzoic acid). Yield: 213 mg (53%). 1H NMR (400 MHz, DMSO) δ: 10.46 (s, NH, 1H), 8.08–8.05 (m, 2H), 7.97–7.95 (d, J = 8 Hz, 2H), 7.82–7.80 (d, J = 8 Hz, 2H), 7.42–7.37 (m, 2H), 2.84–2.76 (m, 4H), 2.03–1.90 (m, 2H). 13C NMR (400 MHz, DMSO): 165.15, 163.39, 140.68, 132.52, 131.01, 129.08, 121.37, 120.86, 115.97, 115.90, 115.75, 28.35, 26.13. ESI-MS: m/z calculated for C16H15AsFNOS2 [M]: 394.9812; [M − H]−: 393.9722; found 393.9777.
- FZ5: N-(2-(1,3,2-dithiarsinan-2-yl)phenyl)-4-fluorobenzamide (F: 4-fluorobenzoic acid). Yield: 124 mg (25%). 1H NMR (400 MHz, DMSO) δ: 10.67 (s, NH, 1H), 8.11–8.09 (d, J = 8 Hz, 1H), 8.05–8.02 (m, 2H), 7.54–7.50 (m, 1H), 7.44–7.37 (m, 4H), 2.94–2.87 (m, 2H), 2.82–2.76 (m, 2H), 2.04–1.97 (m, 1H), 1.84–1.75 (m, 1H). 13C NMR (400 MHz, DMSO): 165.58, 140.47, 131.08, 126.34, 116.03, 115.82, 29.12, 28.11. ESI-MS: m/z calculated for C16H15AsFNOS2 [M]: 394.9812; [M − H]−: 393.9722; found 393.9781.
- PFZ2: N-(4-(1,3,2-dithiarsinan-2-yl)phenyl)-2,2-difluorobenzo[d][1,3]dioxole-5-carboxamide (PF:2,2-difluorobenzo[d][1,3]dioxole-5-carboxylic acid). Yield: 231 mg (25%). 1H NMR (400 MHz, DMSO) δ: 10.48 (s, NH, 1H), 8.01–8.00 (s, 1H), 7.96–7.94 (d, J = 8 Hz, 2H), 7.90–7.87 (m, 1H),7.83–7.81 (d, J = 8 Hz, 2H),7.62–7.60 (d, J = 8 Hz, 1H), 2.85–2.73 (m, 4H), 2.04–1.88 (m, 2H). 13C NMR (400 MHz, DMSO): 164.49, 143.19, 140.51, 133.22, 132.74, 131.92, 125.55, 121.38, 110.45, 110.23, 28.33, 26.01. ESI-MS: m/z calculated for C17H14AsF2NO3S2 [M]: 456.9621; [M − H]−: 455.9526; found 455.9585.
- AZ2: 4-((4-(1,3,2-dithiarsinan-2-yl)phenyl)carbamoyl)phenyl acetate (A: 4-acetoxybenzoic acid). Yield: 44 mg (10%). 1H NMR (400 MHz, DMSO) δ: 10.47 (s, NH, 1H), 8.04–8.01 (d, J = 8 Hz, 2H), 7.98–7.96 (d, J = 8 Hz, 2H), 7.82–7.80 (d, J = 8 Hz, 2H), 7.33–7.31 (d, J = 8 Hz, 2H), 2.85–2.77 (m, 4H), 2.32 (s, 3H), 2.01–1.88 (m, 2H). 13C NMR (400 MHz, DMSO): 169.47, 165.52, 153.50, 133.19, 132.76, 129.76, 122.37, 121.32, 28.35, 26.12, 21.36. ESI-MS: m/z calculated for C18H18AsNO3S2 [M]: 434.9901; [M − H]−: 433.9871; found 433.9917; [M + Cl]−: 469.9638; found 469.9688.
