
PARK7/DJ-1 as a Therapeutic Target in Gut-Brain Axis Diseases


Abstract
1. Introduction
2. Gut-Brain Axis
2.1. Epidemiological Evidence of a Gut-Brain Axis
2.2. Experimental Evidences of Gut Brain-Axis
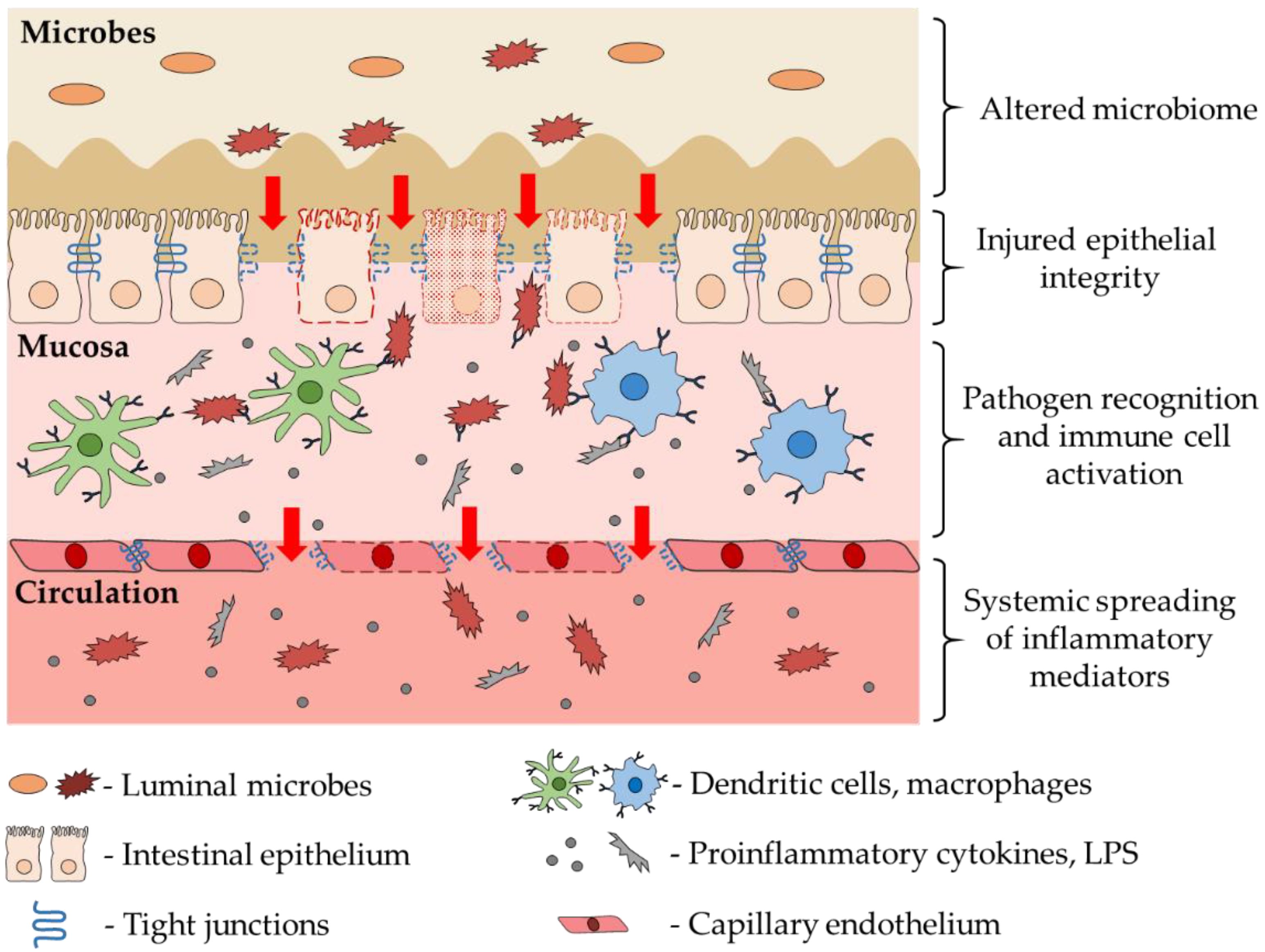
3. Role of Intestinal Dysbiosis and Inflammation in CNS Diseases
4. PARK7/DJ-1
4.1. Regulation of PARK7/DJ-1
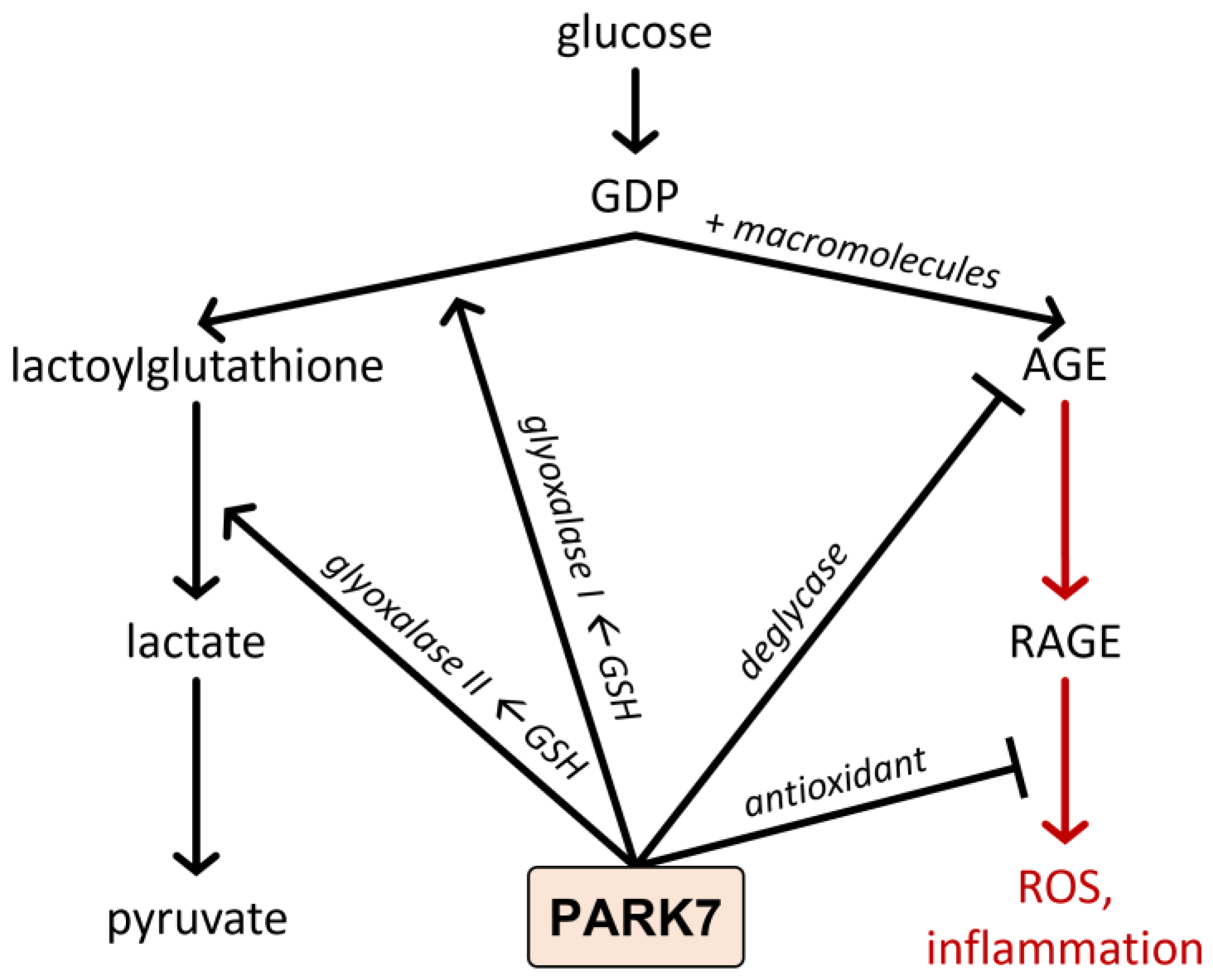
4.2. Functions of PARK7/DJ-1
5. Role of PARK7/dj-1 in the Pathogenesis of Neurodegenerative Diseases
5.1. Genetic Evidence for the Role of PARK7/DJ-1 in Parkinson’s Disease
5.2. Role of PARK7/DJ-1 in Parkinson’s Disease
5.3. Role of PARK7/DJ-1 in Alzheimer’s Disease
5.4. Role of PARK7/DJ-1 in Huntington’s Disease
5.5. Role of PARK7/DJ-1 in Ischemia-Reperfusion Induced Brain Injury
6. Role of PARK7/DJ-1 in the Pathogenesis of Gastrointestinal Diseases
6.1. Genetic Evidence of the Role of PARK7/DJ-1 in Gastrointestinal Diseases
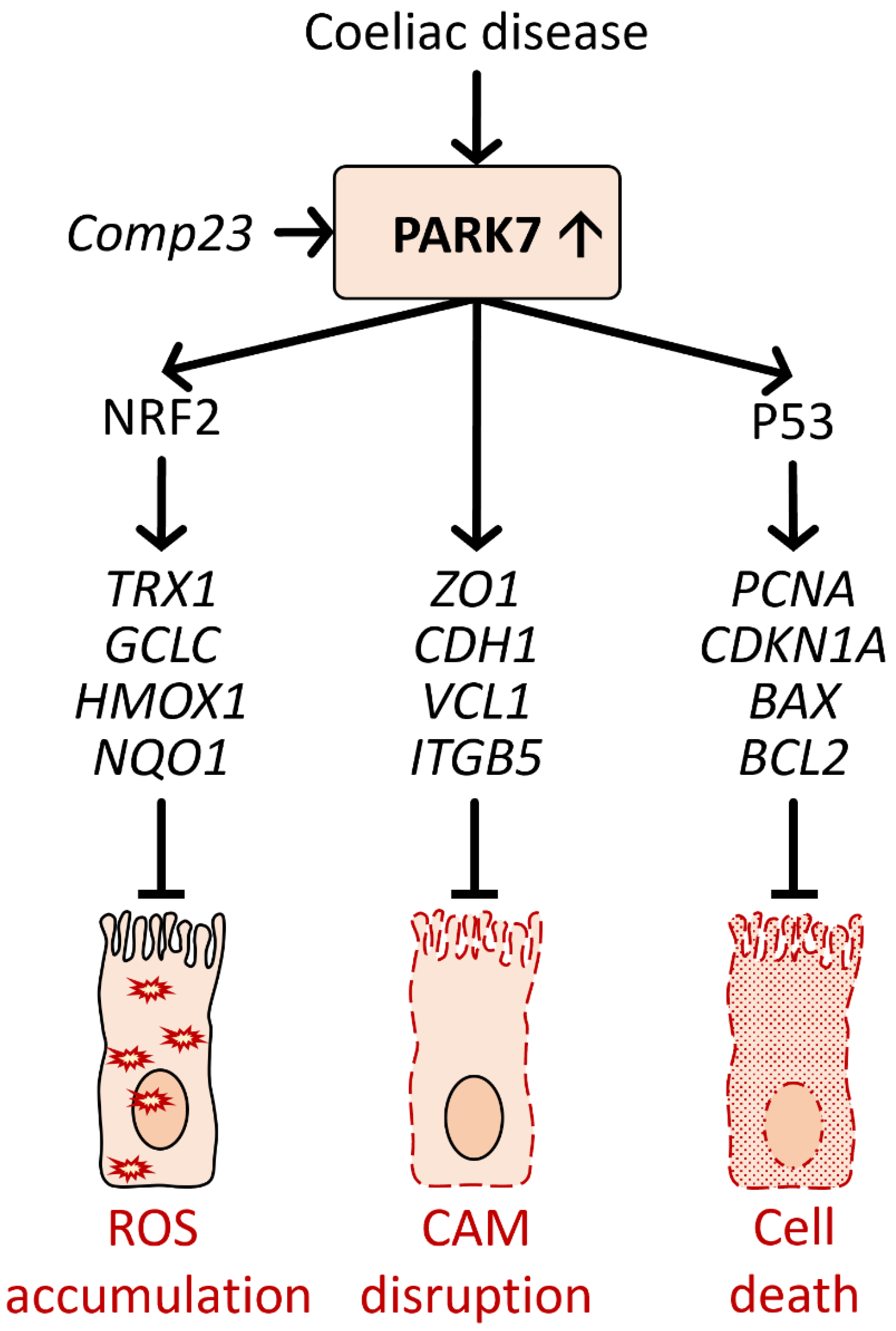
6.2. Role of PARK7/DJ-1 in Coeliac Disease
6.3. Role of PARK7/DJ-1 in Inflammatory Bowel Disease
6.4. The Role of PARK7/DJ-1 in Intestinal Dysbiosis
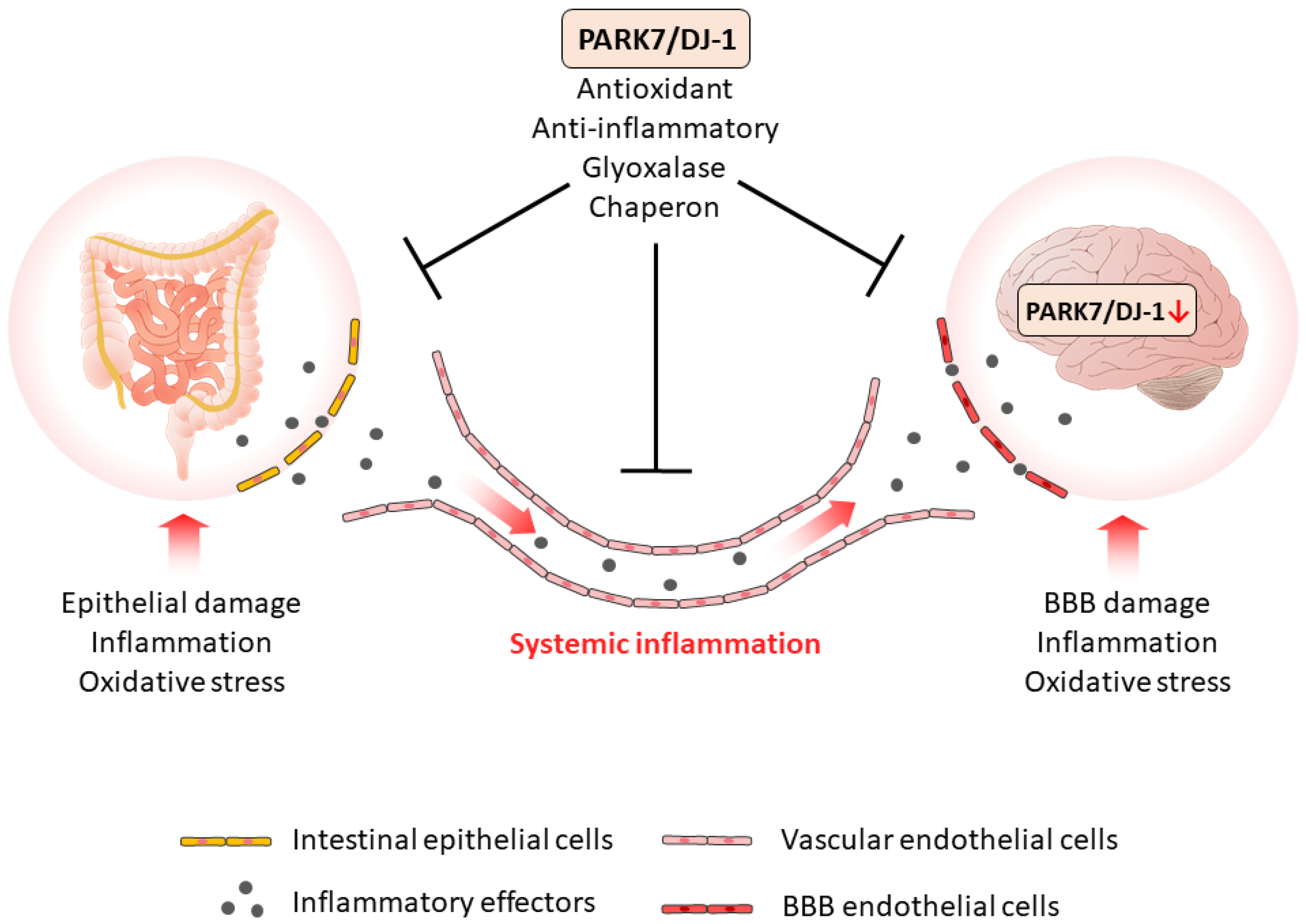
7. Role of PARK7/DJ-1 in GBA Diseases
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brudek, T. Inflammatory Bowel Diseases and Parkinson’s Disease. J. Parkinson’s Dis. 2019, 9, S331–S344. [Google Scholar] [CrossRef]
