
The Regulatory Roles of Mitochondrial Calcium and the Mitochondrial Calcium Uniporter in Tumor Cells

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
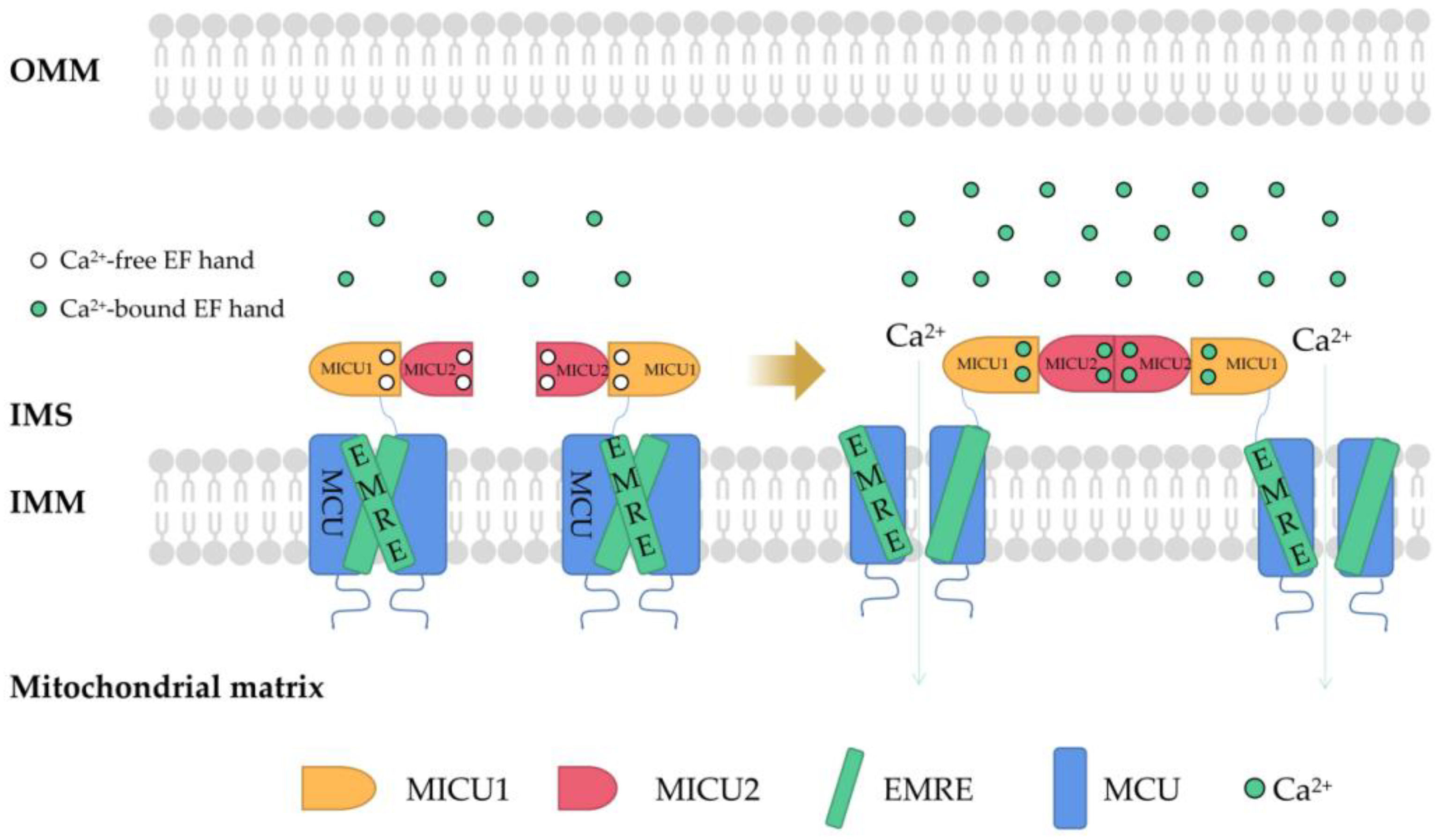
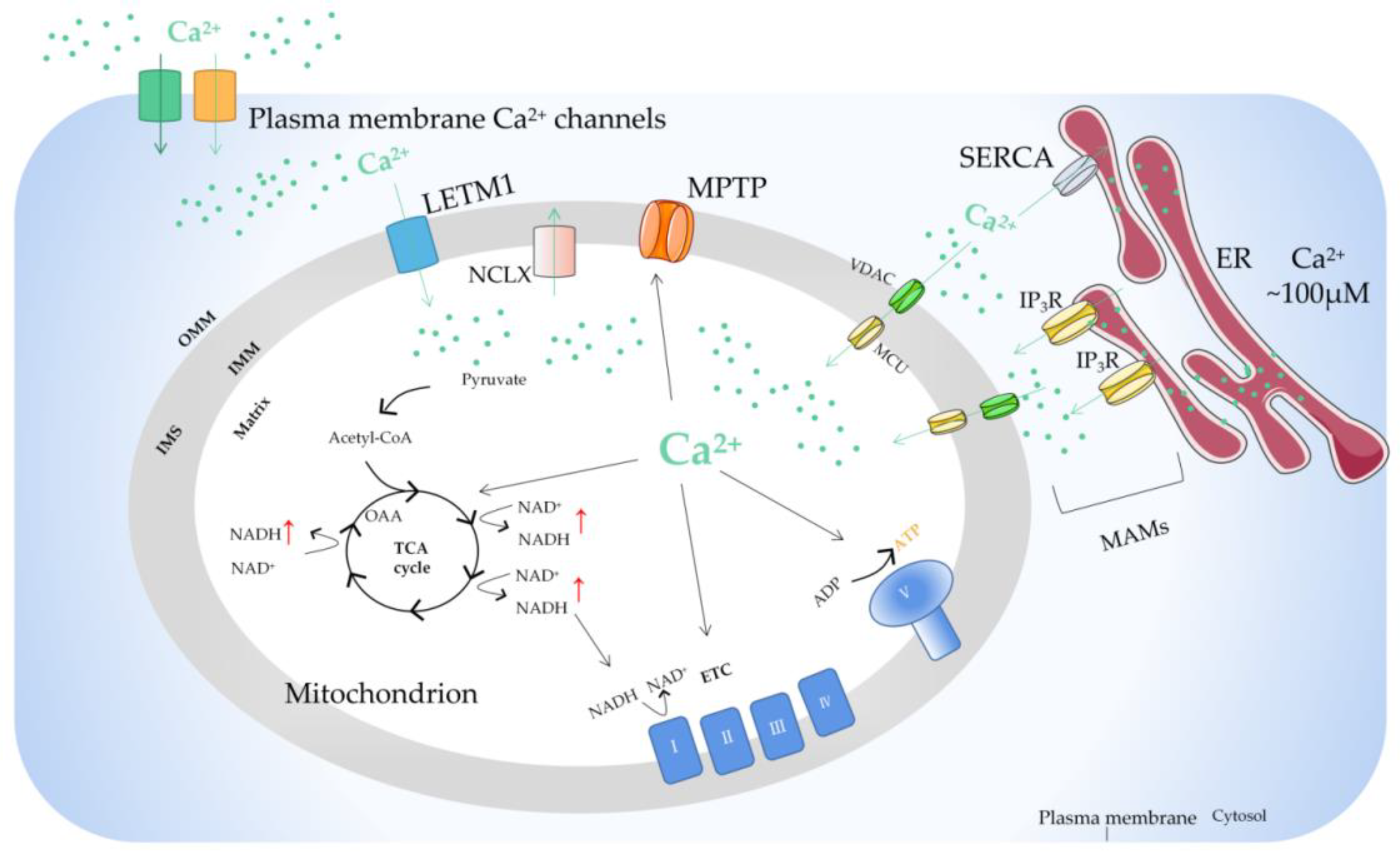
2. Regulation of Mitochondrial Ca2+
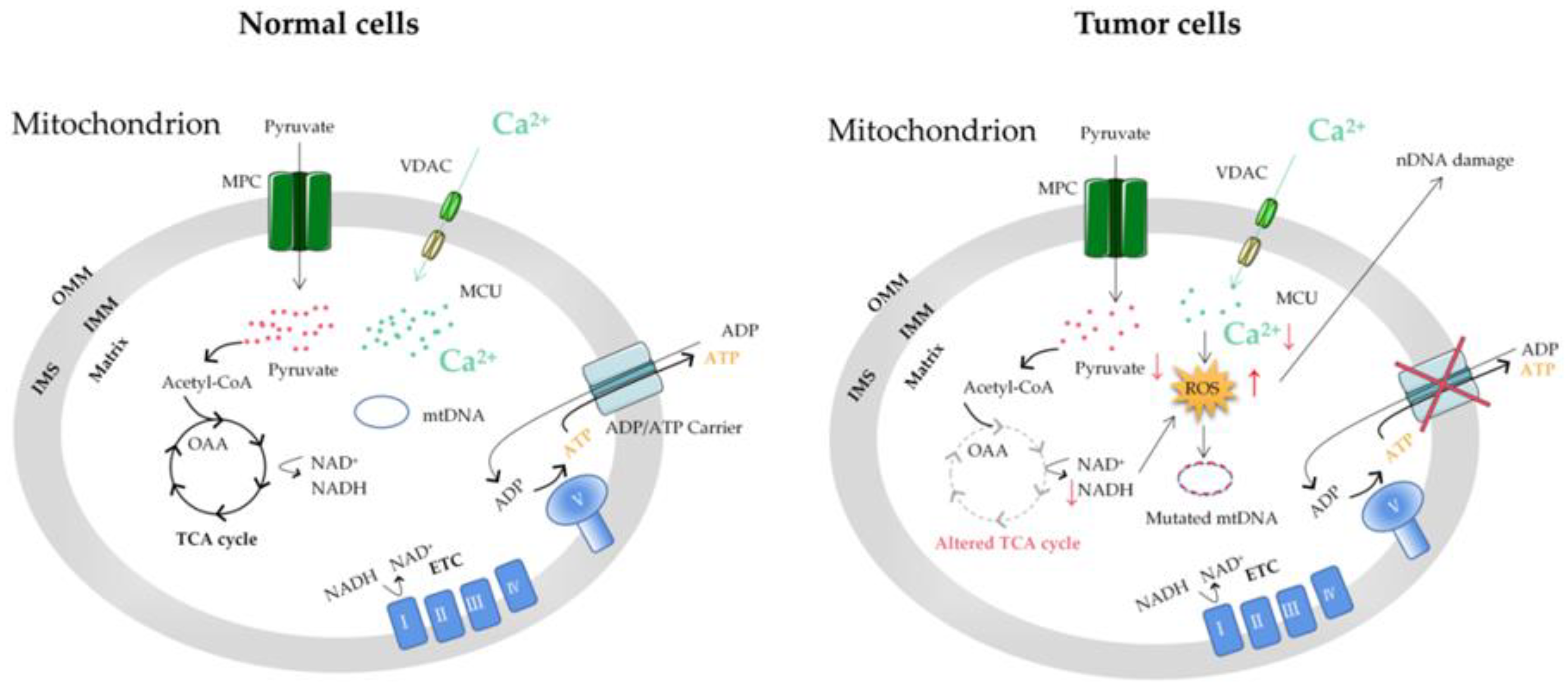
3. Mitochondrial Ca2+ and Energy Metabolism of Tumor Cells
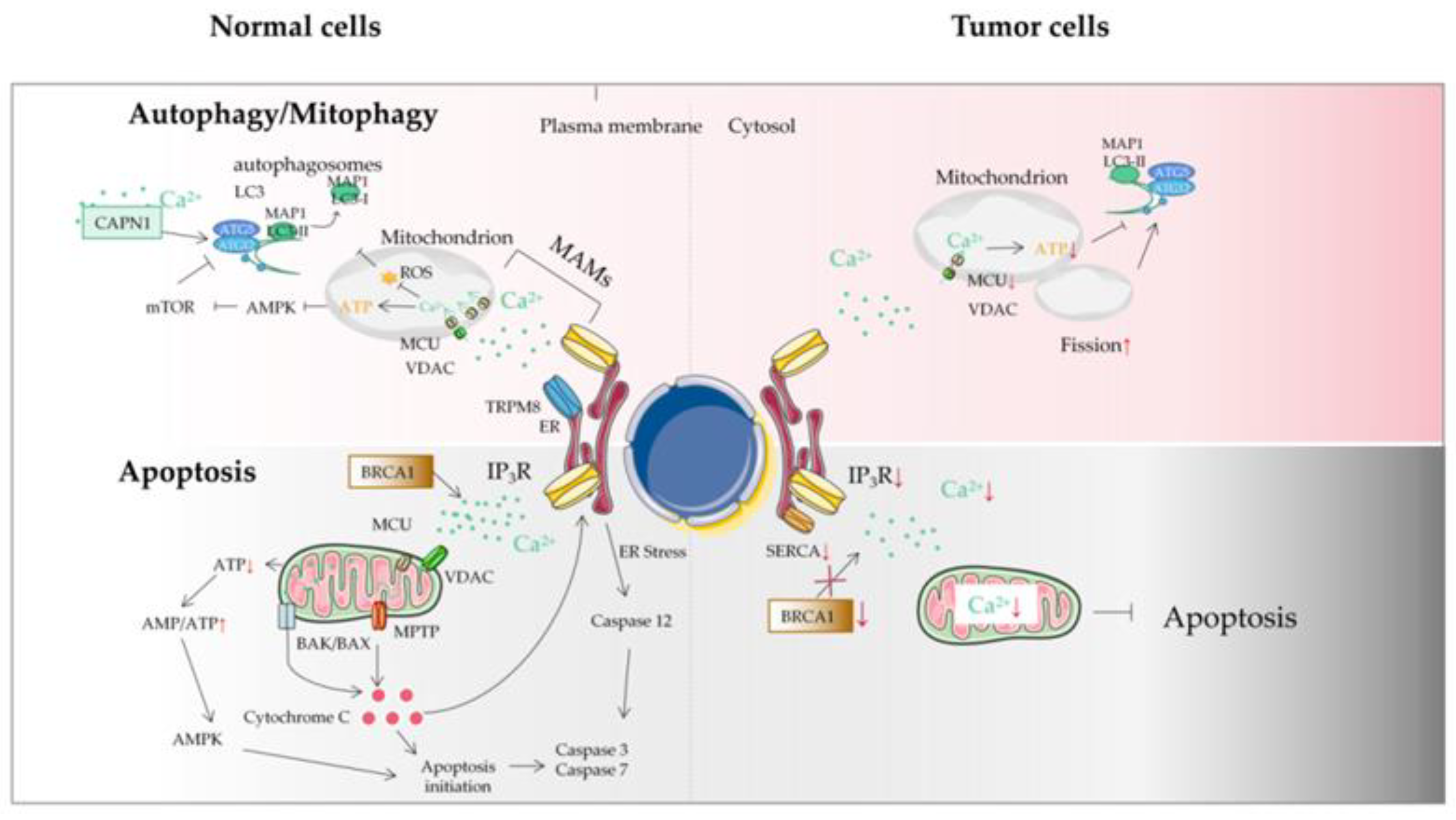
4. Mitochondrial Ca2+ and the MCU in Autophagy/Mitophagy of Tumor Cells
5. Mitochondrial Ca2+ and Tumor Cell Apoptosis
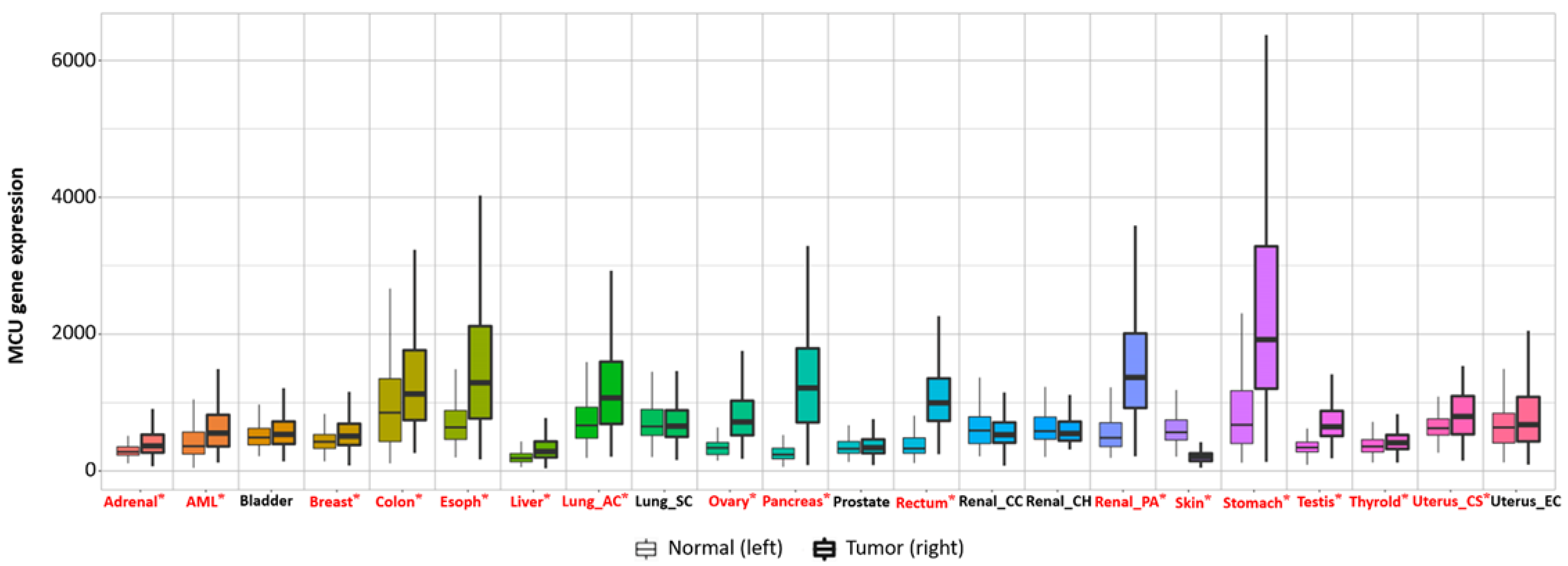
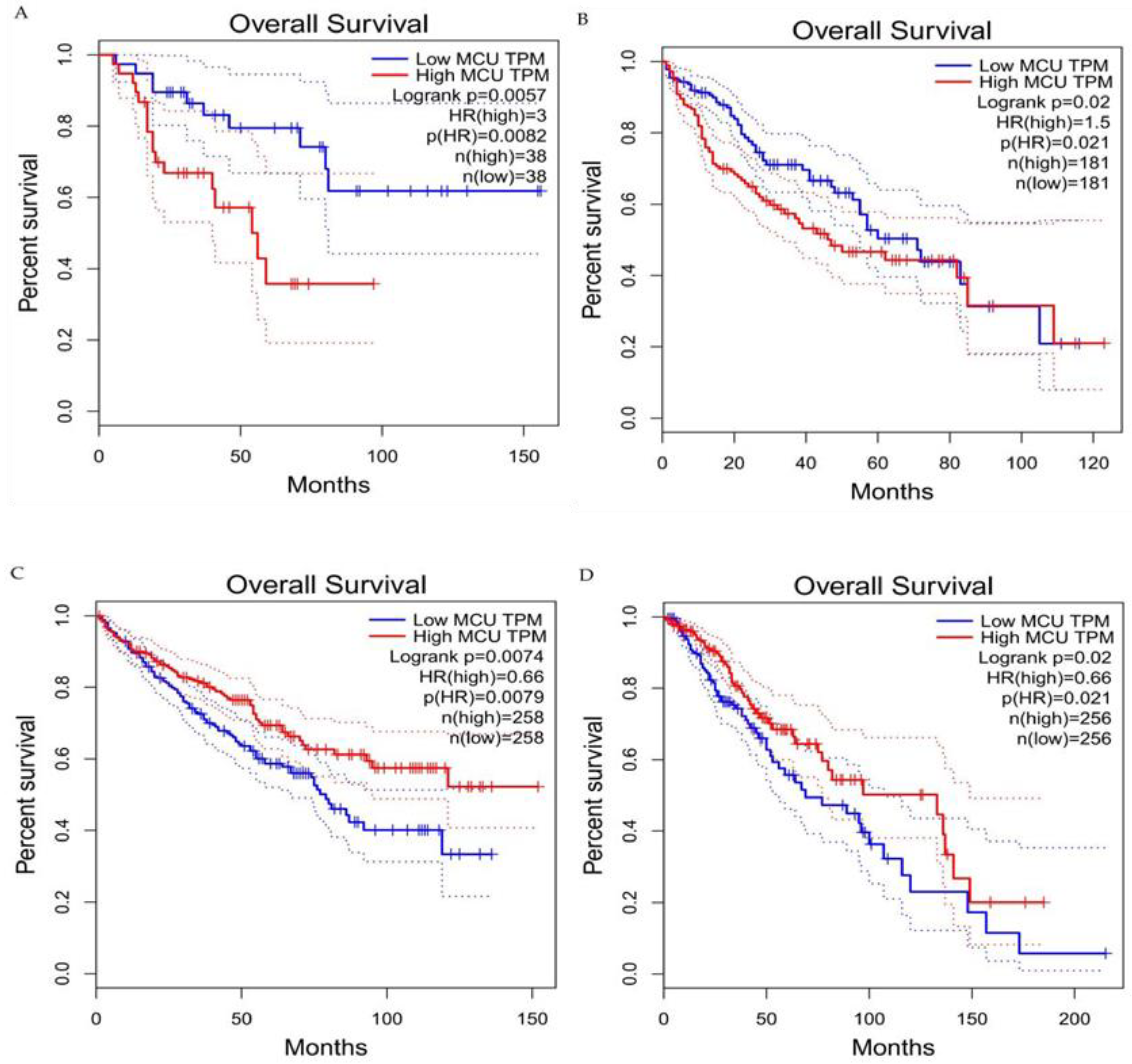
6. The Relationship between the MCU and the Tumor
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Chem. Commun. 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. Compr. Physiol. 2017, 7, 623–634. [Google Scholar]
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Intracellular calcium homeostasis and signaling. Met. Ions Life Sci. 2013, 12, 119–168. [Google Scholar]
- Carafoli, E.; Krebs, J. Why Calcium? How Calcium Became the Best Communicator. JBC 2016, 40, 20849–20857. [Google Scholar] [CrossRef]
- Zavodnik, I.B. Mitochondria, calcium homeostasis and calcium signaling. Biomed. Khim. 2016, 62, 311–317. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium. 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Magalhães, P.J.; Rizzuto, R. Mitochondria and calcium homeostasis: A tale of three luminescent proteins. Luminescence 2001, 16, 67–71. [Google Scholar] [CrossRef]
- Godoy, J.A.; Rios, J.A.; Picón-Pagès, P.; Herrera-Fernández, V.; Swaby, B.; Crepin, G.; Vicente, R.; Fernández-Fernández, J.M.; Muñoz, F.J. Mitostasis, Calcium and Free Radicals in Health, Aging and Neurodegeneration. Biomolecules 2021, 11, 1012. [Google Scholar] [CrossRef]
- Robb-Gaspers, L.; Burnett, P.; Rutter, G.; Denton, R.; Rizzuto, R.; Thomas, A. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 1998, 17, 4987–5000. [Google Scholar] [CrossRef]
- Uhlén, P.; Fritz, N. Biochemistry of calcium oscillations. Biochem. Biophys. Res. Commun. 2010, 396, 28–32. [Google Scholar] [CrossRef]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Hajnóczky, G.; Robb-Gaspers, L.D.; Seitz, M.B.; Thomas, A.P. Decoding of cytosolic calcium oscillations in the mitochondria. Cell 1995, 82, 415–424. [Google Scholar] [CrossRef]
- Sheu, S.S.; Jou, M.J. Mitochondrial free Ca2+ concentration in living cells. J. Bioenerg. Biomembr. 1994, 26, 487–493. [Google Scholar] [CrossRef]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef]
- Paudel, S.; Sindelar, R.; Saha, M. Calcium Signaling in Vertebrate Development and Its Role in Disease. Int. J. Mol. Sci. 2018, 19, 3390. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Stewart, T.A.; Yapa, K.T.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta 2015, 1848, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Yang, J.; Fu, L.; Wang, M.; Wang, X. Progress in understanding mitochondrial calcium uniporter complex-mediated calcium signalling: A potential target for cancer treatment. Br. J. Pharmacol. 2019, 176, 1190–1205. [Google Scholar] [CrossRef]
- Ali, E.S.; Rychkov, G.Y.; Barritt, G.J. Deranged hepatocyte intracellular Ca2+ homeostasis and the progression of non-alcoholic fatty liver disease to hepatocellular carcinoma. Cell Calcium. 2019, 82, 102057. [Google Scholar] [CrossRef]
- Tosatto, A.; Sommaggio, R.; Kummerow, C.; Bentham, R.B.; Blacker, T.S.; Berecz, T.; Duchen, M.R.; Rosato, A.; Bogeski, I.; Szabadkai, G.; et al. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1α. EMBO Mol. Med. 2016, 8, 569–585. [Google Scholar] [CrossRef]
- Leinonen, P.; Aaltonen, V.; Koskela, S.; Lehenkari, P.; Korkiamäki, T.; Peltonen, J. Impaired Gap Junction Formation and Intercellular Calcium Signaling in Urinary Bladder Cancer Cells can be Improved by Gö6976. Cell Commun. Adhes. 2007, 14, 125–136. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef]
