
Immunotherapy as a Therapeutic Strategy for Gastrointestinal Cancer—Current Treatment Options and Future Perspectives


 ,
,  , ,
, ,  ,
,  ,
,  ,
,  and
and 
Abstract
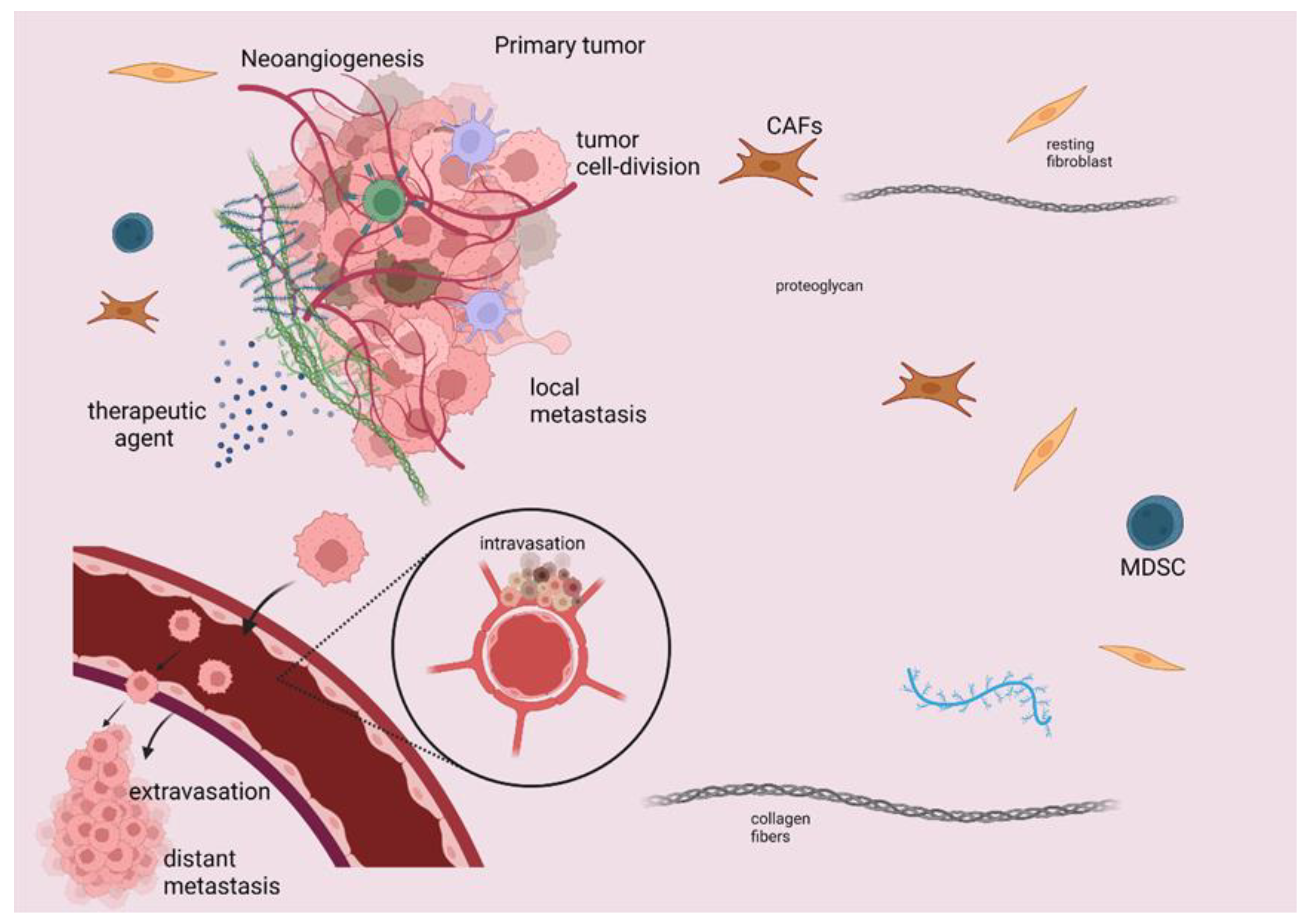
1. Introduction
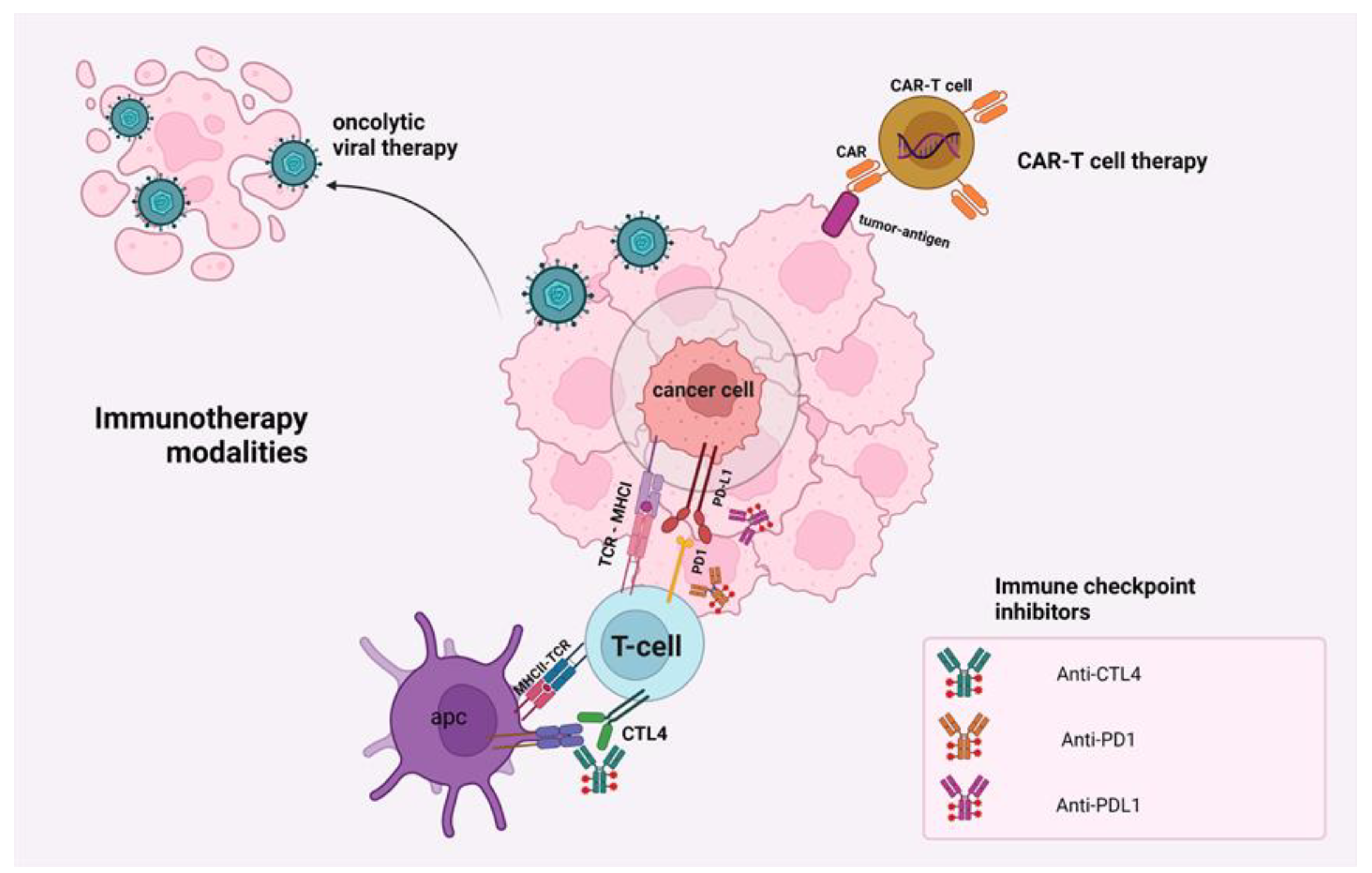
2. Mechanisms of Action of Immunotherapeutic Modalities
3. Immunotherapy in Gastrointestinal Cancers
4. Immunotherapy in Pancreatic Adenocarcinoma
4.1. Immune Checkpoints Inhibition in PAC
4.2. Oncolytic Viral Therapy in PAC
4.3. Cancer Vaccines in PAC
4.4. Adoptive Cell Therapy in PAC
4.5. Other Treatments That Target the TME Components
4.6. Future Approaches for Treating PAC
5. Current Immunotherapy in Gastric Cancer
5.1. Immune Checkpoints Modulation in GC
5.2. Cancer Vaccines in GC
5.3. Future Approaches for Treating GC
6. Current Immunotherapy in Hepatocellular Carcinoma
6.1. Immune Checkpoint Blockade for HCC
6.2. Utilization of Oncolytic Viral Therapy in HCC
6.3. Cancer Vaccine in HCC Management
6.4. Adaptive Cell Therapy in HCC
6.5. Ongoing Clinical Trials and Future Approaches to HCC Management
6.6. Predictors of Immunotherapy Response in HCC
7. Immunotherapy in Colorectal Cancer
7.1. The Importance of the Tumor Microenvironment in CRC
7.2. Current Immune Checkpoint Inhibitors in CRC
7.3. Ongoing Trials for Cancer Vaccines in CRC
7.4. Future Approaches for Treating CRC
8. Immunotherapy in Cholangiocarcinoma
8.1. Immune Checkpoint Inhibitors in CCA
8.2. Cell-Based Therapies and Cancer Vaccines for CCA
8.3. Future Approaches for Treating CCA
9. The Pros and Cons of the Immunotherapeutic Modalities
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| (CB1)/(CB2) | cannabinoid receptor 1 and receptor2 |
| (ceRNA) | competing endogenous RNA |
| (CMA) | chaperon-mediated autophagy |
| (HCQ) | hydroxychloroquine |
| (HIF-1a) | hypoxia-inducible factor-1 |
| (HMGB1) | high-mobility group box 1 |
| (LAMP-2A) | lysosomal membrane protein 2A |
| (lncRNA) | long noncoding RNA |
| (microRNAs) | small non-coding RNA molecules |
| (MSI-H) | microsatellite instability |
| (mTOR) | mechanistic target of rapamycin |
| (PADC) | pancreatic adenocarcinoma |
| (PE) | lipid phosphatidylethanolamine |
| (PVT1) | plasmacytoma variant translocation 1 |
| (Pygo2) | pygopus2 |
| (RAGE) | receptors for advanced glycation end products |
| (ROS) | reactive oxygen species |
| (siRNA) | small interfering RNA |
| (TME) | tumor microenvironment |
| (UA) | ursolic acid |
| (ULK1) | Unc-51-like kinase1 complex |
| (VMP1) | vacuole membrane protein 1 |
References
- Hazama, S.; Tamada, K.; Yamaguchi, Y.; Kawakami, Y.; Nagano, H. Current Status of Immunotherapy against Gastrointestinal Cancers and Its Biomarkers: Perspective for Precision Immunotherapy. Ann. Gastroenterol. Surg. 2018, 2, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer Immunotherapy Comes of Age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Donninger, H.; Li, C.; Eaton, J.W.; Yaddanapudi, K. Cancer Vaccines: Promising Therapeutics or an Unattainable Dream. Vaccines 2021, 9, 668. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, D.S.; Kichloo, A.; Singh, J.; Albosta, M.; Lekkala, M. Current Immunotherapy in Gastrointestinal Malignancies A Review. J. Investig. Med. 2021, 69, 689–696. [Google Scholar] [CrossRef]
- Koi, M.; Carethers, J.M. The Colorectal Cancer Immune Microenvironment and Approach to Immunotherapies. Future Oncol. 2017, 13, 1633–1647. [Google Scholar] [CrossRef]
- Wang, D.-K.; Zuo, Q.; He, Q.-Y.; Li, B. Targeted Immunotherapies in Gastrointestinal Cancer: From Molecular Mechanisms to Implications. Front. Immunol. 2021, 12, 705999. [Google Scholar] [CrossRef]
- Tang, S.; Ning, Q.; Yang, L.; Mo, Z.; Tang, S. Mechanisms of Immune Escape in the Cancer Immune Cycle. Int. Immunopharmacol. 2020, 86, 106700. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the Tumor Microenvironment in PD-L1/PD-1-Mediated Tumor Immune Escape. Mol. Cancer 2019, 18, 10. [Google Scholar] [CrossRef]
- Pio, R.; Ajona, D.; Ortiz-Espinosa, S.; Mantovani, A.; Lambris, J.D. Complementing the Cancer-Immunity Cycle. Front. Immunol. 2019, 10, 774. [Google Scholar] [CrossRef]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 636568. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Koustas, E.; Sarantis, P.; Papavassiliou, A.G.; Karamouzis, M.V. The Resistance Mechanisms of Checkpoint Inhibitors in Solid Tumors. Biomolecules 2020, 10, 666. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pascual, J.; Ayuso-Sacido, A.; Belda-Iniesta, C. Drug Resistance in Cancer Immunotherapy: New Strategies to Improve Checkpoint Inhibitor Therapies. Cancer Drug Resist. 2019, 2, 980–993. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Gadir, A.; Massoud, A.H.; Chatila, T.A. Antigen-Specific Treg Cells in Immunological Tolerance: Implications for Allergic Diseases. F1000Research 2018, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Kim, B.S.; Lee, S.-K. Regulatory T Cells in Tumor Microenvironment and Approach for Anticancer Immunotherapy. Immune Netw. 2020, 20, e4. [Google Scholar] [CrossRef]
- Frankel, T.; Lanfranca, M.P.; Zou, W. The Role of Tumor Microenvironment in Cancer Immunotherapy. Adv. Exp. Med. Biol. 2017, 1036, 51–64. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) Cells in Cancer: Can Treg Cells Be a New Therapeutic Target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef]
- Jacob, J.B.; Jacob, M.K.; Parajuli, P. Review of Immune Checkpoint Inhibitors in Immuno-Oncology. Adv. Pharmacol. 2021, 91, 111–139. [Google Scholar] [CrossRef]
- Young, K.; Hughes, D.J.; Cunningham, D.; Starling, N. Immunotherapy and Pancreatic Cancer: Unique Challenges and Potential Opportunities. Ther. Adv. Med. Oncol. 2018, 10, 1758835918816281. [Google Scholar] [CrossRef]
