
Exploration of Somatostatin Binding Mechanism to Somatostatin Receptor Subtype 4

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
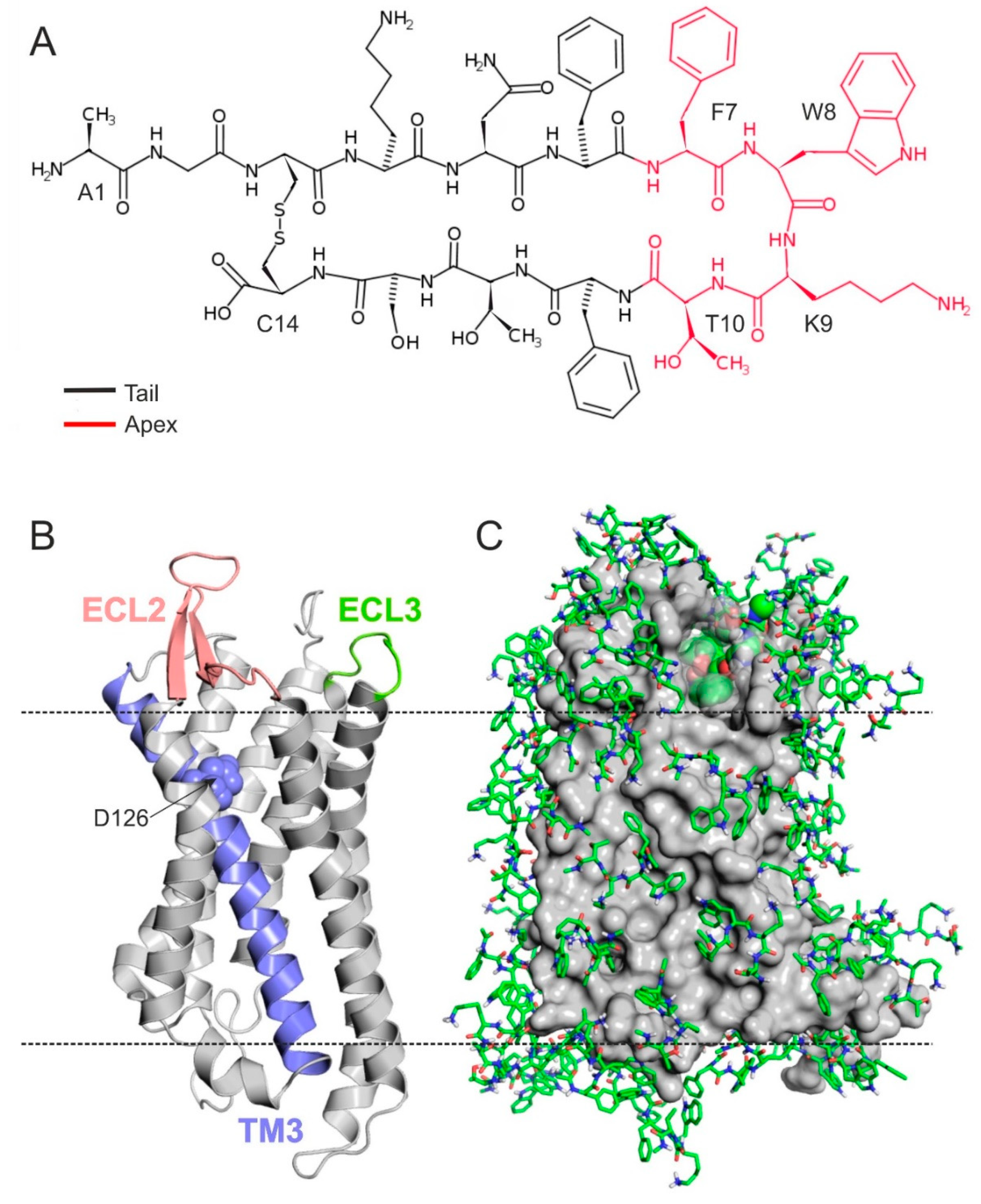
2.1. The Structure of SSTR4
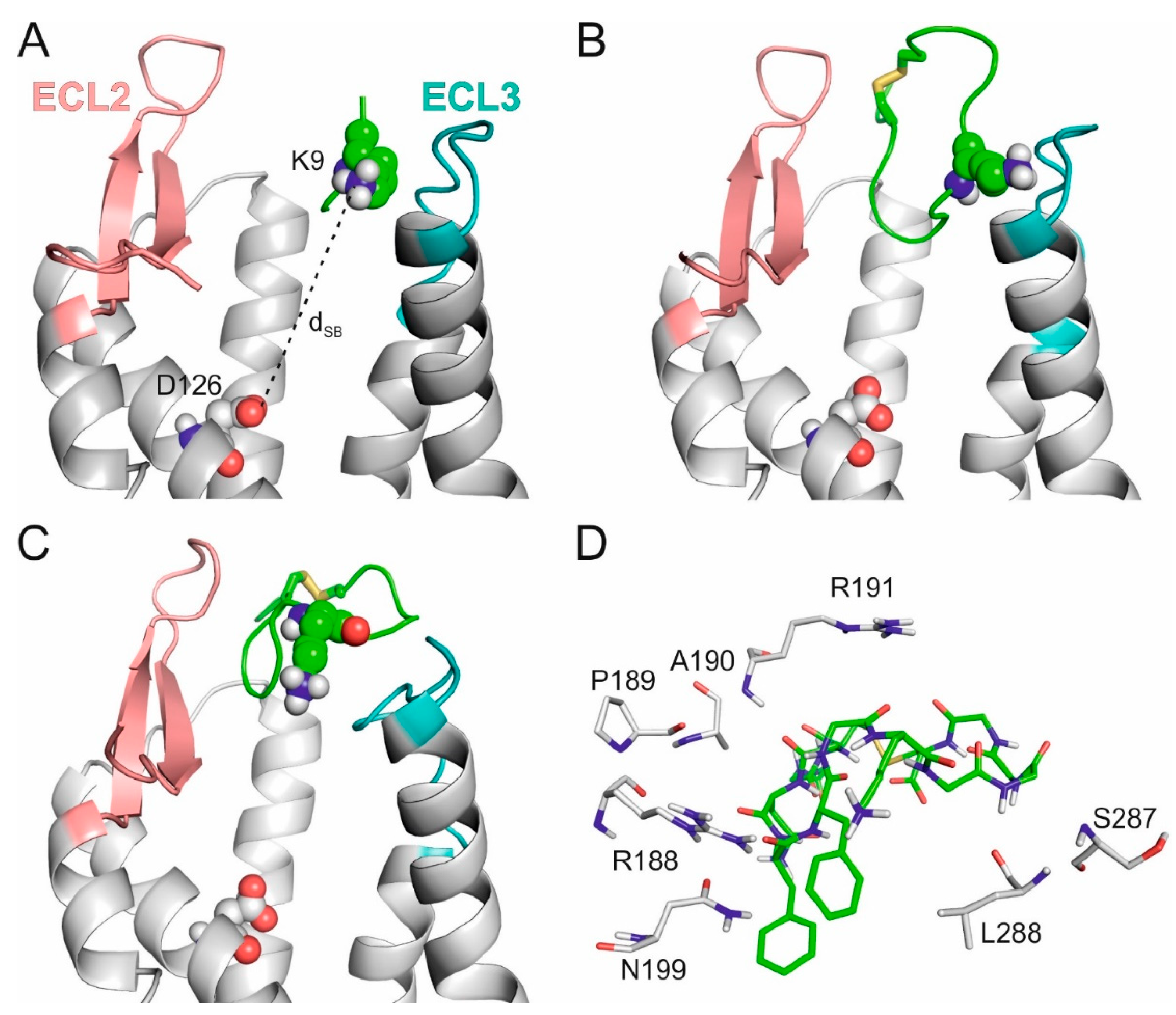
2.2. The External Binding Mode
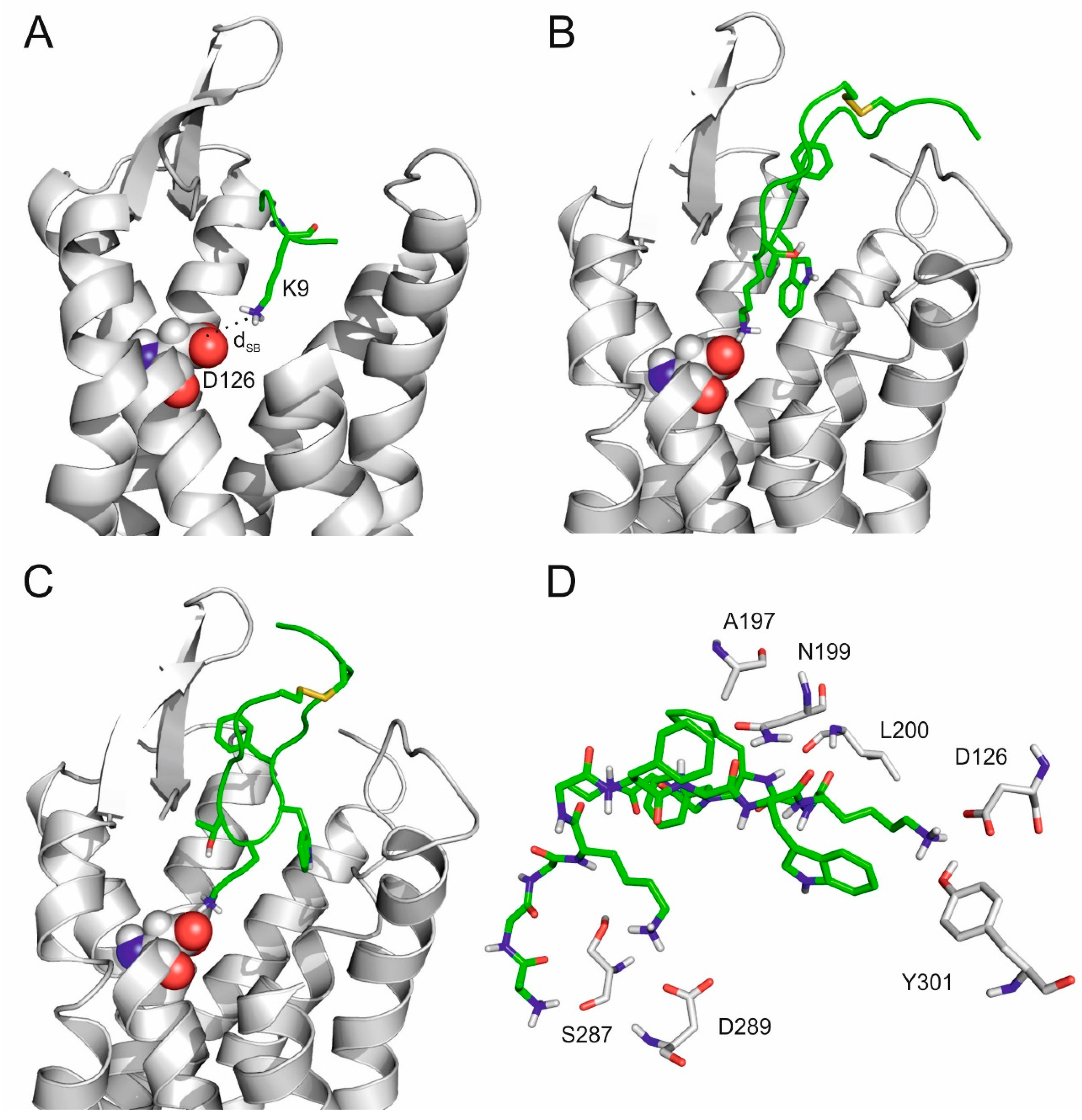
2.3. The Internal Binding Mode
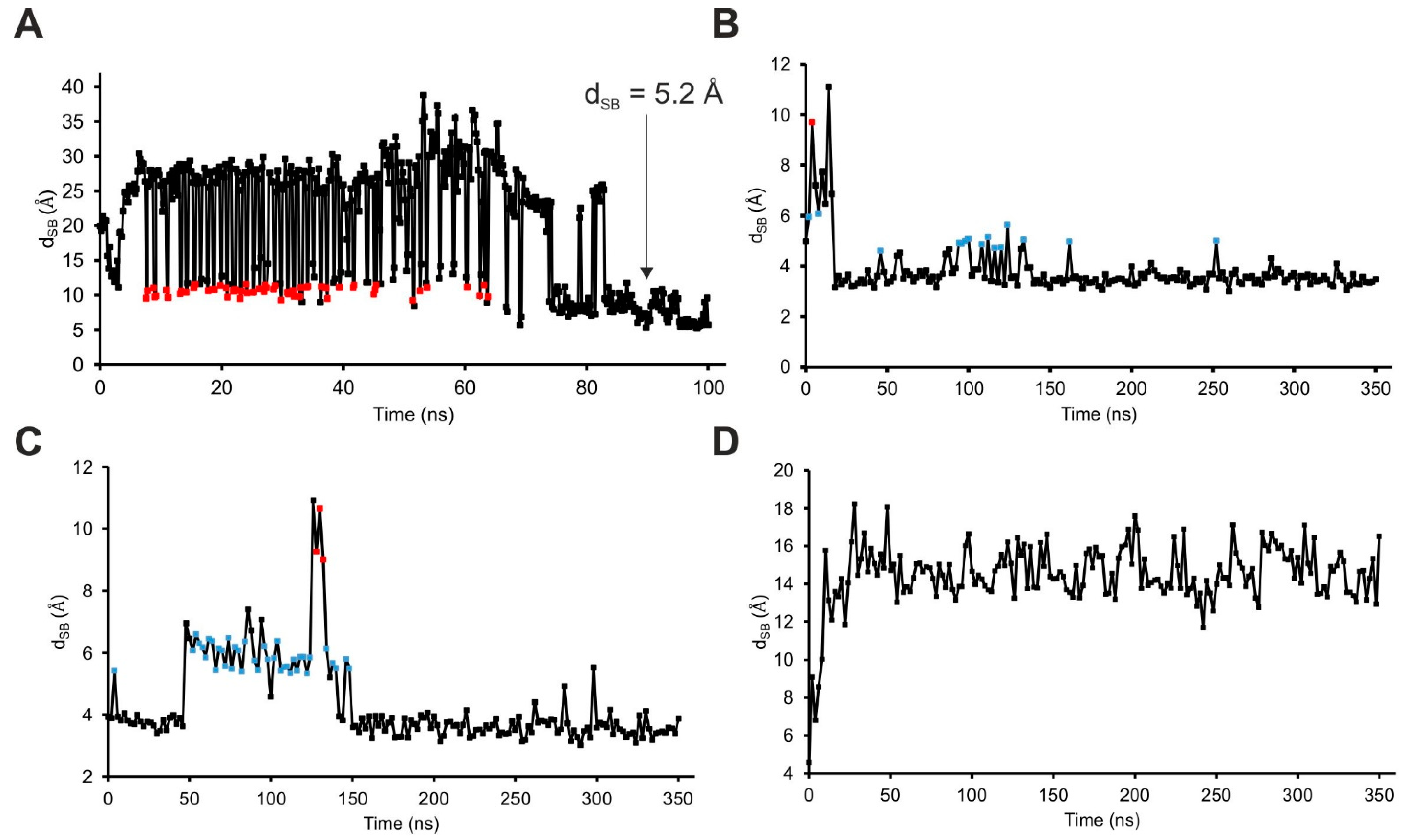
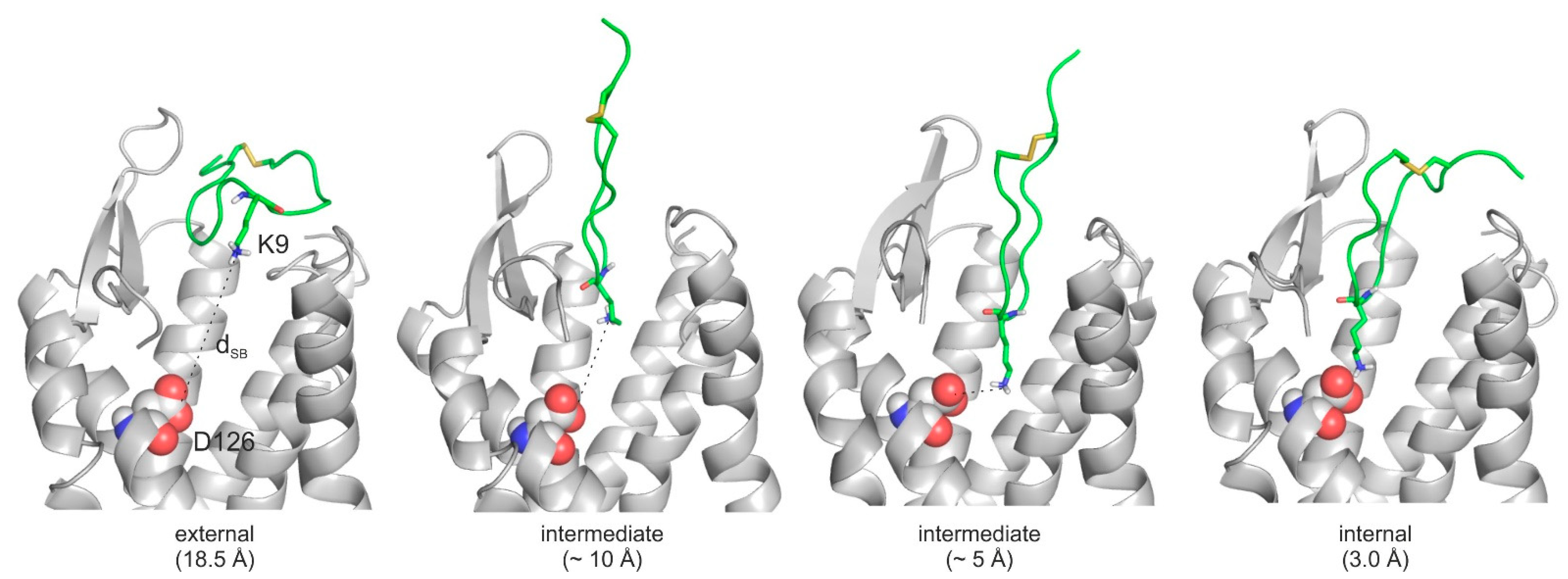
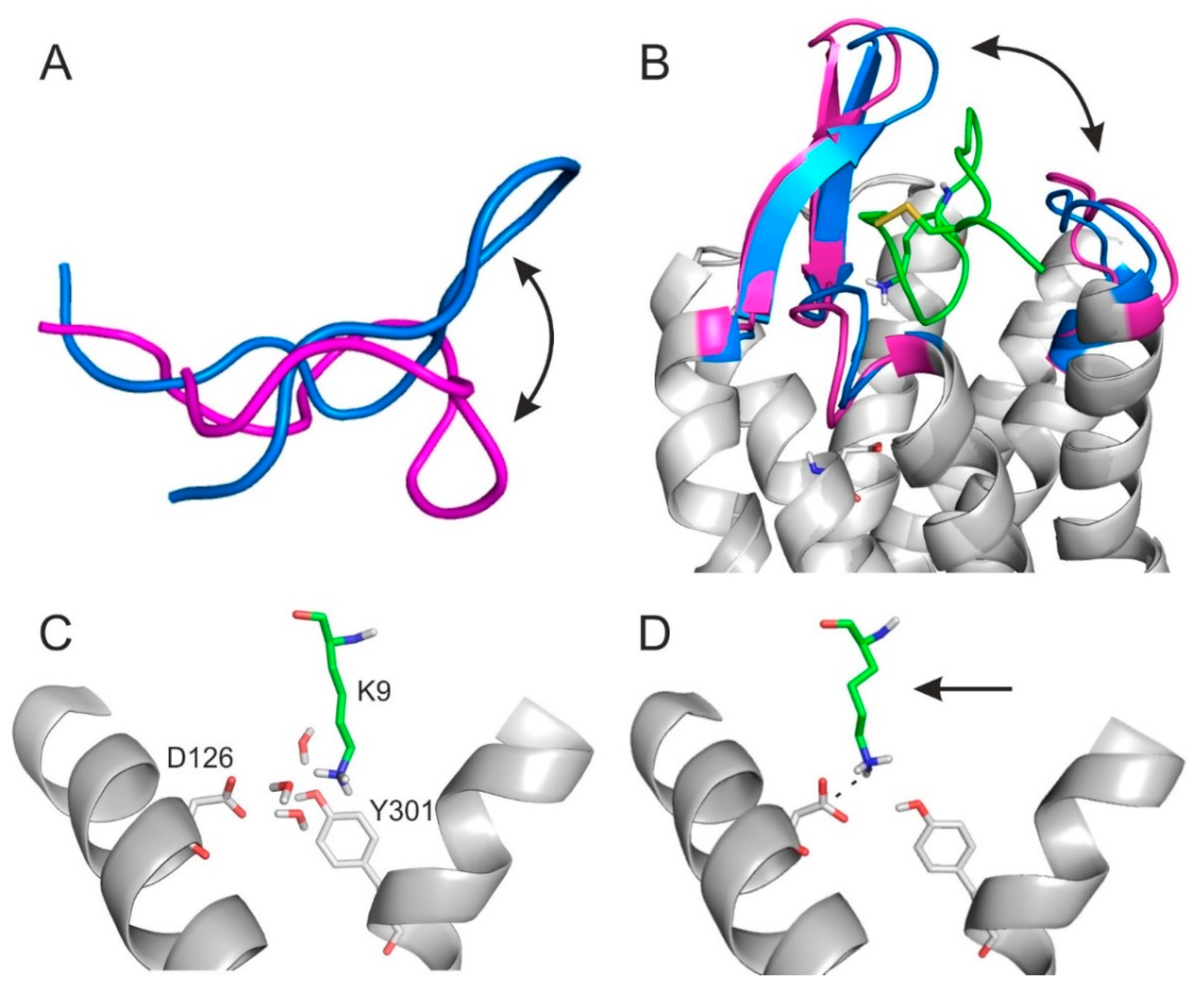
2.4. The Binding Mechanism
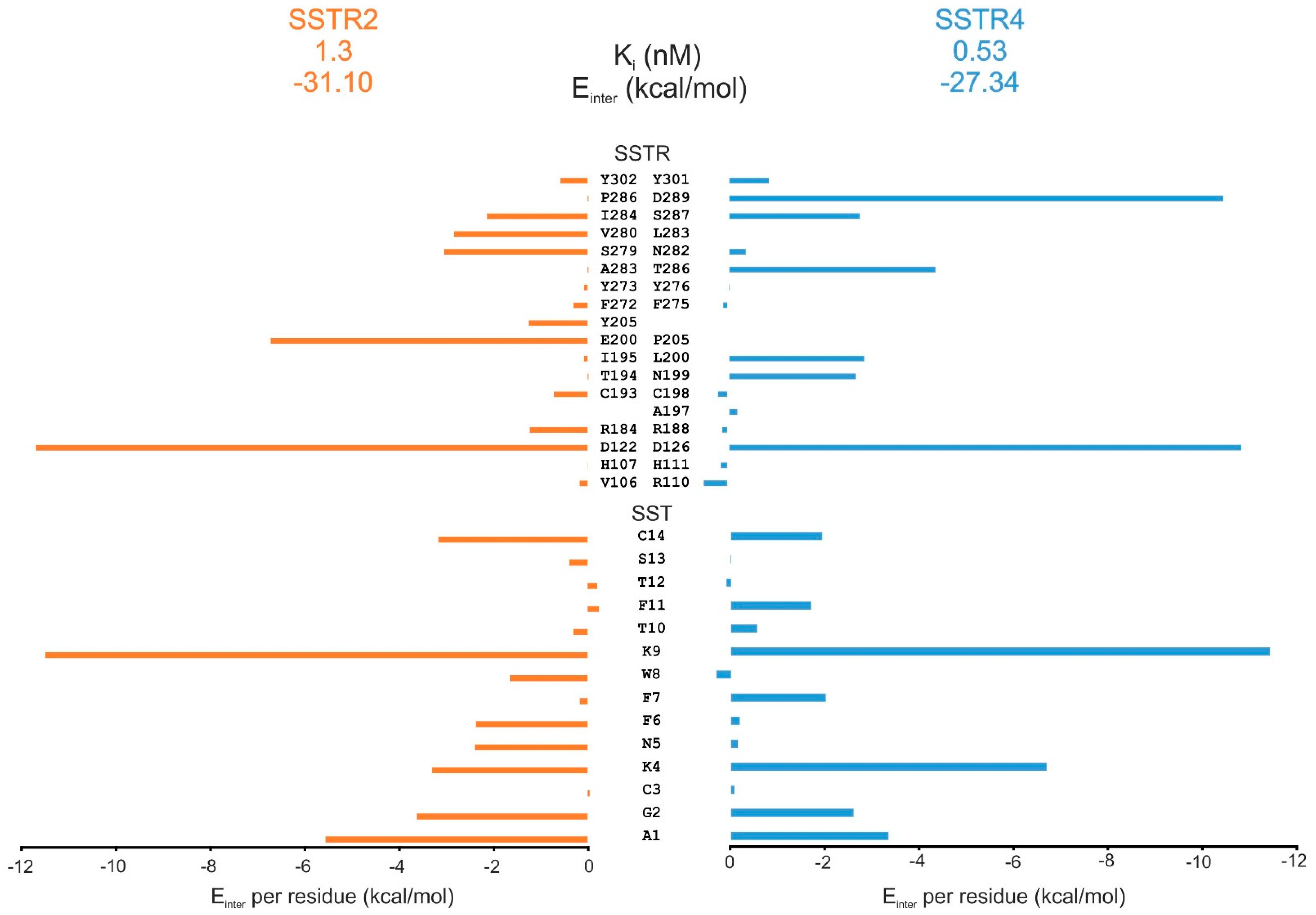
2.5. Comparison of SST Subtype Binding
3. Methods
3.1. The Structure of SSTR4
3.2. The External Binding Mode
3.2.1. Fragment of SST
3.2.2. Energy Minimization
3.2.3. Docking Calculations
3.2.4. Growing of SST into the Binding cleft
3.2.5. Molecular Dynamics Simulation
3.2.6. MD Refinement in the External Binding Cleft
3.3. The Internal Binding Mode
3.3.1. Molecular Simulation for Exploring the Internal Binding Cleft
3.3.2. Growing the Full Length SST into the Receptor
3.3.3. MD Refinement in the Internal Binding Cleft
3.4. Comparison of SST Subtype Binding
