
Mitochondrial Dysfunction and Therapeutic Perspectives in Cardiovascular Diseases


 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Mitochondrial Dysfunctions and CVDs

{kind=link}
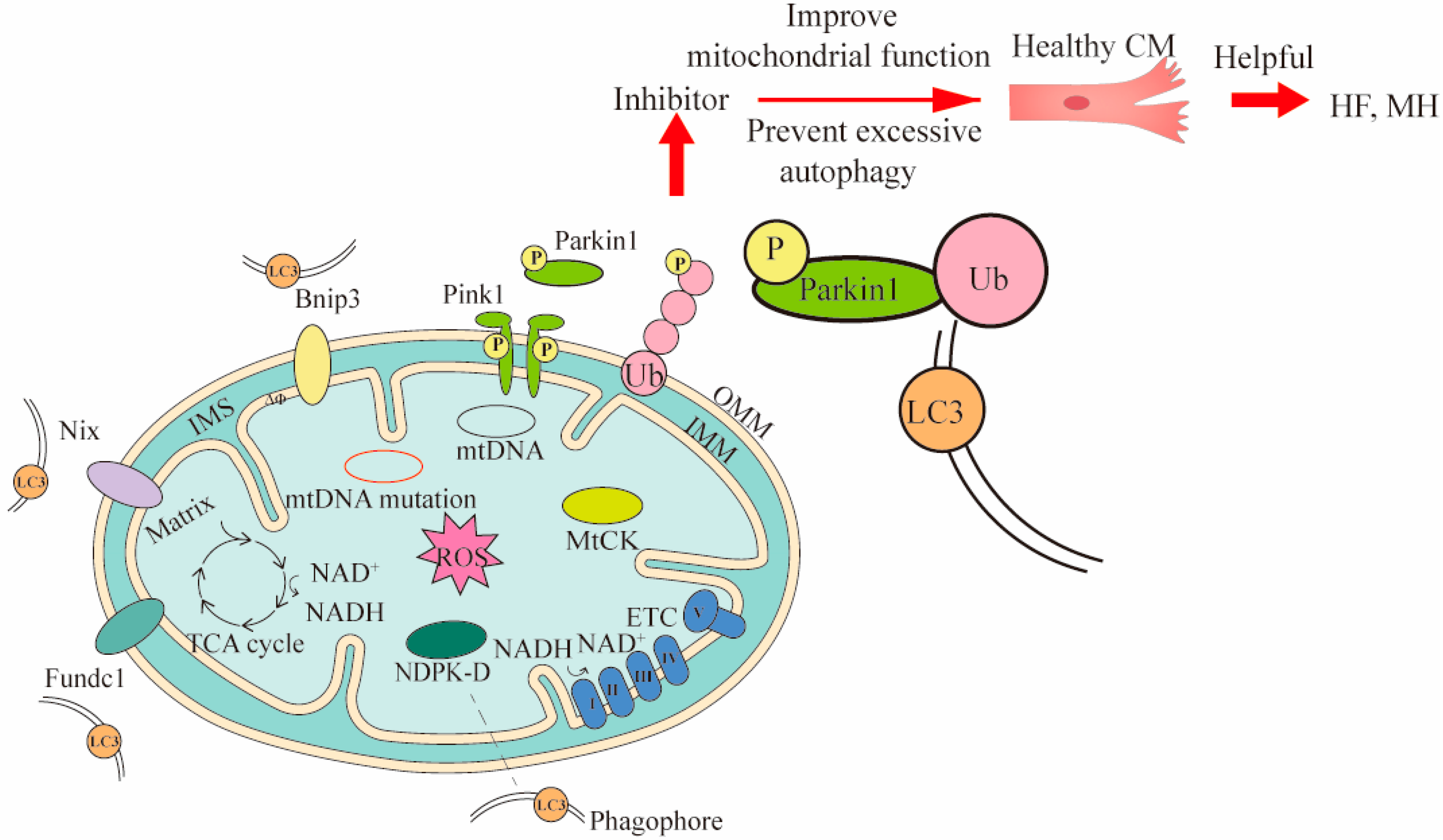{kind=link}
| CVDs | Mitochondrial Dysfunction | References |
|---|---|---|
| Heart failure | Impaired mitophagy ↓OXPHOS ↑ROS | [10] review, 2019 [12] experimental, 2010 [13] experimental, 2011 [14] review, 2022 [15] experimental, 2018 [16] experimental, 2020 [17] experimental, 2011 [18] experimental, 2015 |
| Myocardial hypertrophy | mtDNA mutation Impaired mitophagy ↑ROS | [10] review, 2019 [17] experimental, 2011 [19] experimental, 2022 [20] experimental, 2019 [21] experimental, 2021 [22] experimental, 2021 [23] experimental, 2015 |
| Atherosclerosis | mtDNA mutation Impaired mitophagy ↓OXPHOS ↑ROS | [24] clinical, 2019 [25] experimental, 2017 [26] experimental, 2018 [27] experimental, 2017 [28] experimental, 2019 [29] experimental, 2021 [30] experimental, 2014 |
| Aortic dissection | ↑ROS | [31] experimental, 2016 [32] experimental, 2020 [33] experimental, 2022 [34] experimental, 2021 |
| Aortic aneurysm | ↑ROS ↓OXPHOS | [32] experimental, 2020 [35] experimental, 2020 [36] experimental, 2018 [37] experimental, 2022 [38] experimental, 2006 [39] experimental, 2015 [40] experimental, 2013 [41] clinical, 2020 [42] clinical, 2021 |
2.1. mtDNA Mutation in CVDs
2.2. Mitophagy Damage in CVDs
2.3. Mitochondrial OXPHOS Reduction in CVDs
2.4. Mitochondrial-Derived ROS Increase in CVDs
3. Strategies for Targeting Mitochondria to Treat CVDs
3.1. mtDNA Mutation and Treatment in CVDs
3.1.1. mtDNA Editing Therapy
3.1.2. Mitochondrial Replacement Therapy
3.2. Mitophagy Therapy in CVDs
3.3. Mitochondrial OXPHOS Reduction and Treatment in CVDs
3.3.1. Small Molecule Compounds Enhance Mitochondrial Function
3.3.2. Nanomaterials Targeted Mitochondria to Improve Mitochondrial Function
3.4. Reduction or Elimination of Mitochondrial-Derived ROS in CVDs
4. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: Executive summary: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 74, 1376–1414. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.A.; Kelly, D.P.; Chirinos, J.A. Mitochondrial Dysfunction in Heart Failure With Preserved Ejection Fraction. Circulation 2019, 139, 1435–1450. [Google Scholar] [CrossRef] [PubMed]
- Ranjbarvaziri, S.; Kooiker, K.B.; Ellenberger, M.; Fajardo, G.; Zhao, M.; Roest, A.S.V.; Woldeyes, R.A.; Koyano, T.T.; Fong, R.; Ma, N.; et al. Altered Cardiac Energetics and Mitochondrial Dysfunction in Hypertrophic Cardiomyopathy. Circulation 2021, 144, 1714–1731. [Google Scholar] [CrossRef]
- Eirin, A.; Lerman, A.; Lerman, L.O. Mitochondrial injury and dysfunction in hypertension-induced cardiac damage. Eur. Hear. J. 2014, 35, 3258–3266. [Google Scholar] [CrossRef]
- Manolis, A.S.; Manolis, T.A.; Apostolaki, N.E.; Apostolopoulos, E.J.; Melita, H.; Katsiki, N. Mitochondrial dysfunction in cardiovascular disease: Current status of translational research/clinical and therapeutic implications. Med. Res. Rev. 2020, 41, 275–313. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac energy metabolism in heart failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Khotina, V.A.; Sukhorukov, V.N.; Kalmykov, V.A.; Mikhaleva, L.M.; Orekhov, A.N. The Role of Mitochondrial DNA Mutations in Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 23, 952. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Zampino, M.; Spencer, R.G.; Fishbein, K.W.; Simonsick, E.M.; Ferrucci, L. Cardiovascular Health and Mitochondrial Function: Testing an Association. J. Gerontol. Ser. A 2021, 76, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Ago, T.; Matsushima, S.; Zhai, P.; Schneider, M.D.; Sadoshima, J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. USA 2010, 107, 15565–15570. [Google Scholar] [CrossRef] [PubMed]
- Riehle, C.; Wende, A.R.; Zaha, V.G.; Pires, K.M.; Wayment, B.; Olsen, C.; Bugger, H.; Buchanan, J.; Wang, X.; Moreira, A.B.; et al. PGC-1β Deficiency Accelerates the Transition to Heart Failure in Pressure Overload Hypertrophy. Circ. Res. 2011, 109, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhang, Z.; Zhang, W.; Liu, X. Mitochondrial dysfunction and mitochondrial therapies in heart failure. Pharmacol. Res. 2021, 175, 106038. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Nie, J.; Wu, L.; Hu, Y.; Wen, Z.; Dong, L.; Zou, M.-H.; Chen, C.; Wang, D.W. AMPKα2 Protects Against the Development of Heart Failure by Enhancing Mitophagy via PINK1 Phosphorylation. Circ. Res. 2018, 122, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Abudureyimu, M.; Yu, W.; Cao, R.Y.; Zhang, Y.; Liu, H.; Zheng, H. Berberine promotes cardiac function by upregulating PINK1/Parkin-mediated mitophagy in heart failure. Front. Physiol. 2020, 11, 565751. [Google Scholar] [CrossRef]