- Naran, K.; Nundalall, T.; Chetty, S.; Barth, S. Principles of Immunotherapy: Implications for Treatment Strategies in Cancer and Infectious Diseases. Front. Microbiol. 2018, 9, 3158. [Google Scholar] [CrossRef] [PubMed]
- Dalgleish, A.G. Vaccines versus Immunotherapy: Overview of Approaches in Deciding between Options. Hum. Vaccin. Immunother. 2014, 10, 3369–3374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Zhong, J.F.; Zhang, X. Engineering CAR-T Cells. Biomark. Res. 2017, 5, 22. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The Fourth Generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Mehrabadi, A.Z.; Ranjbar, R.; Farzanehpour, M.; Shahriary, A.; Dorostkar, R.; Hamidinejad, M.A.; Ghaleh, H.E.G. Therapeutic Potential of CAR T Cell in Malignancies: A Scoping Review. Biomed. Pharmacother. 2022, 146, 112512. [Google Scholar] [CrossRef]
- Runcie, K.; Budman, D.R.; John, V.; Seetharamu, N. Bi-Specific and Tri-Specific Antibodies- the next Big Thing in Solid Tumor Therapeutics. Mol. Med. 2018, 24, 50. [Google Scholar] [CrossRef]
- Ellis, G.I.; Riley, J.L. How to Kill Treg Cells for Immunotherapy. Nat Cancer 2020, 1, 1134–1135. [Google Scholar] [CrossRef]
- Miller, K.D.; Siegel, R.L.; Lin, C.C.; Mariotto, A.B.; Kramer, J.L.; Rowland, J.H.; Stein, K.D.; Alteri, R.; Jemal, A. Cancer Treatment and Survivorship Statistics, 2016. CA Cancer J. Clin. 2016, 66, 271–289. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018: Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Schizas, D.; Charalampakis, N.; Kole, C.; Economopoulou, P.; Koustas, E.; Gkotsis, E.; Ziogas, D.; Psyrri, A.; Karamouzis, M.V. Immunotherapy for Pancreatic Cancer: A 2020 Update. Cancer Treat. Rev. 2020, 86, 102016. [Google Scholar] [CrossRef]
- Pourshams, A.; Sepanlou, S.G.; Ikuta, K.S.; Bisignano, C.; Safiri, S.; Roshandel, G.; Sharif, M.; Khatibian, M.; Fitzmaurice, C.; Nixon, M.R.; et al. The Global, Regional, and National Burden of Pancreatic Cancer and Its Attributable Risk Factors in 195 Countries and Territories, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2019, 4, 934–947. [Google Scholar] [CrossRef]
- Klein, A.P. Pancreatic Cancer Epidemiology: Understanding the Role of Lifestyle and Inherited Risk Factors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Gandini, S.; Lowenfels, A.B.; Jaffee, E.M.; Armstrong, T.D.; Maisonneuve, P. Allergies and the Risk of Pancreatic Cancer: A Meta-Analysis with Review of Epidemiology and Biological Mechanisms. Cancer Epidemiol. Biomark. Prev. 2005, 14, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Corbo, V.; Tortora, G.; Scarpa, A. Molecular Pathology of Pancreatic Cancer: From Bench-to-Bedside Translation. Curr. Drug Targets 2012, 13, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Shadhu, K.; Xi, C. Inflammation and Pancreatic Cancer: An Updated Review. Saudi J. Gastroenterol. 2019, 25, 3. [Google Scholar] [CrossRef]
- Manohar, M.; Verma, A.K.; Venkateshaiah, S.U.; Sanders, N.L.; Mishra, A. Pathogenic Mechanisms of Pancreatitis. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 10. [Google Scholar] [CrossRef]
- Hidalgo, M.; Álvarez, R.; Gallego, J.; Guillén-Ponce, C.; Laquente, B.; Macarulla, T.; Muñoz, A.; Salgado, M.; Vera, R.; Adeva, J.; et al. Consensus Guidelines for Diagnosis, Treatment and Follow-up of Patients with Pancreatic Cancer in Spain. Clin. Transl. Oncol. 2017, 19, 667–681. [Google Scholar] [CrossRef]
- Thomas, D.; Radhakrishnan, P. Tumor-Stromal Crosstalk in Pancreatic Cancer and Tissue Fibrosis. Mol. Cancer 2019, 18. [Google Scholar] [CrossRef]
- Hamada, S.; Masamune, A. Elucidating the Link between Collagen and Pancreatic Cancer: What’s next? Expert Rev. Gastroenterol. Hepatol. 2018, 12, 315–317. [Google Scholar] [CrossRef]
- Orhan, A.; Vogelsang, R.P.; Andersen, M.B.; Madsen, M.T.; Hölmich, E.R.; Raskov, H.; Gögenur, I. The Prognostic Value of Tumour-Infiltrating Lymphocytes in Pancreatic Cancer: A Systematic Review and Meta-Analysis. Eur. J. Cancer 2020, 132, 71–84. [Google Scholar] [CrossRef]
- Kole, C.; Charalampakis, N.; Tsakatikas, S.; Frountzas, M.; Apostolou, K.; Schizas, D. Immunotherapy in Combination with Well-Established Treatment Strategies in Pancreatic Cancer: Current Insights. Cancer Manag. Res. 2022, 14, 1043–1061. [Google Scholar] [CrossRef] [PubMed]
- Schmiechen, Z.C.; Stromnes, I.M. Mechanisms Governing Immunotherapy Resistance in Pancreatic Ductal Adenocarcinoma. Front. Immunol. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A. Combination of CTLA-4 and PD-1 Blockers for Treatment of Cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef] [PubMed]
- Rahma, O.E.; Katz, M.H.G.; Wolpin, B.M.; Dias-Costa, A.; Nowak, J.; Rodig, S.J.; Dougan, S.; Bekaii-Saab, T.S.; Stucky, C.-C.H.; Elias, R.; et al. Randomized Multicenter Phase Ib/II Study of Neoadjuvant Chemoradiation Therapy (CRT) Alone or in Combination with Pembrolizumab in Patients with Resectable or Borderline Resectable Pancreatic Cancer. J. Clin. Oncol. 2021, 39, 4128. [Google Scholar] [CrossRef]
- Davelaar, J.; Brown, Z.; Linden, S.; Rodriguez, C.; Elmadbouh, O.; Pachter, J.A.; Gong, J.; Hendifar, A.E.; Lo, S.; Gaddam, S.; et al. Trial in Progress: A Randomized Phase II Study of Pembrolizumab with or without Defactinib, a Focal Adhesion Kinase Inhibitor, Following Chemotherapy as a Neoadjuvant and Adjuvant Treatment for Resectable Pancreatic Ductal Adenocarcinoma (PDAC). J. Clin. Oncol. 2022, 40. [Google Scholar] [CrossRef]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schütz, E.; Khemka, V. Phase Ib/II Study of Gemcitabine, Nab-Paclitaxel, and Pembrolizumab in Metastatic Pancreatic Adenocarcinoma. Investig. New Drugs 2018, 36, 96–102. [Google Scholar] [CrossRef]