3.4.1. Determination of Interacting Energy per Residues in SSTR4/SSTR2–SST Complexes
3.4.2. Energy Analysis of Alternative Binding Mode
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
| SST | somatostatin |
| TM | transmembrane |
| ECL | extracellular loop |
| ICL | intracellular loop |
| SSTR1–5 | somatostatin receptor subtype 1–5 |
| GPCR | G-protein coupled receptor |
| WNS | Wrap ‘n‘ Shake |
| MD | molecular dynamics |
| PDB | Protein Data Bank |
| dSB | Distance between the amino N atom of SST:K9 and the carboxylate C atom of SSTR4:D126 |
| BLAST | Basic Local Alignment Search Tool |
| FBD | Fragment blind docking |
References
- Esch, F.; Böhlen, P.; Ling, N.; Benoit, R.; Brazeau, P.; Guillemin, R. Primary Structure of Ovine Hypothalamic Somatostatin-28 and Somatostatin-25. Proc. Natl. Acad. Sci. USA 1980, 77, 6827–6831. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, D.; Bell, G.I.; Berelowitz, M.; Epelbaum, J.; Feniuk, W.; Humphrey, P.P.A.; O’Carroll, A.-M.; Patel, Y.C.; Schonbrunn, A.; Taylor, J.E.; et al. Classification and Nomenclature of Somatostatin Receptors. Trends Pharmacol. Sci. 1995, 16, 86–88. [Google Scholar] [CrossRef]