- Dai, D.-F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintrón, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W.; Kang, Y.J.; Prolla, T.A. Mitochondrial oxidative stress mediates angiotensin II–induced cardiac hypertrophy and Gαq overexpression–induced heart failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef]
- Wu, S.-P.; Kao, C.-Y.; Wang, L.; Creighton, C.J.; Yang, J.; Donti, T.R.; Harmancey, R.; Vasquez, H.G.; Graham, B.H.; Bellen, H.J.; et al. Increased COUP-TFII expression in adult hearts induces mitochondrial dysfunction resulting in heart failure. Nat. Commun. 2015, 6, 8245. [Google Scholar] [CrossRef]
- Yan, M.; Li, Y.; Luo, Q.; Zeng, W.; Shao, X.; Li, L.; Wang, Q.; Wang, D.; Zhang, Y.; Diao, H.; et al. Mitochondrial damage and activation of the cytosolic DNA sensor cGAS–STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell Death Discov. 2022, 8, 1–12. [Google Scholar] [CrossRef]
- Kumar, V.; Sanawar, R.; Jaleel, A.; Kumar, T.R.S.; Kartha, C.C. Chronic Pressure Overload Results in Deficiency of Mitochondrial Membrane Transporter ABCB7 Which Contributes to Iron Overload, Mitochondrial Dysfunction, Metabolic Shift and Worsens Cardiac Function. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Zou, R.; Tao, J.; Qiu, J.; Shi, W.; Zou, M.; Chen, W.; Li, W.; Zhou, N.; Wang, S.; Ma, L.; et al. Ndufs1 Deficiency Aggravates the Mitochondrial Membrane Potential Dysfunction in Pressure Overload-Induced Myocardial Hypertrophy. Oxidative Med. Cell. Longev. 2021, 2021, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-L.; Tao, L.; Peng, F.-H.; Zheng, N.-Z.; Lin, Q.; Cai, S.-Y.; Wang, Q. GJA1-20k attenuates Ang II-induced pathological cardiac hypertrophy by regulating gap junction formation and mitochondrial function. Acta Pharmacol. Sin. 2021, 42, 536–549. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Umemoto, S.; Yoshimura, K.; Itoh, S.; Murata, T.; Fukai, T.; Matsuzaki, M. Angiotensin Ⅱ Activates MCP-1 and Induces Cardiac Hypertrophy and Dysfunction via Toll-like Receptor 4. J. Atheroscler. Thromb. 2015, 22, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Yokota, T.; Shingu, Y.; Yamada, A.; Iba, Y.; Ujihira, K.; Wakasa, S.; Ooka, T.; Takada, S.; Shirakawa, R.; et al. Impaired mitochondrial oxidative phosphorylation capacity in epicardial adipose tissue is associated with decreased concentration of adiponectin and severity of coronary atherosclerosis. Sci. Rep. 2019, 9, 3535. [Google Scholar] [CrossRef]
- Vilne, B.; Skogsberg, J.; Foroughi Asl, H.; Talukdar, H.A.; Kessler, T.; Björkegren, J.L.; Schunkert, H. Network analysis reveals a causal role of mitochondrial gene activity in atherosclerotic lesion formation. Arteriosclerosis 2017, 267, 39–48. [Google Scholar] [CrossRef]
- Jacinto, T.A.; Meireles, G.S.; Dias, A.T.; Aires, R.; Porto, M.L.; Gava, A.L.; Vasquez, E.C.; Pereira, T.M.C.; Campagnaro, B.P.; Meyrelles, S.S. Increased ROS production and DNA damage in monocytes are biomarkers of aging and atherosclerosis. Biol. Res. 2018, 51, 1–13. [Google Scholar] [CrossRef]
- Sergin, I.; Evans, T.D.; Zhang, X.; Bhattacharya, S.; Stokes, C.J.; Song, E.; Ali, S.; Dehestani, B.; Holloway, K.B.; Micevych, P.S.; et al. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat. Commun. 2017, 8, 15750. [Google Scholar] [CrossRef]
- Nahapetyan, H.; Moulis, M.; Grousset, E.; Faccini, J.; Grazide, M.-H.; Mucher, E.; Elbaz, M.; Martinet, W.; Vindis, C. Altered mitochondrial quality control in Atg7-deficient VSMCs promotes enhanced apoptosis and is linked to unstable atherosclerotic plaque phenotype. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Qiu, J.; Fu, Y.; Chen, Z.; Zhang, L.; Li, L.; Liang, D.; Wei, F.; Wen, Z.; Wang, Y.; Liang, S. BTK Promotes Atherosclerosis by Regulating Oxidative Stress, Mitochondrial Injury, and ER Stress of Macrophages. Oxidative Med. Cell. Longev. 2021, 2021, 1–15. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, G.Z.; Rabinovitch, P.S.; Tabas, I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-κB–mediated inflammation in macrophages. Circ. Res. 2014, 114, 421–433. [Google Scholar] [CrossRef]