- Umemura, M.; Baljinnyam, E.; Feske, S.; De Lorenzo, M.S.; Xie, L.H.; Feng, X.; Oda, K.; Makino, A.; Fujita, T.; Yokoyama, U.; et al. Store-Operated Ca2+ Entry (SOCE) Regulates Melanoma Proliferation and Cell Migration. PLoS ONE 2014, 9, e89292. [Google Scholar] [CrossRef]
- Tennakoon, S.; Aggarwal, A.; Kállay, E. The calcium-sensing receptor and the hallmarks of cancer. Biochim. Biophys. Acta 2016, 1863, 1398–1407. [Google Scholar] [CrossRef]
- Giampazolias, E.; Tait, S. Mitochondria and the hallmarks of cancer. FEBS J. 2016, 283, 803–814. [Google Scholar] [CrossRef]
- Proietti, S.; Cucina, A.; Minini, M.; Bizzarri, M. Melatonin, mitochondria, and the cancer cell. Cell Mol. Life Sci. 2017, 74, 4015–4025. [Google Scholar] [CrossRef]
- Tajada, S.; Villalobos, C. Calcium Permeable Channels in Cancer Hallmarks. Front. Pharmacol. 2020, 11, 968. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; Prevarskaya, N.; Roberts-Thomson, S.J. The calcium-cancer signalling nexus. Nat. Rev. Cancer 2017, 17, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Corricelli, M.; Branchini, A.; Vitto, V.; Missiroli, S.; Morciano, G.; Perrone, M.; Ferrarese, M.; Giorgi, C.; Pinotti, M.; et al. Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca levels and tumor growth. EMBO J. 2019, 38, e99435. [Google Scholar] [CrossRef]
- Modesti, L.; Danese, A.; Angela Maria Vitto, V.; Ramaccini, D.; Aguiari, G.; Gafà, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca Signaling in Health, Disease and Therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef]
- Pézier, A.; Acquistapace, A.; Renou, M.; Rospars, J.P.; Lucas, P. Ca2+ stabilizes the membrane potential of moth olfactory receptor neurons at rest and is essential for their fast repolarization Chem. Senses 2007, 32, 305–317. [Google Scholar] [CrossRef]
- Pendin, D.; Greotti, E.; Filadi, R.; Pozzan, T. Spying on organelle Ca2+ in living cells: The mitochondrial point of view. J. Endocrinol. Investig. 2015, 38, 39–45. [Google Scholar] [CrossRef]
- Yamamoto, T. The Molecular Mechanisms of Mitochondrial Calcium Uptake by Calcium Uniporter. Yakugaku Zasshi J. Pharm. Jpn. 2021, 141, 491–499. [Google Scholar] [CrossRef]
- Stefani, D.D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochemistry 2019, 84, 593–607. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef]
- Garbincius, J.; Elrod, J. Mitochondrial calcium exchange in physiology and disease. Physiol. Rev. 2022, 102, 893–992. [Google Scholar] [CrossRef]
- Chen, L.; Sun, Q.; Zhou, D.; Song, W.; Yang, Q.; Ju, B.; Zhang, L.; Xie, H.; Zhou, L.; Hu, Z.; et al. HINT2 triggers mitochondrial Ca2+ influx by regulating the mitochondrial Ca2+ uniporter (MCU) complex and enhances gemcitabine apoptotic effect in pancreatic cancer. Cancer Lett. 2017, 411, 106–116. [Google Scholar] [CrossRef]
- Curry, M.C.; Peters, A.A.; Kenny, P.A.; Roberts-Thomson, S.J.; Monteith, G.R. Mitochondrial calcium uniporter silencing potentiates caspase-independent cell death in MDA-MB-231 breast cancer cells. Biochem. Biophys. Res. Commun. 2013, 434, 695–700. [Google Scholar] [CrossRef]
- Sancak, Y.; Markhard, A.; Kitami, T.; Kovacs-Bogdan, E.; Kamer, K.; Udeshi, N.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef]
- Wang, W.; Xie, Q.; Zhou, X.; Yao, J.; Zhu, X.; Huang, P.; Zhang, L.; Wei, J.; Xie, H.; Zhou, L.; et al. Mitofusin-2 triggers mitochondria Ca2+ influx from the endoplasmic reticulum to induce apoptosis in hepatocellular carcinoma cells. Cancer Lett. 2015, 358, 47–58. [Google Scholar] [CrossRef]
- Zhuo, W.; Zhou, H.; Guo, R.; Yi, J.; Zhang, L.; Yu, L.; Sui, Y.; Zeng, W.; Wang, P.; Yang, M. Structure of intact human MCU supercomplex with the auxiliary MICU subunits. Protein Cell. 2020, 12, 220–229. [Google Scholar]