- Patel, Y.C. Somatostatin and Its Receptor Family. Front. Neuroendocr. 1999, 20, 157–198. [Google Scholar] [CrossRef] [PubMed]
- Pradayrol, L.; Jörnvall, H.; Mutt, V.; Ribet, A. N-Terminally Extended Somatostatin: The Primary Structure of Somatostatin-28. FEBS Lett. 1980, 109, 55–58. [Google Scholar] [CrossRef]
- Olias, G.; Viollet, C.; Kusserow, H.; Epelbaum, J.; Meyerhof, W. Regulation and Function of Somatostatin Receptors. J. Neurochem. 2004, 89, 1057–1091. [Google Scholar] [CrossRef]
- Rai, U.; Thrimawithana, T.R.; Valery, C.; Young, S.A. Therapeutic Uses of Somatostatin and Its Analogues: Current View and Potential Applications. Pharm. Ther. 2015, 152, 98–110. [Google Scholar] [CrossRef]
- Shamsi, B.H.; Chatoo, M.; Xu, X.K.; Xu, X.; Chen, X.Q. Versatile Functions of Somatostatin and Somatostatin Receptors in the Gastrointestinal System. Front. Endocrinol. 2021, 12, 652363. [Google Scholar] [CrossRef]
- Helyes, Z.; Pintér, E.; Németh, J.; Kéri, G.; Thán, M.; Oroszi, G.; Horváth, A.; Szolcsányi, J. Anti-Inflammatory Effect of Synthetic Somatostatin Analogues in the Rat. Br. J. Pharmacol. 2001, 134, 1571–1579. [Google Scholar] [CrossRef]