- Liu, W.; Wang, B.; Wang, T.; Liu, X.; He, X.; Liu, Y.; Li, Z.; Zeng, H. Ursodeoxycholic Acid Attenuates Acute Aortic Dissection Formation in Angiotensin II-Infused Apolipoprotein E-Deficient Mice Associated with Reduced ROS and Increased Nrf2 Levels. Cell. Physiol. Biochem. 2016, 38, 1391–1405. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Sun, C.; Zhu, H.; Zhai, M.; Zhang, L.; Jiang, L.; Hou, P.; Li, J.; Li, K.; Liu, Z.; et al. Melatonin protects against thoracic aortic aneurysm and dissection through SIRT1-dependent regulation of oxidative stress and vascular smooth muscle cell loss. J. Pineal Res. 2020, 69, e12661. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Wang, Z.; Wu, Q.; Zhong, X.; Zhang, M.; Li, B.; Ren, W.; Yuan, S.; Chen, Y. Iron deficiency promotes aortic media degeneration by activating endoplasmic reticulum stress-mediated IRE1 signaling pathway. Pharmacol. Res. 2022, 183, 106366. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Yi, S.; Yu, T.; Hao, Y. Sirt3 Protects Against Thoracic Aortic Dissection Formation by Reducing Reactive Oxygen Species, Vascular Inflammation, and Apoptosis of Smooth Muscle Cells. Front. Cardiovasc. Med. 2021, 8, 380. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.-H.; Hsu, L.-A.; Tsai, H.-Y.; Yeh, Y.-H.; Lu, C.-Y.; Chen, P.-C.; Wang, J.-C.; Chiu, Y.-L.; Lin, C.-Y.; Hsu, Y.-J. Aldehyde dehydrogenase 2 protects against abdominal aortic aneurysm formation by reducing reactive oxygen species, vascular inflammation, and apoptosis of vascular smooth muscle cells. FASEB J. 2020, 34, 9498–9511. [Google Scholar] [CrossRef] [PubMed]
- Van Der Pluijm, I.; Burger, J.; Van Heijningen, P.M.; Ijpma, A.; Van Vliet, N.; Milanese, C.; Schoonderwoerd, K.; Sluiter, W.; Ringuette, L.-J.; Dekkers, D.H.W.; et al. Decreased mitochondrial respiration in aneurysmal aortas of Fibulin-4 mutant mice is linked to PGC1A regulation. Cardiovasc. Res. 2018, 114, 1776–1793. [Google Scholar] [CrossRef]
- Landis, B.J.; Lai, D.; Guo, D.-C.; Corvera, J.S.; Idrees, M.T.; Stadler, H.W.; Cuevas, C.; Needler, G.U.; Vujakovich, C.E.; Milewicz, D.M.; et al. Identification of a common polymorphism in COQ8B acting as a modifier of thoracic aortic aneurysm severity. Hum. Genet. Genom. Adv. 2022, 3, 100057. [Google Scholar] [CrossRef]
- Thomas, M.; Gavrila, D.; McCormick, M.L.; Miller, F.J., Jr.; Daugherty, A.; Cassis, L.A.; Dellsperger, K.C.; Weintraub, N.L. Deletion of p47 phox attenuates angiotensin II–induced abdominal aortic aneurysm formation in apolipoprotein E–deficient mice. Circulation 2006, 114, 404–413. [Google Scholar] [CrossRef]
- Yuan, K.; Liang, W.; Zhang, J. A comprehensive analysis of differentially expressed genes and pathways in abdominal aortic aneurysm. Mol. Med. Rep. 2015, 12, 2707–2714. [Google Scholar] [CrossRef]
- Guzik, B.; Sagan, A.; Ludew, D.; Mrowiecki, W.; Chwała, M.; Bujak-Gizycka, B.; Filip, G.; Grudzien, G.; Kapelak, B.; Żmudka, K.; et al. Mechanisms of oxidative stress in human aortic aneurysms—Association with clinical risk factors for atherosclerosis and disease severity. Int. J. Cardiol. 2013, 168, 2389–2396. [Google Scholar] [CrossRef]
- Li, Y.; Ren, P.; Dawson, A.; Vasquez, H.G.; Ageedi, W.; Zhang, C.; Luo, W.; Chen, R.; Li, Y.; Kim, S.; et al. Single-Cell Transcriptome Analysis Reveals Dynamic Cell Populations and Differential Gene Expression Patterns in Control and Aneurysmal Human Aortic Tissue. Circulation 2020, 142, 1374–1388. [Google Scholar] [CrossRef] [PubMed]
- Abudupataer, M.; Zhu, S.; Yan, S.; Xu, K.; Zhang, J.; Luo, S.; Ma, W.; Alam, F.; Tang, Y.; Huang, H.; et al. Aorta smooth muscle-on-a-chip reveals impaired mitochondrial dynamics as a therapeutic target for aortic aneurysm in bicuspid aortic valve disease. Elife 2021, 10, 69310. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Shen, S.; Xia, J.; Wang, J.; Gu, Z. Mitochondria as the Essence of Yang Qi in the Human Body. Phenomics 2022, 2, 336–348. [Google Scholar] [CrossRef]
- Zhu, Z.; Wang, X. Significance of Mitochondria DNA Mutations in Diseases. In Mitochondrial DNA and Diseases; Advances in Experimental Medicine and Biology Book Series; Springer: Singapore, 2017; Volume 1038, pp. 219–230. [Google Scholar] [CrossRef]
- Zhu, Y.; You, J.; Xu, C.; Gu, X. Associations of mitochondrial DNA 3777–4679 region mutations with maternally inherited essential hypertensive subjects in China. BMC Med. Genet. 2020, 21, 1–9. [Google Scholar] [CrossRef]
- Siva, M.A.; Mahalakshmi, R.; Bhakta-Guha, D.; Guha, G. Gene therapy for the mitochondrial genome: Purging mutations, pacifying ailments. Mitochondrion 2018, 46, 195–208. [Google Scholar] [CrossRef]