- Aglietta, M.; Barone, C.; Sawyer, M.B.; Moore, M.J.; Miller, W.H., Jr.; Bagalà, C.; Colombi, F.; Cagnazzo, C.; Gioeni, L.; Wang, E.; et al. A Phase I Dose Escalation Trial of Tremelimumab (CP-675,206) in Combination with Gemcitabine in Chemotherapy-Naive Patients with Metastatic Pancreatic Cancer. Ann. Oncol. 2014, 25, 1750–1755. [Google Scholar] [CrossRef]
- Kernel Networks Inc. Combination of Anti-PD-1 Antibody and Chemotherapy in Metastatic Pancreatic Cancer. Case Med. Res. 2019. [Google Scholar] [CrossRef]
- Kamath, S.D.; Kalyan, A.; Kircher, S.; Nimeiri, H.; Fought, A.J.; Benson, A., III; Mulcahy, M. Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A Phase Ib Study. Oncologist 2020, 25, e808–e815. [Google Scholar] [CrossRef]
- Fong, Y.; Ady, J.; Heffner, J.; Klein, E. Oncolytic Viral Therapy for Pancreatic Cancer: Current Research and Future Directions. Oncolytic Virother. 2014, 3, 35–46. [Google Scholar] [CrossRef]
- Nisar, M.; Paracha, R.Z.; Adil, S.; Qureshi, S.N.; Janjua, H.A. An Extensive Review on Preclinical and Clinical Trials of Oncolytic Viruses Therapy for Pancreatic Cancer. Front. Oncol. 2022, 12, 875188. [Google Scholar] [CrossRef] [PubMed]
- Bear, A.S.; Vonderheide, R.H.; O’Hara, M.H. Challenges and Opportunities for Pancreatic Cancer Immunotherapy. Cancer Cell 2020, 38, 788–802. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; You, L.; Dai, M.; Zhao, Y. Mucins in Pancreatic Cancer: A Well-Established but Promising Family for Diagnosis, Prognosis and Therapy. J. Cell. Mol. Med. 2020, 24, 10279–10289. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.A.; Bever, K.M.; Ho, W.J.; Fertig, E.J.; Niu, N.; Zheng, L.; Parkinson, R.M.; Durham, J.N.; Onners, B.; Ferguson, A.K.; et al. A Phase II Study of Allogeneic GM-CSF–Transfected Pancreatic Tumor Vaccine (GVAX) with Ipilimumab as Maintenance Treatment for Metastatic Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 5129–5139. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, J.-A.; Zhang, H.-X.; Jiang, Y.-N.; Luo, W.-H. Cancer Vaccines: Targeting KRAS-Driven Cancers. Expert Rev. Vaccines 2020, 19, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Dodson, L.F.; Hawkins, W.G.; Goedegebuure, P. Potential Targets for Pancreatic Cancer Immunotherapeutics. Immunotherapy 2011, 3, 517–537. [Google Scholar] [CrossRef]
- Luo, W.; Yang, G.; Luo, W.; Cao, Z.; Liu, Y.; Qiu, J.; Chen, G.; You, L.; Zhao, F.; Zheng, L.; et al. Novel Therapeutic Strategies and Perspectives for Metastatic Pancreatic Cancer: Vaccine Therapy Is More than Just a Theory. Cancer Cell Int. 2020, 20, 66. [Google Scholar] [CrossRef]
- Hartley, M.L.; Bade, N.A.; Prins, P.A.; Ampie, L.; Marshall, J.L. Pancreatic Cancer, Treatment Options, and GI-4000. Hum. Vaccin. Immunother. 2014, 10, 3347–3353. [Google Scholar] [CrossRef]
- Ali, A.I.; Oliver, A.J.; Samiei, T.; Chan, J.D.; Kershaw, M.H.; Slaney, C.Y. Genetic Redirection of T Cells for the Treatment of Pancreatic Cancer. Front. Oncol. 2019, 9, 56. [Google Scholar] [CrossRef]
- Akce, M.; Zaidi, M.Y.; Waller, E.K.; El-Rayes, B.F.; Lesinski, G.B. The Potential of CAR T Cell Therapy in Pancreatic Cancer. Front. Immunol. 2018, 9, 2166. [Google Scholar] [CrossRef]
- Yeo, D.; Giardina, C.; Saxena, P.; Rasko, J.E.J. The next Wave of Cellular Immunotherapies in Pancreatic Cancer. Mol. Ther. Oncolytics 2022, 24, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Wolpin, B.M.; Hezel, A.F.; Abrams, T.; Blaszkowsky, L.S.; Meyerhardt, J.A.; Chan, J.A.; Enzinger, P.C.; Allen, B.; Clark, J.W.; Ryan, D.P.; et al. Oral MTOR Inhibitor Everolimus in Patients with Gemcitabine-Refractory Metastatic Pancreatic Cancer. J. Clin. Oncol. 2009, 27, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.M.; Shroff, R.T.; Xiong, H.; Varadhachary, G.A.; Fogelman, D.; Reddy, S.A.; Davis, D.; Zhang, Y.; Wolff, R.A.; Abbruzzese, J.L. Inhibition of the Mammalian Target of Rapamycin (MTOR) in Advanced Pancreatic Cancer: Results of Two Phase II Studies. BMC Cancer 2010, 10, 368. [Google Scholar] [CrossRef] [PubMed]
- Fearon, D.T. The Carcinoma-Associated Fibroblast Expressing Fibroblast Activation Protein and Escape from Immune Surveillance. Cancer Immunol. Res. 2014, 2, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.M.; Horton, K.J.; Coveler, A.L.; Hingorani, S.R.; Harris, W.P. Targeting the Tumor Stroma: The Biology and Clinical Development of Pegylated Recombinant Human Hyaluronidase (PEGPH20). Curr. Oncol. Rep. 2017, 19, 47. [Google Scholar] [CrossRef] [PubMed]
- DuFort, C.C.; DelGiorno, K.E.; Hingorani, S.R. Mounting Pressure in the Microenvironment: Fluids, Solids, and Cells in Pancreatic Ductal Adenocarcinoma. Gastroenterology 2016, 150, 1545–1557. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes with Anti–PD-L1 Immunotherapy in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Tamura, R.; Tanaka, T.; Akasaki, Y.; Murayama, Y.; Yoshida, K.; Sasaki, H. The Role of Vascular Endothelial Growth Factor in the Hypoxic and Immunosuppressive Tumor Microenvironment: Perspectives for Therapeutic Implications. Med. Oncol. 2020, 37, 2. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 Comes of Age as an Inhibitory Receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef]
- Meireson, A.; Devos, M.; Brochez, L. IDO Expression in Cancer: Different Compartment, Different Functionality? Front. Immunol. 2020, 11, 531491. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Pan, Y.; Fei, Q.; Lin, Y.; Zhou, Y.; Liu, Y.; Guan, H.; Yu, X.; Lin, X.; Lu, F.; et al. Prognostic Significance and Therapeutic Potential of the Immune Checkpoint VISTA in Pancreatic Cancer. J. Cancer Res. Clin. Oncol. 2021, 147, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Biasci, D.; Smoragiewicz, M.; Connell, C.M.; Wang, Z.; Gao, Y.; Thaventhiran, J.E.D.; Basu, B.; Magiera, L.; Johnson, T.I.; Bax, L.; et al. CXCR4 Inhibition in Human Pancreatic and Colorectal Cancers Induces an Integrated Immune Response. Proc. Natl. Acad. Sci. USA 2020, 117, 28960–28970. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Guo, S.; Zhang, Y.; Ma, Y.; Guo, X.; Zhou, X.; Yu, Q. Efficacy and Safety of KN046 plus Nab-Paclitaxel/Gemcitabine as First-Line Treatment for Unresectable Locally Advanced or Metastatic Pancreatic Ductal Adenocarcinoma (PDAC). J. Clin. Oncol. 2021, 39, 4138. [Google Scholar] [CrossRef]