- Pintér, E.; Helyes, Z.; Szolcsányi, J. Inhibitory Effect of Somatostatin on Inflammation and Nociception. Pharmacol. Ther. 2006, 112, 440–456. [Google Scholar] [CrossRef]
- Gastambide, F.; Lepousez, G.; Viollet, C.; Loudes, C.; Epelbaum, J.; Guillou, J.-L. Cooperation between Hippocampal Somatostatin Receptor Subtypes 4 and 2: Functional Relevance in Interactive Memory Systems. Hippocampus 2010, 20, 745–757. [Google Scholar] [CrossRef]
- Prasoon, P.; Kumar, R.; Gautam, M.; Sebastian, E.K.; Reeta, K.H.; Ray, S.B. Role of Somatostatin and Somatostatin Receptor Type 2 in Postincisional Nociception in Rats. Neuropeptides 2015, 49, 47–54. [Google Scholar] [CrossRef]
- Schuelert, N.; Just, S.; Kuelzer, R.; Corradini, L.; Gorham, L.C.J.; Doods, H. The Somatostatin Receptor 4 Agonist J-2156 Reduces Mechanosensitivity of Peripheral Nerve Afferents and Spinal Neurons in an Inflammatory Pain Model. Eur. J. Pharmacol. 2015, 746, 274–281. [Google Scholar] [CrossRef]