- HDZ2: N-(4-(1,3,2-dithiarsinan-2-yl)phenyl)-4-hydroxy-3,5-dimethoxybenzamide (HD: 4-hydroxy-3,5-dimethoxybenzoic acid). Yield: 231 mg (60%). 1H NMR (400 MHz, DMSO) δ: 10.50 (s, –OH, 1H), 10.29 (s, NH, 1H), 7.97–7.93 (m, 2H), 7.38–7.37 (m, 2H), 7.32 (s, 2H), 3.85 (s, 6H), 2.82–2.79 (m, 4H), 1.98–1.90 (m, 2H). 13C NMR (400 MHz, DMSO): 165.67, 165.55, 163.73, 152.34, 147.95, 139.77, 132.69, 129.01, 121.50, 118.13, 108.41, 107.90, 106.20, 105.24, 56.67, 28.39, 26.20, 21.23, 14.55. ESI-MS: m/z calculated for C18H20AsNO4S2 [M]:453.0012; [M − H]−: 451.9977; found 452.0033; [M + Cl]−: 487.9744; found 487.9800.
- MZ2: N-(4-(1,3,2-dithiarsinan-2-yl)phenyl)-4-methylbenzamide (M: 4-methylbenzoic acid). Yield: 124 mg (35%). 1H NMR (400 MHz, DMSO) δ: 10.36 (s, NH, 1H), 7.98–7.96 (d, J = 8 Hz, 2H), 7.91–7.89 (d, J = 8 Hz, 2H), 7.81–7.79 (d, J = 8 Hz, 2H), 7.37–7.35 (d, J = 8 Hz, 2H), 2.85–2.74 (m, 4H), 2.51 (s, 3H), 2.04–1.89 (m, 2H). 13C NMR (400 MHz, DMSO): 166.06, 142.26, 140.87, 133.13, 132.32, 132.25, 129.79, 129.58, 129.42, 128.24, 121.33, 28.37, 26.15, 21.50. ESI-MS: m/z calculated for C17H18AsNOS2 [M]:391.3812; [M − H]−: 390.1222; found 390.1273.
- EZ2: N-(4-(1,3,2-dithiarsinan-2-yl)phenyl)-4-ethylbenzamide (E: 4-ethylbenzoic acid). Yield: 124 mg (45%). 1H NMR (400 MHz, DMSO) δ: 10.36 (s, NH, 1H), 7.98–7.96 (d, J = 8 Hz, 2H), 7.92–7.90 (d, J = 8 Hz, 2H), 7.81–7.79 (d, J = 8 Hz, 2H), 7.40–7.38 (d, J = 8 Hz, 2H), 2.85–2.77 (m, 4H), 2.74–2.67 (m, 2H), 2.05–1.88 (m, 2H), 1.24–1.21 (m, 3H), 13C NMR (400 MHz, DMSO): 166.14, 148.41, 140.88, 133.14, 132.66, 132.24, 128.33, 128.26, 121.30, 28.56, 28.36, 26.14, 15.87. ESI-MS: m/z calculated for C18H20AsNOS2 [M]:405.0121; [M − H]−: 404.0133; found 404.1486.