- Houser, M.C.; Tansey, M.G. The gut-brain axis: Is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? NPJ Parkinson’s Dis. 2017, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Z.; Li, Y.-Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Galea, I. The blood–brain barrier in systemic infection and inflammation. Cell. Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Björklund, T.; Wang, Z.-Y.; Roybon, L.; Melki, R.; Li, J.-Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef]
- Huang, M.; Chen, S. DJ-1 in neurodegenerative diseases: Pathogenesis and clinical application. Prog. Neurobiol. 2021, 204, 102114. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; Van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Kitamura, Y.; Watanabe, S.; Taguchi, M.; Takagi, K.; Kawata, T.; Takahashi-Niki, K.; Yasui, H.; Maita, H.; Iguchi-Ariga, S.M.; Ariga, H. Neuroprotective effect of a new DJ-1-binding compound against neurodegeneration in Parkinson’s disease and stroke model rats. Mol. Neurodegener. 2011, 6, 48. [Google Scholar] [CrossRef]
- Kitamura, Y.; Inden, M.; Kimoto, Y.; Takata, K.; Yanagisawa, D.; Hijioka, M.; Ashihara, E.; Tooyama, I.; Shimohama, S.; Ariga, H. Effects of a DJ-1-Binding Compound on Spatial Learning and Memory Impairment in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 55, 67–72. [Google Scholar] [CrossRef]