- Yeo, X.Y.; Cunliffe, G.; Ho, R.C.; Lee, S.S.; Jung, S. Potentials of Neuropeptides as Therapeutic Agents for Neurological Diseases. Biomedicines 2022, 10, 343. [Google Scholar] [CrossRef] [PubMed]
- Boehm, B.O.; Lustig, R.H. Use of Somatostatin Receptor Ligands in Obesity and Diabetic Complications. Best Pract. Res. Clin. Gastroenterol. 2002, 16, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Chanson, P. Medical Treatment of Acromegaly with Dopamine Agonists or Somatostatin Analogs. Neuroendocrinology 2016, 103, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Hofland, L.J. Somatostatin and Somatostatin Receptors in Cushing’s Disease. Mol. Cell. Endocrinol. 2008, 286, 199–205. [Google Scholar] [CrossRef]
- Sun, L.; Coy, D.H. Somatostatin and Its Analogs. Curr. Drug. Targets 2016, 17, 529–537. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F. Treating Pain with Somatostatin Receptor Subtype 4 Agonists. ACS Med. Chem. Lett. 2015, 6, 110–111. [Google Scholar] [CrossRef]
- Clinical Development Pipeline | Science | Eli Lilly and Company. Available online: https://www.lilly.com/discovery/clinical-development-pipeline#/ (accessed on 3 May 2021).