4.4. Cell Culture
4.5. MTT Assay
4.6. Molecular Docking
4.7. Interaction between Peptide AGCUG and MAZ2
4.8. TrxR Activity
4.9. siRNA
4.10. PCR
4.11. ROS Level
4.12. Mitochondrial Membrane Potential
4.13. Caspase 3 Activity
4.14. Apoptosis
4.15. Mice Experiments
4.16. Western Blotting
4.17. As Concentration
4.18. Pharmaceutic Kinetics
4.19. Viscera Hematoxylin and Eosin (H & E)
4.20. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Eklund, H.; Gleason, F.K.; Holmgren, A. Structural and functional relations among thioredoxins of different species. Proteins Struct. Funct. Bioinform. 1991, 11, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef]
- Xie, W.; Ma, W.; Liu, P.; Zhou, F. Overview of thioredoxin system and targeted therapies for acute leukemia. Mitochondrion 2019, 47, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Li, D.; Feng, X.; Zhao, F.; Li, C.; Zheng, S.; Lyu, J. A pan-cancer study of selenoprotein genes as promising targets for cancer therapy. BMC Med. Genom. 2021, 14, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Xu, I.M.J.; Chiu, D.K.C.; Leibold, J.; Tse, A.P.W.; Bao, M.H.R.; Yuen, V.W.H.; Chan, C.Y.K.; Lai, R.K.H.; Chin, D.W.C.; et al. Induction of Oxidative Stress Through Inhibition of Thioredoxin Reductase 1 Is an Effective Therapeutic Approach for Hepatocellular Carcinoma. Hepatology 2019, 69, 1768–1786. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, Y.; Li, X.; Xu, J.; Fang, J. Small Molecules to Target the Selenoprotein Thioredoxin Reductase. Chem. Asian J. 2018, 13, 3593–3600. [Google Scholar] [CrossRef]
- Karunanithi, S.; Liu, R.; Hou, Y.; Gonzalez, G.; Oldford, N.; Roe, A.J.; Idipilly, N.; Gupta, K.; Amara, C.S.; Putluri, S.; et al. Thioredoxin reductase is a major regulator of metabolism in leukemia cells. Oncogene 2021, 40, 5236–5246. [Google Scholar] [CrossRef]
- Zhou, F.; Shen, Q.; Claret, F.X. Novel roles of reactive oxygen species in the pathogenesis of acute myeloid leukemia. J. Leukoc. Biol. 2013, 94, 423–429. [Google Scholar] [CrossRef] [Green Version]
- Hole, P.S.; Darley, R.L.; Tonks, A. Do reactive oxygen species play a role in myeloid leukemias? Blood 2011, 117, 5816–5826. [Google Scholar] [CrossRef] [Green Version]
- Haendeler, J.; Hoffmann, J.; Tischler, V.; Berk, B.C.; Zeiher, A.M.; Dimmeler, S. Redox regulatory and anti-apoptotic functions of thioredoxin depend on S-nitrosylation at cysteine 69. Nat. Cell Biol. 2002, 4, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.; Marletta, M.A. Thioredoxin catalyzes the S-nitrosation of the caspase-3 active site cysteine. Nat. Chem. Biol. 2005, 1, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Bishopric, N.H.; Webster, K.A. Preventing apoptosis with thioredoxin. Circ. Res. 2002, 90, 1237–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Pan, Y.; Wei, Y.; Zhang, R.; Bai, G.; Shen, Q.; Meng, S.; Le, X.-F.; Andreeff, M.; Claret, F.X. Jab1/Csn5–Thioredoxin Signaling in Relapsed Acute Monocytic Leukemia under Oxidative Stress. Clin. Cancer Res. 2017, 23, 4450–4462. [Google Scholar] [CrossRef] [Green Version]