- Vörös, P.; Sziksz, E.; Himer, L.; Ónody, A.; Pap, D.; Frivolt, K.; Szebeni, B.; Lippai, R.; Győrffy, H.; Fekete, A.; et al. Expression of PARK7 is increased in celiac disease. Virchows Arch. 2013, 463, 401–408. [Google Scholar] [CrossRef]
- Lippai, R.; Veres-Székely, A.; Sziksz, E.; Iwakura, Y.; Pap, D.; Rokonay, R.; Szebeni, B.; Lotz, G.; Béres, N.J.; Cseh, Á.; et al. Immunomodulatory role of Parkinson’s disease 7 in inflammatory bowel disease. Sci. Rep. 2021, 11, 14582. [Google Scholar] [CrossRef]
- Veres-Székely, A.; Bernáth, M.; Pap, D.; Rokonay, R.; Szebeni, B.; Takács, I.M.; Lippai, R.; Cseh, Á.; Szabó, A.J.; Vannay, Á. PARK7 Diminishes Oxidative Stress-Induced Mucosal Damage in Celiac Disease. Oxid. Med. Cell. Longev. 2020, 2020, 4787202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, M.; Zhou, W.; Li, D.; Zhang, H.; Chen, Y.; Ning, L.; Zhang, Y.; Li, S.; Yu, M.; et al. Deficiency in the anti-apoptotic protein DJ-1 promotes intestinal epithelial cell apoptosis and aggravates inflammatory bowel disease via p53. J. Biol. Chem. 2020, 295, 4237–4251. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.A.-O.; Trautwein, C.; Dhariwal, A.; Salker, M.A.