- Banno, Y.; Sasaki, S.; Kamata, M.; Kunitomo, J.; Miyamoto, Y.; Abe, H.; Taya, N.; Oi, S.; Watanabe, M.; Urushibara, T.; et al. Design and Synthesis of a Novel Series of Orally Active, Selective Somatostatin Receptor 2 Agonists for the Treatment of Type 2 Diabetes. Bioorganic Med. Chem. 2017, 25, 5995–6006. [Google Scholar] [CrossRef]
- Fattah, S.; Brayden, D.J. Progress in the Formulation and Delivery of Somatostatin Analogs for Acromegaly. Ther. Deliv. 2017, 8, 867–878. [Google Scholar] [CrossRef]
- Giovannini, R.; Cui, Y.; Doods, H.; Ferrara, M.; Just, S.; Kuelzer, R.; Lingard, I.; Mazzaferro, R.; Rudolf, K. New Somatostatin Receptor Subtype 4 (Sstr4) Agonists. Patent International Publication Number WO 2014/184275 A1, 20 November 2014. [Google Scholar]
- Johnson, M.L.; Meyer, T.; Halperin, D.M.; Fojo, A.T.; Cook, N.; Blaszkowsky, L.S.; Schlechter, B.L.; Yao, J.C.; Jemiai, Y.; Kriksciukaite, K.; et al. First in Human Phase 1/2a Study of PEN-221 Somatostatin Analog (SSA)-DM1 Conjugate for Patients (PTS) with Advanced Neuroendocrine Tumor (NET) or Small Cell Lung Cancer (SCLC): Phase 1 Results. J. Clin. Oncol. 2018, 36, 4097. [Google Scholar] [CrossRef]
- Liu, W.; Shao, P.P.; Liang, G.-B.; Bawiec, J.; He, J.; Aster, S.D.; Wu, M.; Chicchi, G.; Wang, J.; Tsao, K.-L.; et al. Discovery and Pharmacology of a Novel Somatostatin Subtype 5 (SSTR5) Antagonist: Synergy with DPP-4 Inhibition. ACS Med. Chem. Lett. 2018, 9, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Mansi, R.; Abid, K.; Nicolas, G.P.; Del Pozzo, L.; Grouzmann, E.; Fani, M. A New 68Ga-Labeled Somatostatin Analog Containing Two Iodo-Amino Acids for Dual Somatostatin Receptor Subtype 2 and 5 Targeting. EJNMMI Res. 2020, 10, 90. [Google Scholar] [CrossRef] [PubMed]
- Kaupmann, K.; Bruns, C.; Raulf, F.; Weber, H.P.; Mattes, H.; Lübbert, H. Two Amino Acids, Located in Transmembrane Domains VI and VII, Determine the Selectivity of the Peptide Agonist SMS 201-995 for the SSTR2 Somatostatin Receptor. EMBO J. 1995, 14, 727–735. [Google Scholar] [CrossRef]
- Leu, F.P.; Nandi, M. GPCR Somatostatin Receptor Extracellular Loop 2 Is a Key Ectodomain for Making Subtype-Selective Antibodies with Agonist-like Activities in the Pancreatic Neuroendocrine Tumor BON Cell Line. Pancreas 2010, 39, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Liapakis, G.; Fitzpatrick, D.; Hoeger, C.; Rivier, J.; Vandlen, R.; Reisine, T. Identification of Ligand Binding Determinants in the Somatostatin Receptor Subtypes 1 and 2. J. Biol. Chem. 1996, 271, 20331–20339. [Google Scholar] [CrossRef]
- Bruns, C.; Raulf, F.; Hoyer, D.; Schloos, J.; Lübbert, H.; Weckbecker, G. Binding Properties of Somatostatin Receptor Subtypes. Metabolism 1996, 45, 17–20. [Google Scholar] [CrossRef]
- Bruns, C.; Lewis, I.; Briner, U.; Meno-Tetang, G.; Weckbecker, G. SOM230: A Novel Somatostatin Peptidomimetic with Broad Somatotropin Release Inhibiting Factor (SRIF) Receptor Binding and a Unique Antisecretory Profile. Eur. J. Endocrinol. 2002, 146, 707–716. [Google Scholar] [CrossRef]
- Lewis, I.; Bauer, W.; Albert, R.; Chandramouli, N.; Pless, J.; Weckbecker, G.; Bruns, C. A Novel Somatostatin Mimic with Broad Somatotropin Release Inhibitory Factor Receptor Binding and Superior Therapeutic Potential. J. Med. Chem. 2003, 46, 2334–2344. [Google Scholar] [CrossRef]
- Patel, Y.C.; Srikant, C.B. Subtype Selectivity of Peptide Analogs for All Five Cloned Human Somatostatin Receptors (Hsstr 1-5). Endocrinology 1994, 135, 2814–2817. [Google Scholar] [CrossRef]
- Rohrer, S.P.; Birzin, E.T.; Mosley, R.T.; Berk, S.C.; Hutchins, S.M.; Shen, D.-M.; Xiong, Y.; Hayes, E.C.; Parmar, R.M.; Foor, F.; et al. Rapid Identification of Subtype-Selective Agonists of the Somatostatin Receptor Through Combinatorial Chemistry. Science 1998, 282, 737–740. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, K.E.; Farr, S.A.; Banks, W.A.; Niehoff, M.L.; Morley, J.E.; Crider, A.M.; Witt, K.A. Chronic Peripheral Administration of Somatostatin Receptor Subtype-4 Agonist NNC 26-9100 Enhances Learning and Memory in SAMP8 Mice. Eur. J. Pharm. 2011, 654, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, K.E.; Witt, K.A.; Crider, A.M.; Kontoyianni, M. Somatostatin Receptor-4 Agonists as Candidates for Treatment of Alzheimer’s Disease. In Drug Design and Discovery in Alzheimer’s Disease; Bentham Science Publishers: Sharjah, United Arab Emirates, 2014; pp. 566–597. ISBN 978-0-12-803959-5. [Google Scholar] [CrossRef]
- Kecskés, A.; Pohóczky, K.; Kecskés, M.; Varga, Z.V.; Kormos, V.; Szőke, É.; Henn-Mike, N.; Fehér, M.; Kun, J.; Gyenesei, A.; et al. Characterization of Neurons Expressing the Novel Analgesic Drug Target Somatostatin Receptor 4 in Mouse and Human Brains. Int. J. Mol. Sci. 2020, 21, 7788. [Google Scholar] [CrossRef] [PubMed]
- Scheich, B.; Gaszner, B.; Kormos, V.; László, K.; Ádori, C.; Borbély, É.; Hajna, Z.; Tékus, V.; Bölcskei, K.; Ábrahám, I.; et al. Somatostatin Receptor Subtype 4 Activation Is Involved in Anxiety and Depression-like Behavior in Mouse Models. Neuropharmacology 2016, 101, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Helyes, Z.; Pintér, E.; Németh, J.; Sándor, K.; Elekes, K.; Szabó, Á.; Pozsgai, G.; Keszthelyi, D.; Kereskai, L.; Engström, M.; et al. Effects of the Somatostatin Receptor Subtype 4 Selective Agonist J-2156 on Sensory Neuropeptide Release and Inflammatory Reactions in Rodents. Br. J. Pharm. 2006, 149, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Lilly Clinical Development Pipeline. Available online: https://www.lilly.com/discovery/pipeline (accessed on 6 August 2020).