- Fan, M.; Zhang, J.; Tsai, C.; Benjamin, J.; Rodriguez, M.; Xu, Y.; Liao, M.; Tsai, M.; Feng, L. Structure and mechanism of the mitochondrial Ca2+ uniporter holocomplex. Nature 2020, 582, 129–133. [Google Scholar] [CrossRef]
- Tomar, D.; Elrod, J.W. Metabolite regulation of the mitochondrial calcium uniporter channel. Cell Calcium. 2020, 92, 102288. [Google Scholar] [CrossRef]
- Gottschalk, B.; Madreiter-Sokolowski, C.T.; Graier, W.F. Cristae junction as a fundamental switchboard for mitochondrial ion signaling and bioenergetics. Cell Calcium. 2022, 101, 102517. [Google Scholar] [CrossRef]
- Gottschalk, B.; Klec, C.; Leitinger, G.; Bernhart, E.; Rost, R.; Bischof, H.; Madreiter-Sokolowski, C.T.; Radulović, S.; Eroglu, E.; Sattler, W.; et al. MICU1 controls cristae junction and spatially anchors mitochondrial Ca2+ uniporter complex. Nat. Commun. 2019, 10, 3732. [Google Scholar] [CrossRef]
- Chakraborty, P.K.; Mustafi, S.B.; Xiong, X.; Dwivedi, S.K.D.; Nesin, V.; Saha, S.; Zhang, M.; Dhanasekaran, D.; Jayaraman, M.; Mannel, R.; et al. MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat. Commun. 2017, 8, 14634. [Google Scholar] [CrossRef]
- Trenker, M.; Malli, R.; Fertschai, I.; Levak-Frank, S.; Graier, W.F. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat. Cell Biol. 2007, 9, 445–452. [Google Scholar] [CrossRef]
- Madreiter-Sokolowski, C.T.; Klec, C.; Parichatikanond, W.; Stryeck, S.; Gottschalk, B.; Pulido, S.; Rost, R.; Eroglu, E.; Hofmann, N.A.; Bondarenko, A.I.; et al. PRMT1-mediated methylation of MICU1 determines the UCP2/3 dependency of mitochondrial Ca(2+) uptake in immortalized cells. Nat. Commun. 2016, 7, 12897. [Google Scholar] [CrossRef]
- Madreiter-Sokolowski, C.T.; Győrffy, B.; Klec, C.; Sokolowski, A.A.; Rost, R.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. UCP2 and PRMT1 are key prognostic markers for lung carcinoma patients. Oncotarget. 2017, 8, 80278–80285. [Google Scholar] [CrossRef]
- Jarrold, J.; Davies, C.C. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol. Med. 2019, 25, 993–1009. [Google Scholar] [CrossRef]
- Payne, R.; Hoff, H.; Roskowski, A.; Foskett, J. MICU2 restricts spatial crosstalk between InsP3R and MCU channels by regulating threshold and gain of MICU1-mediated inhibition and activation of MCU. Cell Reports. 2017, 21, 3141–3154. [Google Scholar] [CrossRef]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019, 26, 179–195. [Google Scholar] [CrossRef]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabò, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef]
- Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.; Thomas, T.; Hoffman, N.; Timbalia, S.; Goldman, S.; Breves, S.; Corbally, D.; et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016, 15, 1673–1685. [Google Scholar] [CrossRef]
- Chaudhuri, D.; Artiga, D.J.; Abiria, S.A.; Clapham, D.E. Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc. Natl. Acad. Sci. USA 2016, 113, E1872–E1880. [Google Scholar] [CrossRef]
- Roy, S.; Dey, K.; Hershfinkel, M.; Ohana, E.; Sekler, I. Identification of residues that control Li+ versus Na+ dependent Ca2+ exchange at the transport site of the mitochondrial NCLX. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 997–1008. [Google Scholar] [CrossRef]
- Starkov, A.A.; Chinopoulos, C.; Starkova, N.N.; Konrad, C.; Kiss, G.; Stepanova, A.; Popov, V.N. Divalent cation chelators citrate and EDTA unmask an intrinsic uncoupling pathway in isolated mitochondria. J. Bioenerg. Biomembr. 2017, 49, 3–11. [Google Scholar] [CrossRef]
- Wilson, J.A.; Lau, Y.S.; Gleeson, J.G.; Wilson, J.S. The action of MPTP on synaptic transmission is affected by changes in Ca2+ concentrations. Brain Res. 1991, 541, 342–346. [Google Scholar] [CrossRef]