- DiMauro, S.; Schon, E.A. Mitochondrial Respiratory-Chain Diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef]
- Bray, A.W.; Ballinger, S.W. Mitochondrial DNA mutations and cardiovascular disease. Curr. Opin. Cardiol. 2017, 32, 267–274. [Google Scholar] [CrossRef]
- Friederich, M.W.; Geddes, G.C.; Wortmann, S.B.; Punnoose, A.; Wartchow, E.; Knight, K.M.; Prokisch, H.; Creadon-Swindell, G.; Mayr, J.A.; Van Hove, J.L. Pathogenic variants in MRPL44 cause infantile cardiomyopathy due to a mitochondrial translation defect. Mol. Genet. Metab. 2019, 133, 362–371. [Google Scholar] [CrossRef]
- Tang, J.; Tang, Q.-X.; Liu, S. METTL3-modified lncRNA-SNHG8 binds to PTBP1 to regulate ALAS2 expression to increase oxidative stress and promote myocardial infarction. Mol. Cell. Biochem. 2022. [Google Scholar] [CrossRef]
- Lioncino, M.; Monda, E.; Caiazza, M.; Fusco, A.; Cirillo, A.; Dongiglio, F.; Simonelli, V.; Sampaolo, S.; Ruggiero, L.; Scarano, G. Cardiovascular involvement in mtDNA disease: Diagnosis, management, and therapeutic options. Heart Fail. Clin. 2022, 18, 51–60. [Google Scholar] [CrossRef]
- Zhu, Y.; Gu, X.; Xu, C. Mitochondrial DNA 7908–8816 region mutations in maternally inherited essential hypertensive subjects in China. BMC Med. Genom. 2018, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Markin, A.; Khotina, V.; Zabudskaya, X.; Bogatyreva, A.; Starodubova, A.; Ivanova, E.; Nikiforov, N.; Orekhov, A. Disturbance of Mitochondrial Dynamics and Mitochondrial Therapies in Atherosclerosis. Life 2021, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Sobenin, I.A.; Sazonova, M.A.; Postnov, A.Y.; Bobryshev, Y.V.; Orekhov, A.N. Changes of mitochondria in atherosclerosis: Possible determinant in the pathogenesis of the disease. Atherosclerosis 2013, 227, 283–288. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Doblado, L.; Lueck, C.; Rey, C.; Samhan-Arias, A.K.; Prieto, I.; Stacchiotti, A.; Monsalve, M. Mitophagy in Human Diseases. Int. J. Mol. Sci. 2021, 22, 3903. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Nikiforov, N.G.; Wu, W.-K.; Kirichenko, T.V.; Orekhov, A.N. Autophagy and Mitophagy as Essential Components of Atherosclerosis. Cells 2021, 10, 443. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.-I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Mitophagy Is Essential for Maintaining Cardiac Function During High Fat Diet-Induced Diabetic Cardiomyopathy. Circ. Res. 2019, 124, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Li, X.; Huang, J.; Xu, C.; Liang, Q.; Ren, K.; Bai, A.; Lu, C.; Qian, R.; Sun, N. BMAL1 regulates mitochondrial fission and mitophagy through mitochondrial protein BNIP3 and is critical in the development of dilated cardiomyopathy. Protein Cell 2020, 11, 661–679. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, T.; Fu, F.; Cui, Z.; Lai, Q.; Zhang, Y.; Yu, B.; Liu, F.; Kou, J.; Li, F. Omentin1 ameliorates myocardial ischemia-induced heart failure via SIRT3/FOXO3a-dependent mitochondrial dynamical homeostasis and mitophagy. J. Transl. Med. 2022, 20, 447. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhu, P.; Wang, J.; Zhu, H.; Ren, J.; Chen, Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018, 25, 1080–1093. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, D.; Zeviani, M. Human diseases associated with defects in assembly of OXPHOS complexes. Essays Biochem. 2018, 62, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Stamerra, C.A.; Di Giosia, P.; Giorgini, P.; Ferri, C.; Sukhorukov, V.N.; Sahebkar, A. Mitochondrial Dysfunction and Cardiovascular Disease: Pathophysiology and Emerging Therapies. Oxidative Med. Cell. Longev. 2022, 2022, 1–16. [Google Scholar] [CrossRef]
- Takada, S.; Maekawa, S.; Furihata, T.; Kakutani, N.; Setoyama, D.; Ueda, K.; Nambu, H.; Hagiwara, H.; Handa, H.; Fumoto, Y. Succinyl-CoA-based energy metabolism dysfunction in chronic heart failure. Proc. Natl. Acad. Sci. USA 2022, 119, e2203628119. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Shao, Y.; Guo, H.C.; Zhi, Y.; Qiao, B.; Ma, K.; Du, J.; Lai, Y.Q.; Li, Y. MicroRNA-27b-3p down-regulates FGF1 and aggravates pathological cardiac remodelling. Cardiovasc. Res. 2021, 118, 2139–2151. [Google Scholar] [CrossRef]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Moghadam, Z.M.; Henneke, P.; Kolter, J. From Flies to Men: ROS and the NADPH Oxidase in Phagocytes. Front. Cell Dev. Biol. 2021, 9, 628991. [Google Scholar] [CrossRef]
- Madamanchi, N.; Runge, M.S. Mitochondrial Dysfunction in Atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef]