- Lum, L.G.; Thakur, A.; Choi, M.; Deol, A.; Kondadasula, V.; Schalk, D.; Fields, K.; Dufrense, M.; Philip, P.; Dyson, G.; et al. Clinical and Immune Responses to Anti-CD3 x Anti-EGFR Bispecific Antibody Armed Activated T Cells (EGFR BATs) in Pancreatic Cancer Patients. Oncoimmunology 2020, 9, 1773201. [Google Scholar] [CrossRef]
- Koustas, E.; Karamouzis, M.V.; Sarantis, P.; Schizas, D.; Papavassiliou, A.G. Inhibition of C-MET Increases the Antitumour Activity of PARP Inhibitors in Gastric Cancer Models. J. Cell. Mol. Med. 2020, 24, 10420–10431. [Google Scholar] [CrossRef]
- Koustas, E.; Trifylli, E.-M.; Sarantis, P.; Kontolatis, N.I.; Damaskos, C.; Garmpis, N.; Vallilas, C.; Garmpi, A.; Papavassiliou, A.G.; Karamouzis, M.V. The Implication of Autophagy in Gastric Cancer Progression. Life 2021, 11, 1304. [Google Scholar] [CrossRef]
- Setia, N.; Clark, J.W.; Duda, D.G.; Hong, T.S.; Kwak, E.L.; Mullen, J.T.; Lauwers, G.Y. Familial Gastric Cancers. Oncologist 2015, 20, 1365–1377. [Google Scholar] [CrossRef]
- Garattini, S.K.; Basile, D.; Cattaneo, M.; Fanotto, V.; Ongaro, E.; Bonotto, M.; Negri, F.V.; Berenato, R.; Ermacora, P.; Cardellino, G.G.; et al. Molecular Classifications of Gastric Cancers: Novel Insights and Possible Future Applications. World J. Gastrointest. Oncol. 2017, 9, 194. [Google Scholar] [CrossRef]
- Zhang, D.; He, W.; Wu, C.; Tan, Y.; He, Y.; Xu, B.; Chen, L.; Li, Q.; Jiang, J. Scoring System for Tumor-Infiltrating Lymphocytes and Its Prognostic Value for Gastric Cancer. Front. Immunol. 2019, 10, 71. [Google Scholar] [CrossRef]
- Tian, C.; Jing, H.; Wang, C.; Wang, W.; Cui, Y.; Chen, J.; Sha, D. Prognostic Role of Tumour-Infiltrating Lymphocytes Assessed by H&E-Stained Section in Gastric Cancer: A Systematic Review and Meta-Analysis. BMJ Open 2021, 11, e044163. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Choi, M.G.; Kim, K.; Kim, K.-M.; Kim, S.T.; Park, S.H.; Cristescu, R.; Peter, S.; Lee, J. High PD-L1 Expression in Gastric Cancer (GC) Patients and Correlation with Molecular Features. Pathol. Res. Pract. 2020, 216, 152881. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Chen, M.; Guo, D.; Zhu, H.; Zhang, W.; Pan, J.; Zhong, X.; Li, X.; Qian, H.; Wang, X. PD-L1 and Gastric Cancer Prognosis: A Systematic Review and Meta-Analysis. PLoS ONE 2017, 12, e0182692. [Google Scholar] [CrossRef] [PubMed]
- Kawazoe, A.; Shitara, K.; Boku, N.; Yoshikawa, T.; Terashima, M. Current Status of Immunotherapy for Advanced Gastric Cancer. Jpn. J. Clin. Oncol. 2021, 51, 20–27. [Google Scholar] [CrossRef]
- Sasaki, S.; Nishikawa, J.; Sakai, K.; Iizasa, H.; Yoshiyama, H.; Yanagihara, M.; Shuto, T.; Shimokuri, K.; Kanda, T.; Suehiro, Y.; et al. EBV-Associated Gastric Cancer Evades T-Cell Immunity by PD-1/PD-L1 Interactions. Gastric Cancer 2019, 22, 486–496. [Google Scholar] [CrossRef]
- Böger, C.; Behrens, H.-M.; Mathiak, M.; Krüger, S.; Kalthoff, H.; Röcken, C. PD-L1 Is an Independent Prognostic Predictor in Gastric Cancer of Western Patients. Oncotarget 2016, 7, 24269–24283. [Google Scholar] [CrossRef]
- Kawazoe, A.; Kuwata, T.; Kuboki, Y.; Shitara, K.; Nagatsuma, A.K.; Aizawa, M.; Yoshino, T.; Doi, T.; Ohtsu, A.; Ochiai, A. Clinicopathological Features of Programmed Death Ligand 1 Expression with Tumor-Infiltrating Lymphocyte, Mismatch Repair, and Epstein–Barr Virus Status in a Large Cohort of Gastric Cancer Patients. Gastric Cancer 2017, 20, 407–415. [Google Scholar] [CrossRef]
- Thompson, E.D.; Zahurak, M.; Murphy, A.; Cornish, T.; Cuka, N.; Abdelfatah, E.; Yang, S.; Duncan, M.; Ahuja, N.; Taube, J.M.; et al. Patterns of PD-L1 Expression and CD8 T Cell Infiltration in Gastric Adenocarcinomas and Associated Immune Stroma. Gut 2017, 66, 794–801. [Google Scholar] [CrossRef]
- Coutzac, C.; Pernot, S.; Chaput, N.; Zaanan, A. Immunotherapy in Advanced Gastric Cancer, Is It the Future? Crit. Rev. Oncol. Hematol. 2019, 133, 25–32. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, F.; Zhou, N.; Gu, Y.-M.; Zhang, Y.-T.; He, Y.-D.; Wang, L.; Yang, L.-X.; Zhao, Y.; Li, Y.-M. Efficacy and Safety of Immune Checkpoint Inhibitors in Advanced Gastric or Gastroesophageal Junction Cancer: A Systematic Review and Meta-Analysis. Oncoimmunology 2019, 8, e1581547. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Kawazoe, A.; Yañez, P.; Li, N.; Lonardi, S.; Kolesnik, O.; Barajas, O.; Bai, Y.; Shen, L.; Tang, Y.; et al. The KEYNOTE-811 Trial of Dual PD-1 and HER2 Blockade in HER2-Positive Gastric Cancer. Nature 2021, 600, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-T.; Satoh, T.; Ryu, M.-H.; Chao, Y.; Kato, K.; Chung, H.C.; Chen, J.-S.; Muro, K.; Kang, W.K.; Yeh, K.-H.; et al. A Phase 3 Study of Nivolumab in Previously Treated Advanced Gastric or Gastroesophageal Junction Cancer (ATTRACTION-2): 2-Year Update Data. Gastric Cancer 2020, 23, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.-P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients with Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.P.; Cao, H.; Chen, W.; Mahmood, K.; Phillips, T.; Sutton, L.; Cato, A. Gastrin Vaccine Alone and in Combination with an Immune Checkpoint Antibody Inhibits Growth and Metastases of Gastric Cancer. Front. Oncol. 2021, 11, 788875. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.A.; Randolph Hecht, J.; Ho, L.; Baker, J.; Oortgiesen, M.; Eduljee, A.; Michaeli, D. An Open-Label, Multinational, Multicenter Study of G17DT Vaccination Combined with Cisplatin and 5-Fluorouracil in Patients with Untreated, Advanced Gastric or Gastroesophageal Cancer: The GC4 Study. Cancer 2006, 106, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Masuzawa, T.; Fujiwara, Y.; Okada, K.; Nakamura, A.; Takiguchi, S.; Nakajima, K.; Miyata, H.; Yamasaki, M.; Kurokawa, Y.; Osawa, R.; et al. Phase I/II Study of S-1 plus Cisplatin Combined with Peptide Vaccines for Human Vascular Endothelial Growth Factor Receptor 1 and 2 in Patients with Advanced Gastric Cancer. Int. J. Oncol. 2012, 41, 1297–1304. [Google Scholar] [CrossRef]