- Kolomiiets’, O.; Danylovych, I.; Danylovych, H. H+-Ca2+-exchanger in the myometrium mitochondria: Modulation of exogenous and endogenous compounds. Fiziol. Zh. 2014, 60, 33–42. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C. The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef]
- Giorgi, C.; Bonora, M.; Sorrentino, G.; Missiroli, S.; Poletti, F.; Suski, J.M.; Galindo Ramirez, F.; Rizzuto, R.; Di Virgilio, F.; Zito, E.; et al. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proc. Natl. Acad. Sci. USA 2015, 112, 1779–1784. [Google Scholar] [CrossRef]
- Betz, C.; Stracka, D.; Prescianotto-Baschong, C.; Frieden, M.; Demaurex, N.; Hall, M.N. Feature Article: mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc. Natl. Acad. Sci. USA 2013, 110, 12526–12534. [Google Scholar] [CrossRef]
- Rimessi, A.; Marchi, S.; Patergnani, S.; Pinton, P. H-Ras-driven tumoral maintenance is sustained through caveolin-1-dependent alterations in calcium signaling. Oncogene 2014, 33, 2329–2340. [Google Scholar] [CrossRef]
- Glitsch, M. Mechano- and pH-sensing convergence on Ca2+-mobilising proteins - A recipe for cancer? Cell Calcium. 2019, 80, 38–45. [Google Scholar] [CrossRef]
- Trapani, V.; Wolf, F.I. Dysregulation of Mg2+ homeostasis contributes to acquisition of cancer hallmarks. Cell Calcium. 2019, 83, 102078. [Google Scholar] [CrossRef]
- Santoni, G.; Morelli, M.B.; Marinelli, O.; Nabissi, M.; Santoni, M.; Amantini, C. Calcium Signaling and the Regulation of Chemosensitivity in Cancer Cells: Role of the Transient Receptor Potential Channels. Adv. Exp. Med. Biol. 2020, 1131, 505–517. [Google Scholar]
- Denton, R. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef]
- Schönekess, B.O.; Brindley, P.G.; Lopaschuk, G.D. Calcium regulation of glycolysis, glucose oxidation, and fatty acid oxidation in the aerobic and ischemic heart. Can. J. Physiol. Pharmacol. 1995, 73, 1632–1640. [Google Scholar] [CrossRef]
- McMillin, J.B.; Pauly, D.F. Control of mitochondrial respiration in muscle. Mol. Cell. Biochem. 1988, 81, 121–129. [Google Scholar] [CrossRef]
- Konji, V.; Montag, A.; Sandri, G.; Nordenbrand, K.; Ernster, L. Transport of Ca2+ and Mn2+ by mitochondria from rat liver, heart and brain. Biochimie 1985, 67, 1241–1250. [Google Scholar] [CrossRef]
- Oliveira, G.L.; Coelho, A.R.; Marques, R.; Oliveira, P.J. Cancer cell metabolism: Rewiring the mitochondrial hub. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166016. [Google Scholar] [CrossRef]
- Iqbal, M.A.; Gupta, V.; Gopinath, P.; Mazurek, S.; Bamezai, R.N. Pyruvate kinase M2 and cancer: An updated assessment. FEBS Lett. 2014, 588, 2685–2692. [Google Scholar] [CrossRef]
- Li, T.; Han, J.; Jia, L.; Hu, X.; Chen, L.; Wang, Y. PKM2 coordinates glycolysis with mitochondrial fusion and oxidative phosphorylation. Protein Cell. 2019, 10, 583–594. [Google Scholar] [CrossRef]
- Mazurek, S. Pyruvate kinase type M2: A key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 2011, 43, 969–980. [Google Scholar] [CrossRef]
- Dey, P.; Kimmelman, A.C.; DePinho, R.A. Metabolic Codependencies in the Tumor Microenvironment. Cancer Discov. 2021, 11, 1067–1081. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Ferro, F.; Servais, S.; Besson, P.; Roger, S.; Dumas, J.F.; Brisson, L. Autophagy and mitophagy in cancer metabolic remodelling. Semin. Cell Dev. Biol. 2020, 98, 129–138. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.S. Autophagy--a key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San, P.J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef]
- Hu, Y.X.; Han, X.S.; Jing, Q. Ca(2+) Ion and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 151–166. [Google Scholar]
- Bootman, M.D.; Chehab, T.; Bultynck, G.; Parys, J.B.; Rietdorf, K. The regulation of autophagy by calcium signals: Do we have a consensus? Cell Calcium. 2018, 70, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.K.; Galione, A. The endoplasmic reticulum and junctional membrane communication during calcium signaling. Biochim. Biophys Acta 2013, 1833, 2542–2559. [Google Scholar] [CrossRef] [PubMed]
- Høyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell. 2007, 25, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, J.; Jiang, P.; Li, K.; Sun, Y.; Huang, Y. ASIC1a promotes acidic microenvironment-induced HCC cells migration and invasion by inducing autophagy. Eur. J. Pharmacol. 2021, 907, 174252. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kondo, T.; Zhao, Q.L.; Li, F.J.; Tanabe, K.; Arai, Y.; Zhou, Z.C.; Kasuya, M. Apoptosis induced by cadmium in human lymphoma U937 cells through Ca2+-calpain and caspase-mitochondria- dependent pathways. J. Biol. Chem. 2000, 275, 39702–39709. [Google Scholar] [CrossRef]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Paudel, R.C.; Parys, J.; Bultynck, G. Intracellular Ca(2+) signaling: A novel player in the canonical mTOR-controlled autophagy pathway. Commun. Integr. Biol. 2013, 6, e25429. [Google Scholar] [CrossRef]
- Bidaux, G.; Gordienko, D.; Shapovalov, G.; Farfariello, V.; Borowiec, A.; Iamshanova, O.; Lemonnier, L.; Gueguinou, M.; Guibon, R.; Fromont, G.; et al. 4TM-TRPM8 channels are new gatekeepers of the ER-mitochondria Ca2+ transfer. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 981–994. [Google Scholar] [CrossRef]
- Cardenas, C.; Lovy, A.; Silva-Pavez, E.; Urra, F.; Mizzoni, C.; Ahumada-Castro, U.; Bustos, G.; Jaňa, F.; Cruz, P.; Farias, P.; et al. Cancer cells with defective oxidative phosphorylation require endoplasmic reticulum-to-mitochondria Ca2+ transfer for survival. Sci. Signal. 2020, 13, eaay1212. [Google Scholar] [CrossRef]
- Kiviluoto, S.; Schneider, L.; Luyten, T.; Vervliet, T.; Missiaen, L.; De Smedt, H.; Parys, J.B.; Methner, A.; Bultynck, G. Bax inhibitor-1 is a novel IP3 receptor-interacting and -sensitizing protein. Cell Death Dis. 2012, 3, e367. [Google Scholar] [CrossRef][Green Version]
- Doghman-Bouguerra, M.; Lalli, E. ER-mitochondria interactions: Both strength and weakness within cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Vultur, A.; Gibhardt, C.S.; Stanisz, H.; Bogeski, I. The role of the mitochondrial calcium uniporter (MCU) complex in cancer. Pflugers Arch. 2018, 470, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Wang, Y.; Peng, J.; Shen, Q.; Chen, M.; Tang, W.; Li, X.; Cai, C.; Wang, B.; Cai, S.; et al. Mitochondrial calcium uniporter as a target of microRNA-340 and promoter of metastasis via enhancing the Warburg effect. Oncotarget 2017, 8, 83831–83844. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Zhivotovsky, B.; Orrenius, S. All along the watchtower: On the regulation of apoptosis regulators. FASEB J. 1999, 13, 1647–1657. [Google Scholar] [CrossRef]
- Allan, L.; Clarke, P. Apoptosis and autophagy: Regulation of caspase-9 by phosphorylation. FEBS J. 2009, 276, 6063–6073. [Google Scholar] [CrossRef]
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin. Cell Dev. Biol. 2020, 98, 139–153. [Google Scholar] [CrossRef]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Jeong, S.; Seol, D. The role of mitochondria in apoptosis. BMB Rep. 2008, 41, 11–22. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, X.; Kubo, H.; Harris, D.M.; Mills, G.D.; Moyer, J.; Berretta, R.; Potts, S.T.; Marsh, J.D.; Houser, S.R. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ. Res. 2005, 97, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- McConkey, D.J. Biochemical determinants of apoptosis and necrosis. Toxicol. Lett. 1998, 99, 157–168. [Google Scholar] [CrossRef]