- Kántás, B.; Börzsei, R.; Szőke, É.; Bánhegyi, P.; Horváth, Á.; Hunyady, Á.; Borbély, É.; Hetényi, C.; Pintér, E.; Helyes, Z. Novel Drug-Like Somatostatin Receptor 4 Agonists Are Potential Analgesics for Neuropathic Pain. Int. J. Mol. Sci. 2019, 20, 6245. [Google Scholar] [CrossRef]
- Szőke, É.; Bálint, M.; Hetényi, C.; Markovics, A.; Elekes, K.; Pozsgai, G.; Szűts, T.; Kéri, G.; Őrfi, L.; Sándor, Z.; et al. Small Molecule Somatostatin Receptor Subtype 4 (Sst4) Agonists Are Novel Anti-Inflammatory and Analgesic Drug Candidates. Neuropharmacology 2020, 178, 108198. [Google Scholar] [CrossRef]
- Tigerstedt, N.-M.; Aavik, E.; Aavik, S.; Savolainen-Peltonen, H.; Hayry, P. Vasculoprotective Effects of Somatostatin Receptor Subtypes. Mol. Cell. Endocrinol. 2007, 279, 34–38. [Google Scholar] [CrossRef][Green Version]
- Dasgupta, P.; Gűnther, T.; Schulz, S. Pharmacological Characterization of Veldoreotide as a Somatostatin Receptor 4 Agonist. Life 2021, 11, 1075. [Google Scholar] [CrossRef]
- Orlewska, E.; Stępień, R.; Orlewska, K. Cost-Effectiveness of Somatostatin Analogues in the Treatment of Acromegaly. Expert Rev. Pharm. Outcomes Res. 2019, 19, 15–25. [Google Scholar] [CrossRef]
- Reynaert, H.; Colle, I. Treatment of Advanced Hepatocellular Carcinoma with Somatostatin Analogues: A Review of the Literature. Int. J. Mol. Sci. 2019, 20, 4811. [Google Scholar] [CrossRef] [PubMed]
- Stueven, A.K.; Kayser, A.; Wetz, C.; Amthauer, H.; Wree, A.; Tacke, F.; Wiedenmann, B.; Roderburg, C.; Jann, H. Somatostatin Analogues in the Treatment of Neuroendocrine Tumors: Past, Present and Future. Int. J. Mol. Sci. 2019, 20, 3049. [Google Scholar] [CrossRef] [PubMed]
- Crider, A.M.; Witt, K.A. Somatostatin Sst4 Ligands: Chemistry and Pharmacology. Mini. Rev. Med. Chem. 2007, 7, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Kumar Nagarajan, S.; Babu, S.; Sohn, H.; Devaraju, P.; Madhavan, T. Toward a Better Understanding of the Interaction between Somatostatin Receptor 2 and Its Ligands: A Structural Characterization Study Using Molecular Dynamics and Conceptual Density Functional Theory. J. Biomol. Struct. Dyn. 2019, 37, 3081–3102. [Google Scholar] [CrossRef]
- Lamberts, S.W.; van der Lely, A.J.; de Herder, W.W.; Hofland, L.J. Octreotide. N. Engl. J. Med. 1996, 334, 246–254. [Google Scholar] [CrossRef]
- Liu, S.; Tang, C.; Ho, B.; Ankersen, M.; Stidsen, C.E.; Crider, A.M. Nonpeptide Somatostatin Agonists with Sst4 Selectivity: Synthesis and Structure-Activity Relationships of Thioureas. J. Med. Chem. 1998, 41, 4693–4705. [Google Scholar] [CrossRef]
- Liu, Z.; Crider, A.M.; Ansbro, D.; Hayes, C.; Kontoyianni, M. A Structure-Based Approach to Understanding Somatostatin Receptor-4 Agonism (Sst4). J. Chem. Inf. Model. 2012, 52, 171–186. [Google Scholar] [CrossRef]
- Nagarajan, S.K.; Babu, S.; Madhavan, T. Theoretical Analysis of Somatostatin Receptor 5 with Antagonists and Agonists for the Treatment of Neuroendocrine Tumors. Mol. Divers. 2017, 21, 367–384. [Google Scholar] [CrossRef]
- Negi, A.; Zhou, J.; Sweeney, S.; Murphy, P.V. Ligand Design for Somatostatin Receptor Isoforms 4 and 5. Eur. J. Med. Chem. 2019, 163, 148–159. [Google Scholar] [CrossRef]
- Veber, D.F.; Freidlinger, R.M.; Perlow, D.S.; Paleveda, W.J.; Holly, F.W.; Strachan, R.G.; Nutt, R.F.; Arison, B.H.; Homnick, C.; Randall, W.C.; et al. A Potent Cyclic Hexapeptide Analogue of Somatostatin. Nature 1981, 292, 55–58. [Google Scholar] [CrossRef]
- Veber, D.F.; Saperstein, R.; Nutt, R.F.; Freidinger, R.M.; Brady, S.F.; Curley, P.; Perlow, D.S.; Paleveda, W.J.; Colton, C.D.; Zacchei, A.G.; et al. A Super Active Cyclic Hexapeptide Analog of Somatostatin. Life Sci. 1984, 34, 1371–1378. [Google Scholar] [CrossRef]
- Chen, L.; Hoeger, C.; Rivier, J.; Fitzpatrick, V.D.; Vandlen, R.L.; Tashjian, A.H. Structural Basis for the Binding Specificity of a SSTR1-Selective Analog of Somatostatin. Biochem. Biophys. Res. Commun. 1999, 258, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Nehring, R.B.; Meyerhof, W.; Richter, D. Aspartic Acid Residue 124 in the Third Transmembrane Domain of the Somatostatin Receptor Subtype 3 Is Essential for Somatostatin-14 Binding. DNA Cell. Biol. 1995, 14, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, M.T.; Hukovic, N.; Kumar, U.; Panetta, R.; Hjorth, S.A.; Srikant, C.B.; Patel, Y.C. Ligand Binding Pocket of the Human Somatostatin Receptor 5: Mutational Analysis of the Extracellular Domains. Mol. Pharm. 1997, 52, 807–814. [Google Scholar] [CrossRef]
- Daryaei, I.; Sandoval, K.; Witt, K.; Kontoyianni, M.; Michael Crider, A. Discovery of a 3,4,5-Trisubstituted-1,2,4-Triazole Agonist with High Affinity and Selectivity at the Somatostatin Subtype-4 (Sst4) Receptor. Medchemcomm 2018, 9, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.J.; Meyerowitz, J.G.; Panova, O.; Borrelli, K.; Skiniotis, G. Plasticity in Ligand Recognition at Somatostatin Receptors. Nat. Struct. Mol. Biol. 2022, 29, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Kántás, B.; Szőke, É.; Börzsei, R.; Bánhegyi, P.; Asghar, J.; Hudhud, L.; Steib, A.; Hunyady, Á.; Horváth, Á.; Kecskés, A.; et al. In Silico, In Vitro and In Vivo Pharmacodynamic Characterization of Novel Analgesic Drug Candidate Somatostatin SST4 Receptor Agonists. Front Pharm. 2021, 11, 601887. [Google Scholar] [CrossRef]