- Bębnowska, D.; Grywalska, E.; Niedźwiedzka-Rystwej, P.; Sosnowska-Pasiarska, B.; Smok-Kalwat, J.; Pasiarski, M.; Góźdź, S.; Roliński, J.; Polkowski, W. CAR-T Cell Therapy—An Overview of Targets in Gastric Cancer. J. Clin. Med. 2020, 9, 1894. [Google Scholar] [CrossRef]
- Giannitrapani, L.; Zerbo, M.; Amodeo, S.; Pipitone, E.; Galia, M.; Li Cavoli, T.V.; Minissale, M.G.; Licata, A.; Schiavone, C.; Brancatelli, G.; et al. The Changing Epidemiology of Hepatocellular Carcinoma: Experience of a Single Center. Biomed Res. Int. 2020, 2020, 5309307. [Google Scholar] [CrossRef]
- Petruzziello, A. Epidemiology of Hepatitis B Virus (HBV) and Hepatitis C Virus (HCV) Related Hepatocellular Carcinoma. Open Virol. J. 2018, 12, 26–32. [Google Scholar] [CrossRef]
- Crissien, A.M.; Frenette, C. Current Management of Hepatocellular Carcinoma. Gastroenterol. Hepatol. (N. Y.) 2014, 10, 153–161. [Google Scholar]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.-L.; Kang, Y.-K.; Chen, Z.; Tsao, C.-J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.-S.; et al. Efficacy and Safety of Sorafenib in Patients in the Asia-Pacific Region with Advanced Hepatocellular Carcinoma: A Phase III Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus Sorafenib in First-Line Treatment of Patients with Unresectable Hepatocellular Carcinoma: A Randomised Phase 3 Non-Inferiority Trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef]
- Vogel, A.; Qin, S.; Kudo, M.; Su, Y.; Hudgens, S.; Yamashita, T.; Yoon, J.-H.; Fartoux, L.; Simon, K.; López, C.; et al. Lenvatinib versus Sorafenib for First-Line Treatment of Unresectable Hepatocellular Carcinoma: Patient-Reported Outcomes from a Randomised, Open-Label, Non-Inferiority, Phase 3 Trial. Lancet Gastroenterol. Hepatol. 2021, 6, 649–658. [Google Scholar] [CrossRef]
- Johnston, M.P.; Khakoo, S.I. Immunotherapy for Hepatocellular Carcinoma: Current and Future. World J. Gastroenterol. 2019, 25, 2977–2989. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.-W.; Finn, R.S.; Cheng, A.-L.; Mathurin, P.; Edeline, J.; Kudo, M.; Harding, J.J.; Merle, P.; Rosmorduc, O.; et al. Nivolumab versus Sorafenib in Advanced Hepatocellular Carcinoma (CheckMate 459): A Randomised, Multicentre, Open-Label, Phase 3 Trial. Lancet Oncol. 2022, 23, 77–90. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.-K.; Kim, T.-Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.-M.; Matilla, A.; et al. Efficacy and Safety of Nivolumab plus Ipilimumab in Patients with Advanced Hepatocellular Carcinoma Previously Treated with Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab as Second-Line Therapy in Patients with Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Ren, Z.; Xu, J.; Bai, Y.; Xu, A.; Cang, S.; Du, C.; Li, Q.; Lu, Y.; Chen, Y.; Guo, Y.; et al. Sintilimab plus a Bevacizumab Biosimilar (IBI305) versus Sorafenib in Unresectable Hepatocellular Carcinoma (ORIENT-32): A Randomised, Open-Label, Phase 2–3 Study. Lancet Oncol. 2021, 22, 977–990. [Google Scholar] [CrossRef]
- Trifylli, E.-M.; Koustas, E.; Papadopoulos, N.; Sarantis, P.; Aloizos, G.; Damaskos, C.; Garmpis, N.; Garmpi, A.; Karamouzis, M.V. An Insight into the Novel Immunotherapy and Targeted Therapeutic Strategies for Hepatocellular Carcinoma and Cholangiocarcinoma. Life 2022, 12, 665. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shen, Y.; Zhao, R.; Samudio, I.; Jia, W.; Bai, X.; Liang, T. Oncolytic Virotherapy in Hepato-bilio-pancreatic Cancer: The Key to Breaking the Log Jam? Cancer Med. 2020, 9, 2943–2959. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kudo, M.; Cheng, A.-L.; Finn, R.S.; Galle, P.R.; Kaneko, S.; Meyer, T.; Qin, S.; Dutcus, C.E.; Chen, E.; et al. Lenvatinib (Len) plus Pembrolizumab (Pembro) for the First-Line Treatment of Patients (Pts) with Advanced Hepatocellular Carcinoma (HCC): Phase 3 LEAP-002 Study. J. Clin. Oncol. 2019, 37. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Kudo, M.; Lau, G.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Dao, T.V.; De Toni, E.N.; et al. Phase 3 Randomized, Open-Label, Multicenter Study of Tremelimumab (T) and Durvalumab (D) as First-Line Therapy in Patients (Pts) with Unresectable Hepatocellular Carcinoma (UHCC): HIMALAYA. J. Clin. Oncol. 2022, 40, 379. [Google Scholar] [CrossRef]
- Guse, K.; Cerullo, V.; Hemminki, A. Oncolytic Vaccinia Virus for the Treatment of Cancer. Expert Opin. Biol. Ther. 2011, 11, 595–608. [Google Scholar] [CrossRef]
- Heo, J.; Breitbach, C.J.; Moon, A.; Kim, C.W.; Patt, R.; Kim, M.K.; Lee, Y.K.; Oh, S.Y.; Woo, H.Y.; Parato, K.; et al. Sequential Therapy with JX-594, A Targeted Oncolytic Poxvirus, Followed by Sorafenib in Hepatocellular Carcinoma: Preclinical and Clinical Demonstration of Combination Efficacy. Mol. Ther. 2011, 19, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Moehler, M.; Heo, J.; Lee, H.C.; Tak, W.Y.; Chao, Y.; Paik, S.W.; Yim, H.J.; Byun, K.S.; Baron, A.; Ungerechts, G.; et al. Vaccinia-Based Oncolytic Immunotherapy Pexastimogene Devacirepvec in Patients with Advanced Hepatocellular Carcinoma after Sorafenib Failure: A Randomized Multicenter Phase IIb Trial (TRAVERSE). Oncoimmunology 2019, 8, 1615817. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Mayer-Mokler, A.; Accolla, R.; Ma, Y.T.; Heidenreich, R.; Avallone, A.; Simeone, E.; Koenigsrainer, A.; Loeffler, M.; Gouttefangeas, C.; et al. HepaVac-101 First-in-Man Therapeutic Cancer Vaccine Phase I/II Clinical Trial for Hepatocellular Carcinoma Patients. J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination with Ipilimumab versus Ipilimumab Alone in Patients with Advanced, Unresectable Melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef]