- Aharoni-Simon, M.; Shumiatcher, R.; Yeung, A.; Shih, A.Z.; Dolinsky, V.W.; Doucette, C.A.; Luciani, D.S. Bcl-2 Regulates Reactive Oxygen Species Signaling and a Redox-Sensitive Mitochondrial Proton Leak in Mouse Pancreatic β-Cells. Endocrinology 2016, 157, 2270–2281. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Selzer, E.; Schlagbauer-Wadl, H.; Okamoto, I.; Pehamberger, H.; Pötter, R.; Jansen, B. Expression of Bcl-2 family members in human melanocytes, in melanoma metastases and in melanoma cell lines. Melanoma Res. 1998, 8, 197–203. [Google Scholar] [CrossRef]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium. 2002, 32, 235–249. [Google Scholar] [CrossRef]
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef]
- Kondratskyi, A.; Kondratska, K.; Skryma, R.; Prevarskaya, N. Ion channels in the regulation of apoptosis. Biochim. Biophys. Acta 2015, 1848, 2532–2546. [Google Scholar] [CrossRef]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef]
- Feissner, R.F.; Skalska, J.; Gaum, W.E.; Sheu, S.S. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. 2009, 14, 1197–1218. [Google Scholar] [CrossRef]
- Yan, X.; Xun, M.; Li, J.; Wu, L.; Dou, X.; Zheng, J. Activation of Na+/K+-ATPase attenuates high glucose-induced H9c2 cell apoptosis via suppressing ROS accumulation and MAPKs activities by DRm217. Acta. Biochim. Biophys. Sin. 2016, 48, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Madreiter-Sokolowski, C.T.; Thomas, C.; Ristow, M. Interrelation between ROS and Ca2+ in aging and age-related diseases. Redox. Biol. 2020, 36, 101678. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, M.; Fontana, F.; Marzagalli, M.; Audano, M.; Beretta, G.; Procacci, P.; Sartori, P.; Mitro, N.; Limonta, P. Ca2+ overload- and ROS-associated mitochondrial dysfunction contributes to δ-tocotrienol-mediated paraptosis in melanoma cells. Apoptosis 2021, 26, 277–292. [Google Scholar] [CrossRef]
- Ataizi, Z.S.; Ertilav, K.; Nazıroğlu, M. Mitochondrial oxidative stress-induced brain and hippocampus apoptosis decrease through modulation of caspase activity, Ca2+ influx and inflammatory cytokine molecular pathways in the docetaxel-treated mice by melatonin and selenium treatments. Metab. Brain Dis. 2019, 34, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, B.; Wang, L.; Liu, J.; Yang, X.; Zheng, J. Melatonin Attenuates Cardiac Ischemia-Reperfusion Injury through Modulation of IP3R-Mediated Mitochondria-ER Contact. Oxid. Med. Cell. Longev. 2021, 1370862. [Google Scholar] [CrossRef] [PubMed]
- Meis, L. Role of the sarcoplasmic reticulum Ca2+-ATPase on heat production and thermogenesis. Biosci. Rep. 2001, 21, 113–137. [Google Scholar] [CrossRef]
- Zhou, D.; Shao, L.; Spitz, D. Reactive oxygen species in normal and tumor stem cells. Adv. Cancer Res. 2014, 122, 1–67. [Google Scholar]
- Hsu, C.C.; Tseng, L.M.; Lee, H.C. Role of mitochondrial dysfunction in cancer progression. Exp. Biol. Med. 2016, 241, 1281–1295. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A. VDAC1 functions in Ca2+ homeostasis and cell life and death in health and disease. Cell Calcium. 2018, 69, 81–100. [Google Scholar] [CrossRef]
- Szatrowski, T.; Nathan, C. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Tsunoda, T.; Koga, H.; Yokomizo, A.; Tatsugami, K.; Eto, M.; Inokuchi, J.; Hirata, A.; Masuda, K.; Okumura, K.; Naito, S. Inositol 1,4,5-trisphosphate (IP3) receptor type1 (IP3R1) modulates the acquisition of cisplatin resistance in bladder cancer cell lines. Oncogene 2005, 24, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Brouland, J.P.; Valleur, P.; Papp, B. Expression of SERCA pumps during cell differentiation and tumorigenesis: Application to colonic carcinogenesis. Ann. Pathol. 2006, 26, 159–172. [Google Scholar] [CrossRef]
- Kuchay, S.; Giorgi, C.; Simoneschi, D.; Pagan, J.; Missiroli, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Collazo-Lorduy, A.; Castillo-Martin, M.; et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature 2017, 546, 554–558. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, S.; Singh, S. Co-delivery of AKT3 siRNA and PTEN Plasmid by Antioxidant Nanoliposomes for Enhanced Antiproliferation of Prostate Cancer Cells. ACS Appl. Bio. Mater. 2020, 3, 3999–4011. [Google Scholar] [CrossRef]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef]