- López-Acosta, O.; Fortis-Barrera, M.D.L.A.; Barrios-Maya, M.A.; Ramírez, A.R.; Aguilar, F.J.A.; El-Hafidi, M. Reactive Oxygen Species from NADPH Oxidase and Mitochondria Participate in the Proliferation of Aortic Smooth Muscle Cells from a Model of Metabolic Syndrome. Oxidative Med. Cell. Longev. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Matsushima, S.; Kuroda, J.; Ago, T.; Zhai, P.; Park, J.Y.; Xie, L.-H.; Tian, B.; Sadoshima, J. Increased Oxidative Stress in the Nucleus Caused by Nox4 Mediates Oxidation of HDAC4 and Cardiac Hypertrophy. Circ. Res. 2013, 112, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Yu, E.; Reinhold, Y.; Starks, L.; Uryga, A.; Foote, K.; Finigan, A.; Figg, N.; Pung, Y.-F.; Logan, A.; et al. Mitochondrial respiration is reduced in atherosclerosis, promoting necrotic core formation and reducing relative fibrous cap thickness. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2322–2332. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.-M.; Hu, Y.-Y.; Yang, T.; Wu, N.; Wang, X.-N. Reactive Oxygen Species and Oxidative Stress in Vascular-Related Diseases. Oxidative Med. Cell. Longev. 2022, 2022, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Jiang, J.; Li, Z.; Liang, J.; Xiang, Y. Strategies for mitochondrial gene editing. Comput. Struct. Biotechnol. J. 2021, 19, 3319–3329. [Google Scholar] [CrossRef]
- Zekonyte, U.; Bacman, S.R.; Moraes, C.T. DNA-editing enzymes as potential treatments for heteroplasmic mtDNA diseases. J. Intern. Med. 2020, 287, 685–697. [Google Scholar] [CrossRef]
- Bacman, S.R.; Williams, S.L.; Garcia, S.; Moraes, C.T. Organ-specific shifts in mtDNA heteroplasmy following systemic delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 2010, 17, 713–720. [Google Scholar] [CrossRef]
- Sendra, L.; García-Mares, A.; Herrero, M.J.; Aliño, S.F. Mitochondrial DNA Replacement Techniques to Prevent Human Mitochondrial Diseases. Int. J. Mol. Sci. 2021, 22, 551. [Google Scholar] [CrossRef]
- Paz, M.V.; Cotán, D.; Garrido-Maraver, J.; Cordero, M.D.; Oropesa-Ávila, M.; de La Mata, M.; Pavón, A.D.; de Lavera, I.; Alcocer-Gómez, E.; Sánchez-Alcázar, J.A. Targeting autophagy and mitophagy for mitochondrial diseases treatment. Expert Opin. Ther. Targets 2016, 20, 487–500. [Google Scholar] [CrossRef]
- Tong, M.; Saito, T.; Zhai, P.; Oka, S.-I.; Mizushima, W.; Nakamura, M.; Ikeda, S.; Shirakabe, A.; Sadoshima, J. Alternative Mitophagy Protects the Heart Against Obesity-Associated Cardiomyopathy. Circ. Res. 2021, 129, 1105–1121. [Google Scholar] [CrossRef]
- Luna-Castillo, K.; Lin, S.; Muñoz-Valle, J.; Vizmanos, B.; López-Quintero, A.; Márquez-Sandoval, F. Functional Food and Bioactive Compounds on the Modulation of the Functionality of HDL-C: A Narrative Review. Nutrients 2021, 13, 1165. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Baur, J.A.; Imai, S.-I. NAD+ Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhao, Z.; Shen, M.; Zhang, Y.; Duan, J.; Guo, Y.; Zhang, D.; Hu, J.; Lin, J.; Man, W.; et al. Polydatin protects cardiomyocytes against myocardial infarction injury by activating Sirt3. Biochim. Et Biophys. Acta Mol. Basis Dis. 2017, 1863, 1962–1972. [Google Scholar] [CrossRef]
- Liu, C.; Chen, L.; Ma, Y.; Hu, K.; Wu, P.; Pan, L.; Chen, H.; Li, L.; Hu, H.; Zhang, J. Pulmonary circulation-mediated heart targeting for the prevention of heart failure by inhalation of intrinsically bioactive nanoparticles. Theranostics 2021, 11, 8550–8569. [Google Scholar] [CrossRef] [PubMed]
- Liew, S.S.; Qin, X.; Zhou, J.; Li, L.; Huang, W.; Yao, S.Q. Smart design of nanomaterials for mitochondria-targeted nanotherapeutics. Angew. Chem. Int. Ed. 2020, 60, 2232–2256. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.D.; Tian, R. Boosting mitochondrial metabolism with dietary supplements in heart failure. Nat. Rev. Cardiol. 2021, 18, 685–686. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, A.; Iannuzzo, G.; Parlato, A.; Cuomo, G.; Testa, C.; Coppola, M.; D’Ambrosio, G.; Oliviero, D.A.; Sarullo, S.; Vitale, G.; et al. Clinical Evidence for Q10 Coenzyme Supplementation in Heart Failure: From Energetics to Functional Improvement. J. Clin. Med. 2020, 9, 1266. [Google Scholar] [CrossRef] [PubMed]
- Pitceathly, R.D.S.; Keshavan, N.; Rahman, J.; Rahman, S. Moving Towards Clinical Trials for Mitochondrial Diseases. J. Inherit. Metab. Dis. 2021, 44, 22–41. [Google Scholar] [CrossRef]