-O.X.; Alauddin, M.; Zizmare, L.A.-O.; Pelzl, L.; Feger, M.; Admard, J.; Casadei, N.A.-O.; et al. DJ-1 (Park7) affects the gut microbiome, metabolites and the development of innate lymphoid cells (ILCs). Sci. Rep. 2020, 10, 16131. [Google Scholar] [CrossRef] [PubMed]
- Alatab, S.; Sepanlou, S.G.; Ikuta, K.; Vahedi, H.; Bisignano, C.; Safiri, S.; Sadeghi, A.; Nixon, M.R.; Abdoli, A.; Abolhassani, H.; et al. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef]
- Levine, J.S.; Burakoff, R. Extraintestinal manifestations of inflammatory bowel disease. Gastroenterol. Hepatol. 2011, 7, 235. [Google Scholar]
- Lin, J.-C.; Lin, C.-S.; Hsu, C.-W.; Lin, C.-L.; Kao, C.-H. Association Between Parkinson’s Disease and Inflammatory Bowel Disease: A Nationwide Taiwanese Retrospective Cohort Study. Inflamm. Bowel Dis. 2016, 22, 1049–1055. [Google Scholar] [CrossRef]
- Villumsen, M.; Aznar, S.; Pakkenberg, B.; Jess, T.; Brudek, T.A.-O. Inflammatory bowel disease increases the risk of Parkinson’s disease: A Danish nationwide cohort study 1977–2014. Gut 2018, 68, 18–24. [Google Scholar] [CrossRef]
- Peter, I.; Dubinsky, M.; Bressman, S.; Park, A.; Lu, C.; Chen, N.; Wang, A. Anti-Tumor Necrosis Factor Therapy and Incidence of Parkinson Disease Among Patients with Inflammatory Bowel Disease. JAMA Neurol. 2018, 75, 939–946. [Google Scholar] [CrossRef]