- Jin, C.; Kumar, P.; Gracia-Sancho, J.; Dufour, J.F. Calcium transfer between endoplasmic reticulum and mitochondria in liver diseases. FEBS Lett. 2021, 595, 1411–1421. [Google Scholar] [CrossRef]
- Boutin, B.; Tajeddine, N.; Monaco, G.; Molgo, J.; Vertommen, D.; Rider, M.; Parys, J.B.; Bultynck, G.; Gailly, P. Endoplasmic reticulum Ca(2+) content decrease by PKA-dependent hyperphosphorylation of type 1 IP3 receptor contributes to prostate cancer cell resistance to androgen deprivation. Cell Calcium. 2015, 57, 312–320. [Google Scholar] [CrossRef]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Prado-Garcia, H. Mitochondrial calcium: Transport and modulation of cellular processes in homeostasis and cancer (Review). Int. J. Oncol. 2019, 54, 1155–1167. [Google Scholar] [CrossRef]
- Colwill, K.; Gräslund, S. A roadmap to generate renewable protein binders to the human proteome. Nat. Methods 2011, 8, 551–558. [Google Scholar] [CrossRef]
- Zeng, F.; Chen, X.; Cui, W.; Wen, W.; Lu, F.; Sun, X.; Ma, D.; Yuan, Y.; Li, Z.; Hou, N.; et al. RIPK1 Binds MCU to Mediate Induction of Mitochondrial Ca2+ Uptake and Promotes Colorectal Oncogenesis. Cancer Res. 2018, 78, 2876–2885. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Zuo, W.; Xu, X.; Bi, L.; Zhang, C.; Chen, H.; Liu, H. MCU That Is Transcriptionally Regulated by Nrf2 Augments Malignant Biological Behaviors in Oral Squamous Cell Carcinoma Cells. BioMed Res. Int. 2021, 6650791. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Wu, R.; Deng, Y.; Zhang, Q. Dihydroartemisinin represses oral squamous cell carcinoma progression through downregulating mitochondrial calcium uniporter. Bioengineered 2022, 13, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Vitto, V.A.M.; Patergnani, S.; Pinton, P. High mitochondrial Ca2+ content increases cancer cell proliferation upon inhibition of mitochondrial permeability transition pore (mPTP). Cell Cycle 2019, 18, 914–916. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Guzzo, S.; Mangolini, A.; dell’Atti, L.; Pinton, P.; Aguiari, G. The induction of AMPK-dependent autophagy leads to P53 degradation and affects cell growth and migration in kidney cancer cells. Exp. Cell Res. 2020, 395, 112190. [Google Scholar] [CrossRef]
- Fei, M.; Zhang, L.; Wang, H.; Zhu, Y.; Niu, W.; Tang, T.; Han, Y. Inhibition of Cathepsin S Induces Mitochondrial Apoptosis in Glioblastoma Cell Lines Through Mitochondrial Stress and Autophagosome Accumulation. Front. Oncol. 2020, 10, 516746. [Google Scholar] [CrossRef]
- Xue, P.; Chen, Q.; Ren, X.; Liu, D.; Yang, X. A novel protoapigenone analog RY10-4 induces apoptosis of breast cancer cells by exacerbating mitochondrial Ca2+ influx through mitochondrial calcium uniporter. Toxicol. Appl. Pharmacol. 2021, 433, 115776. [Google Scholar] [CrossRef]
- Ren, T.; Wang, J.; Zhang, H.; Yuan, P.; Zhu, J.; Wu, Y.; Huang, Q.; Guo, X.; Zhang, J.; Ji, L.; et al. MCUR1-Mediated Mitochondrial Calcium Signaling Facilitates Cell Survival of Hepatocellular Carcinoma via Reactive Oxygen Species-Dependent P53 Degradation. Antioxid. Redox Signal. 2018, 28, 1120–1136. [Google Scholar] [CrossRef]
- Marchi, S.; Pinton, P. Mitochondrial calcium uniporter, MiRNA and cancer: Live and let die. Commun. Integr. Biol. 2013, 6, e23818. [Google Scholar] [CrossRef]