- Martelli, A.; Testai, L.; Colletti, A.; Cicero, A.F.G. Coenzyme Q10: Clinical Applications in Cardiovascular Diseases. Antioxidants 2020, 9, 341. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X. New potentials of mitochondrial DNA editing. Cell Biol. Toxicol. 2020, 36, 391–393. [Google Scholar] [CrossRef]
- Peeva, V.; Blei, D.; Trombly, G.; Corsi, S.; Szukszto, M.J.; Rebelo-Guiomar, P.; Gammage, P.A.; Kudin, A.P.; Becker, C.; Altmüller, J.; et al. Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bacman, S.R.; Williams, S.L.; Duan, N.; Moraes, C.T. Manipulation of mtDNA heteroplasmy in all striated muscles of newborn mice by AAV9-mediated delivery of a mitochondria-targeted restriction endonuclease. Gene Ther. 2012, 19, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Gammage, P.A.; Rorbach, J.; Vincent, A.I.; Rebar, E.J.; Minczuk, M. Mitochondrially targeted ZFN s for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations. EMBO Mol. Med. 2014, 6, 458–466. [Google Scholar] [CrossRef]
- Pereira, C.V.; Bacman, S.R.; Arguello, T.; Zekonyte, U.; Williams, S.L.; Edgell, D.R.; Moraes, C.T. mitoTev-TALE: A monomeric DNA editing enzyme to reduce mutant mitochondrial DNA levels. EMBO Mol. Med. 2018, 10, e8084. [Google Scholar] [CrossRef] [PubMed]
- Bacman, S.R.; Williams, S.L.; Pinto, M.; Peralta, S.; Moraes, C.T. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat. Med. 2013, 19, 1111–1113. [Google Scholar] [CrossRef]
- Wang, B.; Lv, X.; Wang, Y.; Wang, Z.; Liu, Q.; Lu, B.; Liu, Y.; Gu, F. CRISPR/Cas9-mediated mutagenesis at microhomologous regions of human mitochondrial genome. Sci. China Life Sci. 2021, 64, 1463–1472. [Google Scholar] [CrossRef]
- Bian, W.-P.; Chen, Y.-L.; Luo, J.-J.; Wang, C.; Xie, S.-L.; Pei, D.-S. Knock-In Strategy for Editing Human and Zebrafish Mitochondrial DNA Using Mito-CRISPR/Cas9 System. ACS Synth. Biol. 2019, 8, 621–632. [Google Scholar] [CrossRef]
- Hussain, S.-R.A.; Yalvac, M.E.; Khoo, B.; Eckardt, S.; McLaughlin, K.J. Adapting CRISPR/Cas9 System for Targeting Mitochondrial Genome. Front. Genet. 2021, 12, 627050. [Google Scholar] [CrossRef]
- Guo, J.; Chen, X.; Liu, Z.; Sun, H.; Zhou, Y.; Dai, Y.; Ma, Y.; He, L.; Qian, X.; Wang, J.; et al. DdCBE mediates efficient and inheritable modifications in mouse mitochondrial genome. Mol. Ther. Nucleic Acids 2022, 27, 73–80. [Google Scholar] [CrossRef]
- Silva-Pinheiro, P.; Nash, P.A.; Van Haute, L.; Mutti, C.D.; Turner, K.; Minczuk, M. In vivo mitochondrial base editing via adeno-associated viral delivery to mouse post-mitotic tissue. Nat. Commun. 2022, 13, 1–9. [Google Scholar] [CrossRef]
- Mok, B.Y.; De Moraes, M.H.; Zeng, J.; Yeh, M.M.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Singh, D.; Mahant, A.; Sohal, S.K.; Kesavan, A.K. Samiksha Development of mitochondrial replacement therapy: A review. Heliyon 2020, 6, e04643. [Google Scholar] [CrossRef] [PubMed]
- Greggains, G.D.; Lister, L.M.; Tuppen, H.A.L.; Zhang, Q.; Needham, L.H.; Prathalingam, N.; Hyslop, L.A.; Craven, L.; Polanski, Z.; Murdoch, A.P.; et al. Therapeutic potential of somatic cell nuclear transfer for degenerative disease caused by mitochondrial DNA mutations. Sci. Rep. 2014, 4, 3844. [Google Scholar] [CrossRef]
- Hyslop, L.A.; Blakeley, P.; Craven, L.; Richardson, J.; Fogarty, N.M.; Fragouli, E.; Lamb, M.; Wamaitha, S.E.; Prathalingam, N.; Zhang, Q.; et al. Towards clinical application of pronuclear transfer to prevent mitochondrial DNA disease. Nature 2016, 534, 383–386. [Google Scholar] [CrossRef]
- Kang, E.; Wu, J.; Gutierrez, N.M.; Koski, A.; Tippner-Hedges, R.; Agaronyan, K.; Platero-Luengo, A.; Martinez-Redondo, P.; Ma, H.; Lee, Y.; et al. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature 2016, 540, 270–275. [Google Scholar] [CrossRef]
- Adashi, E.Y.; Caplan, A.L.; Capron, A.; Chapman, A.R.; Cho, M.; Clayton, E.W.; Cohen, I.G.; Cook-Deegan, R.; Faden, R.R.; Friedmann, T.; et al. In support of mitochondrial replacement therapy. Nat. Med. 2019, 25, 870–871. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Kuno, T.; Yaegashi, N. Mitochondrial replacement therapy and assisted reproductive technology: A paradigm shift toward treatment of genetic diseases in gametes or in early embryos. Reprod. Med. Biol. 2018, 17, 421–433. [Google Scholar] [CrossRef]
- Qiu, Z.; Wei, Y.; Song, Q.; Du, B.; Wang, H.; Chu, Y.; Hu, Y. The Role of Myocardial Mitochondrial Quality Control in Heart Failure. Front. Pharmacol. 2019, 10, 1404. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Muqit, M.M. PINK1 and Parkin: Emerging themes in mitochondrial homeostasis. Curr. Opin. Cell Biol. 2017, 45, 83–91. [Google Scholar] [CrossRef]