- Weimers, P.; Halfvarson, J.; Sachs, M.C.; Saunders-Pullman, R.; Ludvigsson, J.F.; Peter, I.; Burisch, J.; Olén, O. Inflammatory Bowel Disease and Parkinson’s Disease: A Nationwide Swedish Cohort Study. Inflamm. Bowel Dis. 2019, 25, 111–123. [Google Scholar] [CrossRef]
- Zhu, F.; Li, C.; Gong, J.; Zhu, W.; Gu, L.; Li, N. The risk of Parkinson’s disease in inflammatory bowel disease: A systematic review and meta-analysis. Dig. Liver Dis. 2019, 51, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.-O.; Kim, J.; Chun, J.A.-O.; Han, K.; Soh, H.A.-O.; Kang, E.A.; Lee, H.J.; Im, J.A.-O.; Kim, J.S. Patients with Inflammatory Bowel Disease Are at an Increased Risk of Parkinson’s Disease: A South Korean Nationwide Population-Based Study. J. Clin. Med. 2019, 8, 1191. [Google Scholar] [CrossRef]
- Fu, P.A.-O.; Gao, M.A.-O.; Yung, K.A.-O. Association of Intestinal Disorders with Parkinson’s Disease and Alzheimer’s Disease: A Systematic Review and Meta-Analysis. ACS Chem. Neurosci. 2019, 11, 395–405. [Google Scholar] [CrossRef]
- Zhang, B.A.-O.; Wang, H.A.-O.; Bai, Y.M.; Tsai, S.J.; Su, T.P.; Chen, T.J.; Wang, Y.A.-O.; Chen, M.A.-O. Inflammatory bowel disease is associated with higher dementia risk: A nationwide longitudinal study. Gut 2021, 70, 85–91. [Google Scholar] [CrossRef]