- Bian, M.; Fan, R.; Zhao, S.; Liu, W. Targeting the Thioredoxin System as a Strategy for Cancer Therapy. J. Med. Chem. 2019, 62, 7309–7321. [Google Scholar] [CrossRef]
- Millet, R.; Urig, S.; Jacob, J.; Amtmann, E.; Moulinoux, J.-P.; Gromer, S.; Becker, K.; Davioud-Charvet, E. Synthesis of 5-Nitro-2-furancarbohydrazides and Their cis-Diamminedichloroplatinum Complexes as Bitopic and Irreversible Human Thioredoxin Reductase Inhibitors. J. Med. Chem. 2005, 48, 7024–7039. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, B.; Li, X.; Han, X.; Liu, R.; Fang, J. Small molecule inhibitors of mammalian thioredoxin reductase as potential anticancer agents: An update. Med. Res. Rev. 2019, 39, 5–39. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Duan, D.; Yao, J.; Zhang, B.; Peng, S.; Ma, H.; Song, Y.; Fang, J. Dithiaarsanes Induce Oxidative Stress-Mediated Apoptosis in HL-60 Cells by Selectively Targeting Thioredoxin Reductase. J. Med. Chem. 2014, 57, 5203–5211. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, Y.; Luo, Z.; Chen, Y.; Xu, A.; Liang, Y.; Wu, B.; Tong, X.; Liu, X.; Shen, H.; et al. The novel thioredoxin reductase inhibitor A-Z2 triggers intrinsic apoptosis and shows efficacy in the treatment of acute myeloid leukemia. Free Radic. Biol. Med. 2019, 146, 275–286. [Google Scholar] [CrossRef]
- Fan, X.-Y.; Liu, Y.-J.; Chen, K.; Jiang, F.-L.; Hu, Y.-J.; Liu, D.; Liu, Y.; Ge, Y.-S. Organic arsenicals target thioredoxin reductase followed by oxidative stress and mitochondrial dysfunction resulting in apoptosis. Eur. J. Med. Chem. 2018, 143, 1090–1102. [Google Scholar] [CrossRef]
- Strzelczyk, A.; Schubert-Bast, S. Therapeutic advances in Dravet syndrome: A targeted literature review. Expert Rev. Neurother. 2020, 20, 1065–1079. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, A.-S.; Ceulemans, B. Dravet syndrome—toward an optimal and disease-specific treatment. Z. Epileptol. 2021, 34, 146–153. [Google Scholar] [CrossRef]
- Pernici, C.D.; Mensah, J.A.; Dahle, E.J.; Johnson, K.J.; Handy, L.; Buxton, L.; Smith, M.D.; West, P.J.; Metcalf, C.S.; Wilcox, K.S. Development of an antiseizure drug screening platform for Dravet syndrome at the NINDS contract site for the Epilepsy Therapy Screening Program. Epilepsia 2021, 62, 1665–1676. [Google Scholar] [CrossRef] [PubMed]
- Trojnar, M.; Wojtal, K.; Trojnar, M.; Czuczwar, S.S. A novel antiepileptic drug. Pharmacol. Rep. 2004, 57, 154–160. [Google Scholar]
- Kamitaki, B.K.; Minacapelli, C.D.; Zhang, P.; Wachuku, C.; Gupta, K.; Catalano, C.; Rustgi, V. Drug-induced liver injury associated with antiseizure medications from the FDA Adverse Event Reporting System (FAERS). Epilepsy Behav. 2021, 117, 107832–107836. [Google Scholar] [CrossRef]