- Bansal, M.; Moharir, S.C.; Swarup, G. Autophagy receptor optineurin promotes autophagosome formation by potentiating LC3-II production and phagophore maturation. Commun. Integr. Biol. 2018, 11, 1–4. [Google Scholar] [CrossRef]
- Lei, S.; Feng, Y.; Huang, P.; Chen, B.; Bao, K.; Wu, Q.; Zhang, H.; Huang, X. Ophiopogonin D’-induced mitophagy and mitochondrial damage are associated with dysregulation of the PINK1/Parkin signaling pathway in AC16 cells. Toxicology 2022, 477, 153275. [Google Scholar] [CrossRef] [PubMed]
- Quiles, J.M.; Gustafsson, B. The role of mitochondrial fission in cardiovascular health and disease. Nat. Rev. Cardiol. 2022, 19, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.-Y.; Wei, X.-X.; Zhi, X.-L.; Wang, X.-H.; Meng, D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol. Sin. 2021, 42, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Losón, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical Inhibition of the Mitochondrial Division Dynamin Reveals Its Role in Bax/Bak-Dependent Mitochondrial Outer Membrane Permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Aishwarya, R.; Alam, S.; Abdullah, C.S.; Morshed, M.; Nitu, S.S.; Panchatcharam, M.; Miriyala, S.; Kevil, C.G.; Bhuiyan, S. Pleiotropic effects of mdivi-1 in altering mitochondrial dynamics, respiration, and autophagy in cardiomyocytes. Redox Biol. 2020, 36, 101660. [Google Scholar] [CrossRef]
- Sun, W.; Liu, C.; Chen, Q.; Liu, N.; Yan, Y.; Liu, B. SIRT3: A New Regulator of Cardiovascular Diseases. Oxidative Med. Cell. Longev. 2018, 2018, 1–11. [Google Scholar] [CrossRef]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.-R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef]
- Bagul, P.K.; Katare, P.B.; Bugga, P.; Dinda, A.K.; Banerjee, S.K. SIRT-3 Modulation by Resveratrol Improves Mitochondrial Oxidative Phosphorylation in Diabetic Heart through Deacetylation of TFAM. Cells 2018, 7, 235. [Google Scholar] [CrossRef]
- Lauritzen, K.H.; Olsen, M.B.; Ahmed, M.S.; Yang, K.; Rinholm, J.E.; Bergersen, L.H.; Esbensen, Q.Y.; Sverkeli, L.J.; Ziegler, M.; Attramadal, H.; et al. Instability in NAD+ metabolism leads to impaired cardiac mitochondrial function and communication. eLife 2021, 10, e59828. [Google Scholar] [CrossRef]
- Yang, B.; Dan, X.; Hou, Y.; Lee, J.; Wechter, N.; Krishnamurthy, S.; Kimura, R.; Babbar, M.; Demarest, T.; McDevitt, R.; et al. NAD + supplementation prevents STING-induced senescence in ataxia telangiectasia by improving mitophagy. Aging Cell 2021, 20, e13329. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Wang, D.D.-H.; Qiu, Y.; Airhart, S.; Liu, Y.; Stempien-Otero, A.; O’Brien, K.D.; Tian, R. Boosting NAD level suppresses inflammatory activation of PBMCs in heart failure. J. Clin. Investig. 2020, 130, 6054–6063. [Google Scholar] [CrossRef] [PubMed]
- Tarantini, S.; Valcarcel-Ares, M.N.; Toth, P.; Yabluchanskiy, A.; Tucsek, Z.; Kiss, T.; Hertelendy, P.; Kinter, M.; Ballabh, P.; Süle, Z.; et al. Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol. 2019, 24, 101192. [Google Scholar] [CrossRef] [PubMed]
- De Picciotto, N.E.; Gano, L.B.; Johnson, L.C.; Martens, C.R.; Sindler, A.L.; Mills, K.F.; Imai, S.; Seals, D.R. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 2016, 15, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Charles, C.; Cohen-Erez, I.; Kazaoka, B.; Melnikov, O.; Stein, D.E.; Sensenig, R.; Rapaport, H.; Orynbayeva, Z. Mitochondrial responses to organelle-specific drug delivering nanoparticles composed of polypeptide and peptide complexes. Nanomedicine 2020, 15, 2917–2932. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Khalique, A.; Du, X.; Gao, Z.; Wu, J.; Zhang, X.; Zhang, R.; Sun, Z.; Liu, Q.; Xu, Z.; et al. Biomimetic Design of Mitochondria-Targeted Hybrid Nanozymes as Superoxide Scavengers. Adv. Mater. 2021, 33, e2006570. [Google Scholar] [CrossRef]
- Cohen-Erez, I.; Rapaport, H. Negatively charged polypeptide-peptide nanoparticles showing efficient drug delivery to the mitochondria. Colloids Surfaces B: Biointerfaces 2018, 162, 186–192. [Google Scholar] [CrossRef]
- Lee, T.-L.; Lee, M.-H.; Chen, Y.-C.; Lee, Y.-C.; Lai, T.-C.; Lin, H.Y.-H.; Hsu, L.-F.; Sung, H.-C.; Lee, C.-W.; Chen, Y.-L. Vitamin D attenuates ischemia/reperfusion-induced cardiac injury by reducing mitochondrial fission and mitophagy. Front. Pharmacol. 2020, 11, 604700. [Google Scholar] [CrossRef]