- Ríos, J.P.; Navarro, C.J.M.; Navarro, M.J.P.; Tapia, M.J.C.; Vera, M.J.P.; Arillo, V.C.; García, M.R.G.; Castellanos, A.M.; Sevilla, F.E. Association of Parkinson’s disease and treatment with aminosalicylates in inflammatory bowel disease: A cross-sectional study in a Spain drug dispensation records. BMJ Open 2019, 9, e025574. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.A.-O.; Benzvi, C. “Let Food Be Thy Medicine”: Gluten and Potential Role in Neurodegeneration. Cells 2021, 10, 756. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, E.; Margonis, G.A.; Angelou, A.; Pikouli, A.; Argiri, P.; Karavokyros, I.; Papalois, A.; Pikoulis, E. The TNBS-induced colitis animal model: An overview. Ann. Med. Surg. 2016, 11, 9–15. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15–25. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Kouli, A.; Torsney, K.M.; Kuan, W.-L. Parkinson’s disease: Etiology, neuropathology, and pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Codon Publications: Singapore, 2018. [Google Scholar]
- Hathaway, C.A.; Appleyard, C.B.; Percy, W.H.; Williams, J.L. Experimental colitis increases blood-brain barrier permeability in rabbits. Am. J. Physiol.-Gastrointest. Liver Physiol. 1999, 276, G1174–G1180. [Google Scholar] [CrossRef] [PubMed]