- Puzanov, I.; Chesney, J.; Collichio, F.; Singh, P.; Milhem, M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. 433 Talimogene Laherparepvec (T-VEC) in Combination with Ipilimumab (IPI) versus IPI Alone for Advanced Melanoma: 4-Year Interim Analysis of a Randomized, Open-Label, Phase 2 Trial. In Regular and Young Investigator Award Abstracts; BMJ Publishing Group Ltd.: London, UK, 2020; Volume 8. [Google Scholar]
- He, Y.; Hong, Y.; Mizejewski, G.J. Engineering α-Fetoprotein-Based Gene Vaccines to Prevent and Treat Hepatocellular Carcinoma: Review and Future Prospects. Immunotherapy 2014, 6, 725–736. [Google Scholar] [CrossRef]
- Rochigneux, P.; Chanez, B.; De Rauglaudre, B.; Mitry, E.; Chabannon, C.; Gilabert, M. Adoptive Cell Therapy in Hepatocellular Carcinoma: Biological Rationale and First Results in Early Phase Clinical Trials. Cancers 2021, 13, 271. [Google Scholar] [CrossRef] [PubMed]
- Roddy, H.; Meyer, T.; Roddie, C. Novel Cellular Therapies for Hepatocellular Carcinoma. Cancers 2022, 14, 504. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Tang, Q. Recent Updates on Chimeric Antigen Receptor T Cell Therapy for Hepatocellular Carcinoma. Cancer Gene Ther. 2021, 28, 1075–1087. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H. Update of Early Phase Clinical Trials in Cancer Immunotherapy. BMB Rep. 2021, 54, 70–88. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Ricci, A.D. PD-L1, TMB, and Other Potential Predictors of Response to Immunotherapy for Hepatocellular Carcinoma: How Can They Assist Drug Clinical Trials? Expert Opin. Investig. Drugs 2022, 31, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Ricci, A.D.; Di Federico, A.; Frega, G.; Palloni, A.; Tavolari, S.; Brandi, G. Predictive Biomarkers for Checkpoint Inhibitor-Based Immunotherapy in Hepatocellular Carcinoma: Where Do We Stand? Front. Oncol. 2021, 11, 803133. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Brandi, G. Biochemical Predictors of Response to Immune Checkpoint Inhibitors in Unresectable Hepatocellular Carcinoma. Cancer Treat. Res. Commun. 2021, 27, 100328. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Greten, T.F. Gut Microbiome in HCC-Mechanisms, Diagnosis and Therapy. J. Hepatol. 2020, 72, 230–238. [Google Scholar] [CrossRef]
- Amaro, A.; Chiara, S.; Pfeffer, U. Molecular Evolution of Colorectal Cancer: From Multistep Carcinogenesis to the Big Bang. Cancer Metastasis Rev. 2016, 35, 63–74. [Google Scholar] [CrossRef]
- Koustas, E.; Karamouzis, M.V.; Mihailidou, C.; Schizas, D.; Papavassiliou, A.G. Co-Targeting of EGFR and Autophagy Signaling Is an Emerging Treatment Strategy in Metastatic Colorectal Cancer. Cancer Lett. 2017, 396, 94–102. [Google Scholar] [CrossRef]
- Koustas, E.; Papavassiliou, A.G.; Karamouzis, M.V. The Role of Autophagy in the Treatment of BRAF Mutant Colorectal Carcinomas Differs Based on Microsatellite Instability Status. PLoS ONE 2018, 13, e0207227. [Google Scholar] [CrossRef]
- Saridaki, Z.; Papadatos-Pastos, D.; Tzardi, M.; Mavroudis, D.; Bairaktari, E.; Arvanity, H.; Stathopoulos, E.; Georgoulias, V.; Souglakos, J. BRAF Mutations, Microsatellite Instability Status and Cyclin D1 Expression Predict Metastatic Colorectal Patients’ Outcome. Br. J. Cancer 2010, 102, 1762–1768. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grassi, E.; Corbelli, J.; Papiani, G.; Barbera, M.A.; Gazzaneo, F.; Tamberi, S. Current Therapeutic Strategies in BRAF-Mutant Metastatic Colorectal Cancer. Front. Oncol. 2021, 11, 601722. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Mullins, C.S.; Linnebacher, M. Colorectal Cancer Vaccines: Tumor-Associated Antigens vs Neoantigens. World J. Gastroenterol. 2018, 24, 5418–5432. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tian, T.; Zhang, J. Tumor-Associated Macrophages (TAMs) in Colorectal Cancer (CRC): From Mechanism to Therapy and Prognosis. Int. J. Mol. Sci. 2021, 22, 8470. [Google Scholar] [CrossRef] [PubMed]
- Sieminska, I.; Baran, J. Myeloid-Derived Suppressor Cells in Colorectal Cancer. Front. Immunol. 2020, 11, 1526. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yao, Z.; Wang, J.; Zhang, W.; Yang, Y.; Zhang, Y.; Qu, X.; Zhu, Y.; Zou, J.; Peng, S.; et al. Correction: Macrophage-Derived CCL5 Facilitates Immune Escape of Colorectal Cancer Cells via the P65/STAT3-CSN5-PD-L1 Pathway. Cell Death Differ. 2020, 27, 2293. [Google Scholar] [CrossRef]
- Pyo, J.; Park, H.-J. Treatment Efficacy of Immune Checkpoint Inhibitors for Patients with Advanced or Metastatic Colorectal Cancer: A Systematic Review and Meta-Analysis. J. Clin. Med. 2021, 10, 3599. [Google Scholar] [CrossRef]
- Lenz, H.-J.; Van Cutsem, E.; Luisa Limon, M.; Wong, K.Y.M.; Hendlisz, A.; Aglietta, M.; García-Alfonso, P.; Neyns, B.; Luppi, G.; Cardin, D.B.; et al. First-Line Nivolumab plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. J. Clin. Oncol. 2022, 40, 161–170. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Azadi, A.; Golchini, A.; Delazar, S.; Abarghooi Kahaki, F.; Dehnavi, S.M.; Payandeh, Z.; Eyvazi, S. Recent Advances on Immune Targeted Therapy of Colorectal Cancer Using Bi-Specific Antibodies and Therapeutic Vaccines. Biol. Proced. Online 2021, 23, 13. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Hoffner, B.; Gasal, E.; Hong, J.; Carvajal, R.D. Oncolytic Immunotherapy: Unlocking the Potential of Viruses to Help Target Cancer. Cancer Immunol. Immunother. 2017, 66, 1249–1264. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.S.; Hecht, J.R.; Chan, E. Talimogene Laherparepvec: Review of Its Mechanism of Action and Clinical Efficacy and Safety. Immunotherapy 2019, 11, 705–723. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Lenz, H.-J.; Marshall, J.; Singh, D.; Garett, C.; Cripps, C.; Moore, M.; von Mehren, M.; Dalfen, R.; Heim, W.J.; et al. Combination Chemotherapy and ALVAC-CEA/B7.1 Vaccine in Patients with Metastatic Colorectal Cancer. Clin. Cancer Res. 2008, 14, 4843–4849. [Google Scholar] [CrossRef] [PubMed]