- Hao, T.; Li, J.; Yao, F.; Dong, D.; Wang, Y.; Yang, B.; Wang, C. Injectable Fullerenol/Alginate Hydrogel for Suppression of Oxidative Stress Damage in Brown Adipose-Derived Stem Cells and Cardiac Repair. ACS Nano 2017, 11, 5474–5488. [Google Scholar] [CrossRef]
- Zhang, M.; Zhu, J.; Qin, X.; Zhou, M.; Zhang, X.; Gao, Y.; Zhang, T.; Xiao, D.; Cui, W.; Cai, X. Cardioprotection of Tetrahedral DNA Nanostructures in Myocardial Ischemia-Reperfusion Injury. ACS Appl. Mater. Interfaces 2019, 11, 30631–30639. [Google Scholar] [CrossRef]
- Napolitano, G.; Fasciolo, G.; Venditti, P. Mitochondrial Management of Reactive Oxygen Species. Antioxidants 2021, 10, 1824. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Mariscal, F.M.; Larriva, A.P.A.-D.; Limia-Perez, L.; Romero-Cabrera, J.L.; Yubero-Serrano, E.M.; López-Miranda, J. Coenzyme Q10 Supplementation for the Reduction of Oxidative Stress: Clinical Implications in the Treatment of Chronic Diseases. Int. J. Mol. Sci. 2020, 21, 7870. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, M.; Ren, J. The Emerging Role of Coenzyme Q-10 in Aging, Neurodegeneration, Cardiovascular Disease, Cancer and Diabetes Mellitus. Curr. Neurovascular Res. 2005, 2, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Szczepańska, E.; Białek-Dratwa, A.; Janota, B.; Kowalski, O. Dietary Therapy in Prevention of Cardiovascular Disease (CVD)—Tradition or Modernity? A Review of the Latest Approaches to Nutrition in CVD. Nutrients 2022, 14, 2649. [Google Scholar] [CrossRef]
- Mortensen, A.L.; Rosenfeldt, F.; Filipiak, K.J. Effect of coenzyme Q10 in Europeans with chronic heart failure: A sub-group analysis of the Q-SYMBIO randomized double-blind trial. Cardiol. J. 2019, 26, 147–156. [Google Scholar] [CrossRef]
- Jafari, M.; Mousavi, S.M.; Asgharzadeh, A.; Yazdani, N. Coenzyme Q10 in the treatment of heart failure: A systematic review of systematic reviews. Indian Hear. J. 2018, 70, S111–S117. [Google Scholar] [CrossRef]
- Renke, G.; Pereira, M.B.; Renke, A. Coenzyme Q10 for Diabetes and Cardiovascular Disease: Useful or Useless? Curr Diabetes Rev. 2022. [Google Scholar] [CrossRef]
- Gioscia-Ryan, R.A.; LaRocca, T.J.; Sindler, A.L.; Zigler, M.C.; Murphy, M.P.; Seals, D.R. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J. Physiol. 2014, 592, 2549–2561. [Google Scholar] [CrossRef]


| Features | Strategies | Advantages | Limitations | Examples as Applied | References |
|---|---|---|---|---|---|
| Mutant mtDNA | mtDNA editing | High specificity; Easy operation | High cost; Limited availability | mito-RE, heart related CVDs | [46,76,77,78] |
| Mitochondrial replacement therapy | Reduce the risk of vertical transmission | Ethical and legal issues | [79] | ||
| Impaired mitophagy | Enhanced mitophagy | Play protective roles | Unclear conditions for mitophagy; A potential cytotoxic | Berberine and the Pink1/Parkin pathway, HF; Active Ulk1/Rab9-dependent, Cardiomyopathy | [16,55,80,81] |
| Decreased OXPHOS | Small molecule compounds to improve OXPHOS | Dietary supplements; Wide range of sources | Lack of clinical trials; Being degraded in advance | Control of the SIRT3 activity, MI | [82,83,84] |
| Nanomaterials to enhance mitochondrial function | Precise targeting; Noninvasive; High load drug | Lack of clinical trials; Collaborative targeting against multiple subcellular organelles is limited | NAD+, HF | [85,86,87] | |
| Increased ROS | Antioxidant | Amounts of clinical trials (CoQ10) | Interaction with statin (CoQ10); Long-term exposure maybe harmful | CoQ10, HF; Melatonin, AA/AD | [32,88,89,90] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Huang, Y.; Xu, C.; An, P.; Luo, Y.; Jiao, L.; Luo, J.; Li, Y. Mitochondrial Dysfunction and Therapeutic Perspectives in Cardiovascular Diseases. Int. J. Mol. Sci. 2022, 23, 16053. https://doi.org/10.3390/ijms232416053
Liu Y, Huang Y, Xu C, An P, Luo Y, Jiao L, Luo J, Li Y. Mitochondrial Dysfunction and Therapeutic Perspectives in Cardiovascular Diseases. International Journal of Molecular Sciences. 2022; 23(24):16053. https://doi.org/10.3390/ijms232416053
Chicago/Turabian StyleLiu, Yu, Yuejia Huang, Chong Xu, Peng An, Yongting Luo, Lei Jiao, Junjie Luo, and Yongzhi Li. 2022. "Mitochondrial Dysfunction and Therapeutic Perspectives in Cardiovascular Diseases" International Journal of Molecular Sciences 23, no. 24: 16053. https://doi.org/10.3390/ijms232416053
APA StyleLiu, Y., Huang, Y., Xu, C., An, P., Luo, Y., Jiao, L., Luo, J., & Li, Y. (2022). Mitochondrial Dysfunction and Therapeutic Perspectives in Cardiovascular Diseases. International Journal of Molecular Sciences, 23(24), 16053. https://doi.org/10.3390/ijms232416053