- Verrotti, A.; Prezioso, G.; Stagi, S.; Paolino, M.C.; Parisi, P. Pharmacological considerations in the use of stiripentol for the treatment of epilepsy. Expert Opin. Drug Metab. Toxicol. 2016, 12, 345–352. [Google Scholar] [CrossRef]
- Devi, N.; Madaan, P.; Asrar, M.M.; Sahu, J.K.; Bansal, D. Comparative short-term efficacy and safety of add-on anti-seizure medications in Dravet syndrome: An indirect treatment comparison. Seizure 2021, 91, 316–324. [Google Scholar] [CrossRef]
- Lin, H.-S.; Levy, R.H. Pharmacokinetic Profile of a New Anticonvulsant, Stiripentol, in the Rhesus Monkey. Epilepsia 1983, 24, 692–702. [Google Scholar] [CrossRef]
- Liu, Y.-J.; Fan, X.-Y.; Wang, A.-D.; Xia, Y.-Z.; Fu, W.-R.; Liu, J.-Y.; Jiang, F.-L.; Liu, Y. LDHA Suppression Altering Metabolism Inhibits Tumor Progress by an Organic Arsenical. Int. J. Mol. Sci. 2019, 20, 6239. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Chen, G.; Wang, Y.; Wang, Q.; Zhong, Y.; Yang, X.-F.; Li, Z.; Li, H. Far-Red Fluorescent Probe for Imaging of Vicinal Dithiol-Containing Proteins in Living Cells Based on a pKa Shift Mechanism. Anal. Chem. 2018, 90, 2946–2953. [Google Scholar] [CrossRef]
- Hu, G.; Jia, H.; Zhao, L.; Cho, D.-H.; Fang, J. Small molecule fluorescent probes of protein vicinal dithiols. Chin. Chem. Lett. 2019, 30, 1704–1716. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Q.; Liu, X.; Zhang, J.; Yang, X.-F.; Li, Z.; Li, H. Sensitive and Selective Fluorescent Probe for Selenol in Living Cells Designed via a pKa Shift Strategy. Anal. Chem. 2018, 90, 4119–4125. [Google Scholar] [CrossRef] [PubMed]
- Joardar, N.; Guevara-Flores, A.; Martínez-González, J.D.J.; Babu, S.P.S. Thiol antioxidant thioredoxin reductase: A prospective biochemical crossroads between anticancer and antiparasitic treatments of the modern era. Int. J. Biol. Macromol. 2020, 165 Pt A, 249–267. [Google Scholar] [CrossRef]
- Wilson, P.; Anastasaki, A.; Owen, M.R.; Kempe, K.; Haddleton, D.M.; Mann, S.K.; Johnston, A.P.R.; Quinn, J.F.; Whittaker, M.R.; Hogg, P.J.; et al. Organic Arsenicals as Efficient and Highly Specific Linkers for Protein/Peptide–Polymer Conjugation. J. Am. Chem. Soc. 2015, 137, 4215–4222. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Li, J.; Liu, Q.; Peng, H.; Popowich, A.; Wang, Z.; Li, X.F.; Le, X.C. p-Azidophenylarsenoxide: An Arsenical “Bait” for the In Situ Capture and Identification of Cellular Arsenic-Binding Proteins. Angew. Chem. Int. Ed. 2016, 55, 14051–14056. [Google Scholar] [CrossRef] [PubMed]
- Chupakhin, E.; Krasavin, M. Thioredoxin reductase inhibitors: Updated patent review (2017-present). Expert Opin. Ther. Patents 2021, 31, 745–758. [Google Scholar] [CrossRef]
- Ndugire, W.; Raviranga, N.G.H.; Lao, J.; Ramström, O.; Yan, M. Gold Nanoclusters as Nanoantibiotic Auranofin Analogues. Adv. Healthc. Mater. 2021, 11, 2101032. [Google Scholar] [CrossRef]