- Sarvizadeh, M.; Ghasemi, F.; Tavakoli, F.; Sadat Khatami, S.; Razi, E.; Sharifi, H.; Biouki, N.M.; Taghizadeh, M. Vaccines for Colorectal Cancer: An Update. J. Cell. Biochem. 2019, 120, 8815–8828. [Google Scholar] [CrossRef]
- Lugini, L.; Valtieri, M.; Federici, C.; Cecchetti, S.; Meschini, S.; Condello, M.; Signore, M.; Fais, S. Exosomes from Human Colorectal Cancer Induce a Tumor-like Behavior in Colonic Mesenchymal Stromal Cells. Oncotarget 2016, 7, 50086–50098. [Google Scholar] [CrossRef]
- Huda, M.N.; Nurunnabi, M. Potential Application of Exosomes in Vaccine Development and Delivery. Pharm. Res. 2022, 1–37. [Google Scholar] [CrossRef]
- Cho, J.-A.; Lee, Y.-S.; Kim, S.-H.; Ko, J.-K.; Kim, C.-W. MHC Independent Anti-Tumor Immune Responses Induced by Hsp70-Enriched Exosomes Generate Tumor Regression in Murine Models. Cancer Lett. 2009, 275, 256–265. [Google Scholar] [CrossRef]
- Li, H.; Yang, C.; Cheng, H.; Huang, S.; Zheng, Y. CAR-T Cells for Colorectal Cancer: Target-Selection and Strategies for Improved Activity and Safety. J. Cancer 2021, 12, 1804–1814. [Google Scholar] [CrossRef]
- Koustas, E.; Trifylli, E.-M.; Sarantis, P.; Papavassiliou, A.G.; Karamouzis, M.V. Role of Autophagy in Cholangiocarcinoma: An Autophagy-Based Treatment Strategy. World J. Gastrointest. Oncol. 2021, 13, 1229–1243. [Google Scholar] [CrossRef]
- Lendvai, G.; Szekerczés, T.; Illyés, I.; Dóra, R.; Kontsek, E.; Gógl, A.; Kiss, A.; Werling, K.; Kovalszky, I.; Schaff, Z.; et al. Cholangiocarcinoma: Classification, Histopathology and Molecular Carcinogenesis. Pathol. Oncol. Res. 2020, 26, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.-W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the Diagnosis and Management of Intrahepatic Cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next Horizon in Mechanisms and Management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef] [PubMed]
- Piha-Paul, S.A.; Oh, D.-Y.; Ueno, M.; Malka, D.; Chung, H.C.; Nagrial, A.; Kelley, R.K.; Ros, W.; Italiano, A.; Nakagawa, K.; et al. Efficacy and Safety of Pembrolizumab for the Treatment of Advanced Biliary Cancer: Results from the KEYNOTE -158 and KEYNOTE -028 Studies. Int. J. Cancer 2020, 147, 2190–2198. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.-Y.; He, A.R.; Qin, S.; Chen, L.-T.; Okusaka, T.; Vogel, A.; Kim, J.W.; Suksombooncharoen, T.; Lee, M.A.; Kitano, M.; et al. A Phase 3 Randomized, Double-Blind, Placebo-Controlled Study of Durvalumab in Combination with Gemcitabine plus Cisplatin (GemCis) in Patients (Pts) with Advanced Biliary Tract Cancer (BTC): TOPAZ-1. J. Clin. Oncol. 2022, 40. [Google Scholar] [CrossRef]
- Kim, R.D.; Chung, V.; Alese, O.B.; El-Rayes, B.F.; Li, D.; Al-Toubah, T.E.; Schell, M.J.; Zhou, J.-M.; Mahipal, A.; Kim, B.H.; et al. A Phase 2 Multi-Institutional Study of Nivolumab for Patients with Advanced Refractory Biliary Tract Cancer. JAMA Oncol. 2020, 6, 888. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Harb, W.; Peer, C.J.; Hua, Q.; Xu, S.; Lu, H.; Lu, N.; He, Y.; Xu, T.; Dong, R.; et al. First-in-Human Phase I Study of Envafolimab, a Novel Subcutaneous Single-Domain Anti-PD-L1 Antibody, in Patients with Advanced Solid Tumors. Oncologist 2021, 26, e1514–e1525. [Google Scholar] [CrossRef]
- Fiste, O.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Liontos, M.; Koutsoukos, K.; Dimopoulos, M.A.; Zagouri, F. The Emerging Role of Immunotherapy in Intrahepatic Cholangiocarcinoma. Vaccines 2021, 9, 422. [Google Scholar] [CrossRef]
- Han, S.; Lee, S.Y.; Wang, W.-W.; Tan, Y.B.; Sim, R.H.Z.; Cheong, R.; Tan, C.; Hopkins, R.; Connolly, J.; Shuen, W.H.; et al. A Perspective on Cell Therapy and Cancer Vaccine in Biliary Tract Cancers (BTCs). Cancers 2020, 12, 3404. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D.; Brandi, G. Durvalumab: An Investigational Anti-PD-L1 Antibody for the Treatment of Biliary Tract Cancer. Expert Opin. Investig. Drugs 2021, 30, 343–350. [Google Scholar] [CrossRef]
- Gutiérrez-Larrañaga, M.; González-López, E.; Roa-Bautista, A.; Rodrigues, P.M.; Díaz-González, Á.; Banales, J.M.; López-Hoyos, M.; Santos-Laso, A.; Crespo, J. Immune Checkpoint Inhibitors: The Emerging Cornerstone in Cholangiocarcinoma Therapy? Liver Cancer 2021, 10, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Akateh, C.; Ejaz, A.M.; Pawlik, T.M.; Cloyd, J.M. Neoadjuvant Treatment Strategies for Intrahepatic Cholangiocarcinoma. World J. Hepatol. 2020, 12, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Woods, E.; Le, D.; Jakka, B.K.; Manne, A. Changing Landscape of Systemic Therapy in Biliary Tract Cancer. Cancers 2022, 14, 2137. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Li, D.; Zhu, X. Cancer Immunotherapy: Pros, Cons and Beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef] [PubMed]
- Haanen, J.B.A.G.; Carbonnel, F.; Robert, C.; Kerr, K.M.; Peters, S.; Larkin, J.; Jordan, K.; ESMO Guidelines Committee. Management of Toxicities from Immunotherapy: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2017, 28, iv119–iv142. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Trial | Drug | Phase | Results |
|---|---|---|---|