- Sun, Y.; Li, M.; Liu, G.; Zhang, X.; Zhi, L.; Zhao, J.; Wang, G. The function of Piezo1 in colon cancer metastasis and its potential regulatory mechanism. J. Cancer Res. Clin. Oncol. 2020, 146, 1139–1152. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Li, Z.; Lin, S.; Wang, H.; Sun, J.; Lan, C.; Wu, L.; Sun, D.; Huang, C.; et al. Mitochondrial calcium uniporter drives metastasis and confers a targetable cystine dependency in pancreatic cancer. Cancer Res. 2022, 3230, 2021. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, X.; Shen, Q.; Yang, X.; Yu, C.; Cai, C.; Cai, G.; Meng, X.; Zou, F. Mitochondrial Ca2+ uniporter is critical for store-operated Ca2+ entry-dependent breast cancer cell migration. Biochem. Biophys. Res. Commun. 2015, 458, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Peng, T.; Lin, S.; Zhi, W.; Cao, C.; Wu, P.; Xi, L.; Wu, P.; Yang, Q.; Ding, W. Key Role of MCUR1 in Malignant Progression of Breast Cancer. OncoTargets Ther. 2021, 14, 4163–4175. [Google Scholar] [CrossRef] [PubMed]
- Che, X.; Wang, Q.; Zhang, B. MCUR1 is a marker for the progression and prognosis of breast cancer. Res. Square 2022. preprint (Version 1). [Google Scholar]
- Ren, T.; Zhang, H.; Wang, J.; Zhu, J.; Jin, M.; Wu, Y.; Guo, X.; Ji, L.; Huang, Q.; Zhang, H.; et al. MCU-dependent mitochondrial Ca2+ inhibits NAD+/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 2017, 36, 5897–5909. [Google Scholar] [CrossRef] [PubMed]
- Bartha, A.; Győrffy, B. TNMplot.com: A Web Tool for the Comparison of Gene Expression in Normal, Tumor and Metastatic Tissues. Int. J. Mol. Sci. 2021, 22, 2622. [Google Scholar] [CrossRef]
- Parekh, A. Calcium signalling in health and disease. Semin Cell Dev Biol. 2019, 94, 1–2. [Google Scholar] [CrossRef]
- Li, C.; Lin, H.; Ko, C.; Lai, J.; Chu, P. A Novel Biomarker Driving Poor-Prognosis Liver Cancer: Overexpression of the Mitochondrial Calcium Gatekeepers. Biomedicines 2020, 8, 451. [Google Scholar] [CrossRef]
- Jin, M.; Wang, J.; Ji, X.; Cao, H.; Zhu, J.; Chen, Y.; Yang, J.; Zhao, Z.; Ren, T.; Xing, J. MCUR1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ROS/Nrf2/Notch pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 136. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Y.; Zhao, J.; Zhang, Z.; Jin, M.; Zhou, F.; Jin, C.; Zhang, J.; Xing, J.; Wang, N.; et al. PDE2 Inhibits PKA-Mediated Phosphorylation of TFAM to Promote Mitochondrial Ca2+-Induced Colorectal Cancer Growth. Front. Oncol. 2021, 11, 663778. [Google Scholar] [CrossRef]
- Liu, Y.; Jin, M.; Wang, Y.; Zhu, J.; Tan, R.; Zhao, J.; Ji, X.; Jin, C.; Jia, Y.; Ren, T.; et al. MCU-induced mitochondrial calcium uptake promotes mitochondrial biogenesis and colorectal cancer growth. Signal Transduct. Target Ther. 2020, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Vitto, V.A.M.; Danese, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial calcium uniporter complex modulation in cancerogenesis. Cell Cycle 2019, 18, 1068–1083. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Qi, J.; Zhang, X.; Zhao, X.; An, P.; Luo, Y.; Luo, J. The Regulatory Roles of Mitochondrial Calcium and the Mitochondrial Calcium Uniporter in Tumor Cells. Int. J. Mol. Sci. 2022, 23, 6667. https://doi.org/10.3390/ijms23126667
Zhang L, Qi J, Zhang X, Zhao X, An P, Luo Y, Luo J. The Regulatory Roles of Mitochondrial Calcium and the Mitochondrial Calcium Uniporter in Tumor Cells. International Journal of Molecular Sciences. 2022; 23(12):6667. https://doi.org/10.3390/ijms23126667
Chicago/Turabian StyleZhang, Linlin, Jingyi Qi, Xu Zhang, Xiya Zhao, Peng An, Yongting Luo, and Junjie Luo. 2022. "The Regulatory Roles of Mitochondrial Calcium and the Mitochondrial Calcium Uniporter in Tumor Cells" International Journal of Molecular Sciences 23, no. 12: 6667. https://doi.org/10.3390/ijms23126667
APA StyleZhang, L., Qi, J., Zhang, X., Zhao, X., An, P., Luo, Y., & Luo, J. (2022). The Regulatory Roles of Mitochondrial Calcium and the Mitochondrial Calcium Uniporter in Tumor Cells. International Journal of Molecular Sciences, 23(12), 6667. https://doi.org/10.3390/ijms23126667