- Serrano, J.J.; Delgado, B.; Medina, M. Control of tumor angiogenesis and metastasis through modulation of cell redox state. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188352. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Ganesan, S.; Alex, A.A.; Chendamarai, E.; Balasundaram, N.; Palani, H.K.; David, S.; Kulkarni, U.; Aiyaz, M.; Mugasimangalam, R.; Korula, A.; et al. Rationale and efficacy of proteasome inhibitor combined with arsenic trioxide in the treatment of acute promyelocytic leukemia. Leukemia 2016, 30, 2169–2178. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50/μM | Molm | NB4 | 4T1 | HeLa | COS-7 | PBMC | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | |
| Z2 | 0.60 | 0.53 | 1.50 | 1.20 | 1.67 | 1.89 | 1.01 | 0.70 | 6.1 | 3.2 | 4.6 | 4.0 |
| Z5 | 0.45 | 0.41 | 0.40 | 0.30 | 0.87 | 0.84 | 0.30 | 0.25 | 1.75 | 0.81 | 3.5 | 2.8 |
| PZ2 | 1.06 | 0.75 | 0.70 | 0.44 | 2.6 | 3.4 | 0.70 | 0.44 | 4.1 | 1.75 | 6.4 | 5.3 |
| PZ5 | 0.27 | 0.24 | 0.26 | 0.14 | 0.40 | 0.83 | 0.26 | 0.14 | 1.01 | 0.82 | 5.0 | 4.5 |
| PFZ2 | 0.80 | 0.70 | 0.50 | 0.30 | 1.00 | 0.80 | 0.50 | 0.15 | 6.0 | 3.5 | 6.3 | 5.7 |
| HDZ2 | 0.60 | 0.60 | 0.80 | 0.50 | 1.00 | 1.01 | 2.0 | 1.01 | 9.1 | 3.5 | 5.8 | 4.9 |
| TAZ2 | 1.60 | 1.50 | 1.00 | 0.60 | 3.5 | 2.2 | 2.0 | 1.04 | 9.1 | 5.2 | 7.3 | 6.5 |
| TAZ5 | 0.71 | 0.70 | 0.20 | 0.15 | 0.70 | 0.70 | 0.80 | 0.50 | 2.0 | 1.80 | 2.2 | 1.54 |
| DAZ2 | 0.90 | 0.75 | 1.71 | 1.14 | 1.50 | 1.20 | 2.3 | 1.06 | 7.1 | 3.3 | 6.2 | 5.7 |
| DAZ5 | 0.45 | 0.32 | 0.80 | 0.51 | 0.60 | 0.50 | 0.81 | 0.55 | 1.61 | 1.27 | 4.1 | 3.5 |
| MAZ2 | 0.20 | 0.10 | 0.40 | 0.30 | 0.80 | 0.40 | 0.40 | 0.30 | 5.2 | 3.3 | 7.6 | 6.9 |
| FZ2 | 1.20 | 1.00 | 0.40 | 0.30 | 2.3 | 2.2 | 1.20 | 0.40 | 3.5 | 2.2 | 5.5 | 4.7 |
| FZ5 | 0.50 | 0.50 | 0.15 | 0.15 | 0.70 | 0.60 | 0.35 | 0.15 | 1.10 | 0.80 | 2.4 | 1.82 |
| MZ2 | 1.20 | 0.80 | 0.40 | 0.30 | 1.20 | 0.91 | 0.61 | 0.40 | 3.4 | 1.21 | 3.4 | 2.7 |
| EZ2 | 1.10 | 1.02 | 0.41 | 0.30 | 1.40 | 1.40 | 1.01 | 0.81 | 4.2 | 2.1 | 3.7 | 2.9 |
| AZ2 | 1.10 | 1.00 | 0.60 | 0.60 | 2.0 | 1.00 | 0.80 | 0.50 | 2.2 | 2.1 | 5.2 | 4.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, X.-M.; She, W.-Y.; Liu, T.-T.; Gao, L.-X.; Liu, Y.-J.; Liu, Y. 1,3-Benzodioxole Derivatives Improve the Anti-Tumor Efficiency of Arsenicals. Int. J. Mol. Sci. 2022, 23, 6930. https://doi.org/10.3390/ijms23136930
Shi X-M, She W-Y, Liu T-T, Gao L-X, Liu Y-J, Liu Y. 1,3-Benzodioxole Derivatives Improve the Anti-Tumor Efficiency of Arsenicals. International Journal of Molecular Sciences. 2022; 23(13):6930. https://doi.org/10.3390/ijms23136930
Chicago/Turabian StyleShi, Xue-Min, Wen-Yan She, Ting-Ting Liu, Lian-Xun Gao, Yu-Jiao Liu, and Yi Liu. 2022. "1,3-Benzodioxole Derivatives Improve the Anti-Tumor Efficiency of Arsenicals" International Journal of Molecular Sciences 23, no. 13: 6930. https://doi.org/10.3390/ijms23136930
APA StyleShi, X.-M., She, W.-Y., Liu, T.-T., Gao, L.-X., Liu, Y.-J., & Liu, Y. (2022). 1,3-Benzodioxole Derivatives Improve the Anti-Tumor Efficiency of Arsenicals. International Journal of Molecular Sciences, 23(13), 6930. https://doi.org/10.3390/ijms23136930