| Oriental | Sorafenib | Phase III, randomized, double-blind, placebo-controlled | 6.5 vs. 4.2 months OS 2.8 vs. 1.4 months TTP |
| Sharp | Sorafenib | Phase III Randomized, double-blind, Placebo-controlled | 10.7 vs. 7.9 months OS 5.5 vs. 2.8 months TTP 43% vs. 32% DCR |
| Reflect | Lenvatinib vs. sorafenib | Phase III, open-label, multicenter, non-inferiority | 13.6 vs. 12.3 months OS 7.4 vs. 3.7 months TTP |
| CheckMate 459 | Nivolumab vs. sorafenib | Phase III, randomized, open-label | 16.4 vs. 14.7 months OS |
| KEYNOTE-224 | Pembrolizumab | Phase II, non-randomized, multicenter, open-label | 13.2 months OS 4.8 months TTP 61.5% DCR |
| IMbrave150 | Atezolizumabplusbevacizumabvssorafenib | Phase III study, randomized, open-label | 19.2 vs. 13.4 months OS 6.9 vs. 4.3 months PFS |
| Clinical Trial | Regimen | Phase | Results |
|---|---|---|---|
| MSB0011359C (M7824) in Subjects With Metastatic or Locally Advanced Solid Tumors | Bintrafuspalfa | Phase I, open-label trial expansion cohort | 12.7 months OS 2.5 months PFS 20% ORR |
| TOPAZ-1 | Durvalumab plus gemcitabine and cisplatin vs. gemcitabine and cisplatin | Phase III, randomized, double-blinded clinical trial | 12.8 vs. 11.5 months OS 7.2 vs. 5.7 months PFS 26.7 vs. 18.7 months ORR |
| INTR@PID BTC 055 | Bintrafuspalfa plus gemcitabine and cisplatin | Phase II, open-label, randomized, double-blinded | 10.1% ORR |
| IMMUNOBIL PRODIGE 57 | Durvalumab and tremelimumab vs. durvalumabplustremelimumab and paclitaxel | Phase II, non-comparative randomized | Raising safety concerns regarding co-administration of paclitaxel with durvalumab and tremelimumab |
| KEYNOTE-158 | Pembrolizumab | Phase II, non-randomized, open-label | 23.5 months OS 4.1 months PFS 34.3 ORR |
| A Phase 2 Clinical Trial of Entinostat in Combination With Nivolumab for Patients With Previously Treated Unresectable or Metastatic Cholangiocarcinoma and Pancreatic Adenocarcinoma | Entinostat plus nivolumab | Phase II, open-label | 6.4 months OS |
| A Randomized Phase 2 Study of Atezolizumab in Combination With Cobimetinib Versus Atezolizumab Monotherapy in Participants With Unresectable Cholangiocarcinoma | Atezolizumab vs. Atezolizumabpluscobimetinib | Phase II, open-label randomized | 3.65 vs. 1.87 months PFS |
| CA209-538 | Nivolumab and ipilimumab | Phase II, non-randomized | 5.7 months OS 2.9 months PFS 23% ORR |
| Immunotherapy Modality. | Agents |
|---|---|
| Immune checkpoint inhibitors | |
| Pancreatic cancer | |
| PD-1 inhibitors | Nivolumab, Pembrolizumab |
| PD-L1 inhibitors | |
| CTLA-4 | Tremelimumab, Ipillimumab |
| Gastric cancer | |
| PD-1 inhibitors | Nivolumab, Pembrolizumab |
| PD-L1 inhibitors | Atezolizumab |
| CTLA-4 | |
| Hepatocellular carcinoma | |
| PD-1 inhibitors | Nivolumab, Pembrolizumab |
| PD-L1 inhibitors | Atezolizumab |
| CTLA-4 | |
| Colorectal cancer | |
| PD-1 inhibitors | Nivolumab, Pembrolizumab |
| PD-L1 inhibitors | |
| CTLA-4 | Ipillimumab |
| Cholangiocarcinoma | |
| PD-1 inhibitors | Nivolumab, Pembrolizumab, Bintrafuspalfa |
| PD-L1 inhibitors | Durvalumab |
| CTLA-4 | Tremelimumab, Ipillimumab |
| Cancer vaccines | |
| Pancreatic cancer | Gvax, Peptide vaccines, mKras vaccine, CV301, GI-4000 |
| Gastric cancer | PAS-vaccination |
| Hepatocellular carcinoma | HEPAVAC, dendritic cell vaccine (DC vaccine), glypican-3 (GPC3) vaccine |
| Colorectal cancer | Talimogene laherparepvec vaccine |
| Cholangiocarcinoma | DC-based vaccines |
| Oncolytic viral therapy | |
| Pancreatic cancer | Adeno-associated viruses (AVV), Herpes Simplex Virus-1 and 2 (HSV-1 and HSV-2), HSV1716, R3616, vaccinia virus, rabbit-MYXV poxvirus |
| Hepatocellular carcinoma | HSV-1-based, adenovirus-based (CNHK500, ONYX-015, AD, ZD55-IFN-β, Smac/ZD55-TRAIL), vaccinia-based (JX-594 therapy) |
| Adaptive cell therapy | |
| Pancreatic cancer | Targets: MUC1, mesothelin, and CEA, FAP, HER2, PSCA, CD24, |
| Hepatocellular carcinoma | cytokine-induced killer cells (CIKS), bone-derived mesenchymal stem cells (MSCs), CAR-T cell treatment |
| Colorectal cancer | CAR-T cells treatment |
| Cholangiocarcinoma | CAR-T cells against epidermal growth factor receptor (EGFR), and CD133 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koustas, E.; Trifylli, E.-M.; Sarantis, P.; Papadopoulos, N.; Karapedi, E.; Aloizos, G.; Damaskos, C.; Garmpis, N.; Garmpi, A.; Papavassiliou, K.A.; et al. Immunotherapy as a Therapeutic Strategy for Gastrointestinal Cancer—Current Treatment Options and Future Perspectives. Int. J. Mol. Sci. 2022, 23, 6664. https://doi.org/10.3390/ijms23126664
Koustas E, Trifylli E-M, Sarantis P, Papadopoulos N, Karapedi E, Aloizos G, Damaskos C, Garmpis N, Garmpi A, Papavassiliou KA, et al. Immunotherapy as a Therapeutic Strategy for Gastrointestinal Cancer—Current Treatment Options and Future Perspectives. International Journal of Molecular Sciences. 2022; 23(12):6664. https://doi.org/10.3390/ijms23126664
Chicago/Turabian StyleKoustas, Evangelos, Eleni-Myrto Trifylli, Panagiotis Sarantis, Nikolaos Papadopoulos, Eleni Karapedi, Georgios Aloizos, Christos Damaskos, Nikolaos Garmpis, Anna Garmpi, Kostas A. Papavassiliou, and et al. 2022. "Immunotherapy as a Therapeutic Strategy for Gastrointestinal Cancer—Current Treatment Options and Future Perspectives" International Journal of Molecular Sciences 23, no. 12: 6664. https://doi.org/10.3390/ijms23126664
APA StyleKoustas, E., Trifylli, E.-M., Sarantis, P., Papadopoulos, N., Karapedi, E., Aloizos, G., Damaskos, C., Garmpis, N., Garmpi, A., Papavassiliou, K. A., Karamouzis, M. V., & Papavassiliou, A. G. (2022). Immunotherapy as a Therapeutic Strategy for Gastrointestinal Cancer—Current Treatment Options and Future Perspectives. International Journal of Molecular Sciences, 23(12), 6664. https://doi.org/10.3390/ijms23126664