- Nagarajan, S.K.; Babu, S.; Sohn, H.; Madhavan, T. Molecular-Level Understanding of the Somatostatin Receptor 1 (SSTR1)-Ligand Binding: A Structural Biology Study Based on Computational Methods. ACS Omega 2020, 5, 21145–21161. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Bálint, M.; Jeszenői, N.; Horváth, I.; van der Spoel, D.; Hetényi, C. Systematic Exploration of Multiple Drug Binding Sites. J. Cheminform. 2017, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Bálint, M.; Horváth, I.; Mészáros, N.; Hetényi, C. Towards Unraveling the Histone Code by Fragment Blind Docking. Int. J. Mol. Sci 2019, 20, 422. [Google Scholar] [CrossRef] [PubMed]
- Bauer, W.; Briner, U.; Doepfner, W.; Haller, R.; Huguenin, R.; Marbach, P.; Petcher, T.J.; Pless, J. SMS 201–995: A Very Potent and Selective Octapeptide Analogue of Somatostatin with Prolonged Action. Life Sci. 1982, 31, 1133–1140. [Google Scholar] [CrossRef]
- Hallenga, K.; Binst, G.V.; Scarso, A.; Michel, A.; Knappenberg, M.; Dremier, C.; Brison, J.; Dirkx, J. The Conformational Properties of the Peptide Hormone Somatostatin (III). FEBS Lett. 1980, 119, 47–52. [Google Scholar] [CrossRef]
- Knappenberg, M.; Michel, A.; Scarso, A.; Brison, J.; Zanen, J.; Hallenga, K.; Deschrijver, P.; Van Binst, G. The Conformational Properties of Somatostatin IV. The Conformers Contributing to the Conformational Equilibrium of Somatostatin in Aqueous Solution as Found by Semi-Empirical Energy Calculations and High-Resolution NMR Experiments. Biochim. Et Biophys. Acta (BBA)—Protein Struct. Mol. Enzymol. 1982, 700, 229–246. [Google Scholar] [CrossRef]
- Simon, Á.; Czajlik, A.; Perczel, A.; Kéri, G.; Nyikos, L.; Emri, Z.; Kardos, J. Binding Crevice for TT-232 in a Homology Model of Type 1 Somatostatin Receptor. Biochem. Biophys. Res. Commun. 2004, 316, 1059–1064. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Strnad, J.; Hadcock, J.R. Identification of a Critical Aspartate Residue in Transmembrane Domain Three Necessary for the Binding of Somatostatin to the Somatostatin Receptor SSTR2. Biochem. Biophys. Res. Commun. 1995, 216, 913–921. [Google Scholar] [CrossRef]
- Pernomian, L.; Gomes, M.S.; de Paula da Silva, C.H.T.; Rosa, J.M.C. Reverse Induced Fit-Driven MAS-Downstream Transduction: Looking for Metabotropic Agonists. Curr. Med. Chem. 2017, 24, 4360–4367. [Google Scholar] [CrossRef]
- Van Regenmortel, M.H. Transcending the Structuralist Paradigm in Immunology-Affinity and Biological Activity Rather than Purely Structural Considerations Should Guide the Design of Synthetic Peptide Epitopes. Biomed Pept. Proteins Nucleic Acids 1995, 1, 109–116. [Google Scholar]
- Ye, L.; Van Eps, N.; Zimmer, M.; Ernst, O.P.; Prosser, R.S. Activation of the A2A Adenosine G-Protein-Coupled Receptor by Conformational Selection. Nature 2016, 533, 265–268. [Google Scholar] [CrossRef]
- Rastelli, G.; Pacchioni, S.; Sirawaraporn, W.; Sirawaraporn, R.; Parenti, M.D.; Ferrari, A.M. Docking and Database Screening Reveal New Classes of Plasmodium Falciparum Dihydrofolate Reductase Inhibitors. J. Med. Chem. 2003, 46, 2834–2845. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hussain, M.; Kuang, G.; Zhang, J.; Tu, Y. Mechanistic Insights into Peptide and Ligand Binding of the ATAD2-Bromodomain via Atomistic Simulations Disclosing a Role of Induced Fit and Conformational Selection. Phys. Chem. Chem. Phys. 2018, 20, 23222–23232. [Google Scholar] [CrossRef] [PubMed]
- Zsidó, B.Z.; Hetényi, C. The Role of Water in Ligand Binding. Curr. Opin. Struct. Biol. 2021, 67, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Hetényi, C.; van der Spoel, D. Toward Prediction of Functional Protein Pockets Using Blind Docking and Pocket Search Algorithms. Protein Sci. 2011, 20, 880–893. [Google Scholar] [CrossRef]
- Horváth, I.; Jeszenői, N.; Bálint, M.; Paragi, G.; Hetényi, C. A Fragmenting Protocol with Explicit Hydration for Calculation of Binding Enthalpies of Target-Ligand Complexes at a Quantum Mechanical Level. Int. J. Mol. Sci. 2019, 20, 4384. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Wang, J.; Cieplak, P.; Li, J.; Cai, Q.; Hsieh, M.; Luo, R.; Duan, Y. Development of Polarizable Models for Molecular Mechanical Calculations IV: Van Der Waals Parameterization. J. Phys. Chem. B 2012, 116, 7088–7101. [Google Scholar] [CrossRef] [PubMed]
- Mehler, E.L.; Solmajer, T. Electrostatic Effects in Proteins: Comparison of Dielectric and Charge Models. Protein Eng. 1991, 4, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and Sequence Analysis Tools Services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, gkac240. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Börzsei, R.; Zsidó, B.Z.; Bálint, M.; Helyes, Z.; Pintér, E.; Hetényi, C. Exploration of Somatostatin Binding Mechanism to Somatostatin Receptor Subtype 4. Int. J. Mol. Sci. 2022, 23, 6878. https://doi.org/10.3390/ijms23136878
Börzsei R, Zsidó BZ, Bálint M, Helyes Z, Pintér E, Hetényi C. Exploration of Somatostatin Binding Mechanism to Somatostatin Receptor Subtype 4. International Journal of Molecular Sciences. 2022; 23(13):6878. https://doi.org/10.3390/ijms23136878
Chicago/Turabian StyleBörzsei, Rita, Balázs Zoltán Zsidó, Mónika Bálint, Zsuzsanna Helyes, Erika Pintér, and Csaba Hetényi. 2022. "Exploration of Somatostatin Binding Mechanism to Somatostatin Receptor Subtype 4" International Journal of Molecular Sciences 23, no. 13: 6878. https://doi.org/10.3390/ijms23136878
APA StyleBörzsei, R., Zsidó, B. Z., Bálint, M., Helyes, Z., Pintér, E., & Hetényi, C. (2022). Exploration of Somatostatin Binding Mechanism to Somatostatin Receptor Subtype 4. International Journal of Molecular Sciences, 23(13), 6878. https://doi.org/10.3390/ijms23136878