- Natah, S.S.; Mouihate, A.; Pittman, Q.J.; Sharkey, K.A. Disruption of the blood-brain barrier during TNBS colitis. Neurogastroenterol. Motil. 2005, 17, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuan, C.-P.; Wang, W.; Yang, Z.-Q.; Cui, W.; Mu, L.-Z.; Yue, Z.-P.; Yin, X.-L.; Hu, Z.-M.; Liu, J.-X. Expression of interleukin 6 in brain and colon of rats with TNBS-induced colitis. World J. Gastroenterol. 2010, 16, 2252–2259. [Google Scholar] [CrossRef]
- Villarán, R.F.; Espinosa-Oliva, A.M.; Sarmiento, M.; De Pablos, R.M.; Argüelles, S.; Delgado-Cortés, M.J.; Sobrino, V.; Van Rooijen, N.; Venero, J.L.; Herrera, A.J. Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: Potential risk factor in Parkinsons disease. J. Neurochem. 2010, 114, 1687–1700. [Google Scholar] [CrossRef]
- Han, Y.; Zhao, T.; Cheng, X.; Zhao, M.; Gong, S.-H.; Zhao, Y.-Q.; Wu, H.-T.; Fan, M.; Zhu, L.-L. Cortical Inflammation is Increased in a DSS-Induced Colitis Mouse Model. Neurosci. Bull. 2018, 34, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Gil, P.; Rodriguez-Perez, A.I.; Dominguez-Meijide, A.; Guerra, M.J.; Labandeira-Garcia, J.L. Bidirectional Neural Interaction Between Central Dopaminergic and Gut Lesions in Parkinson’s Disease Models. Mol. Neurobiol. 2018, 55, 7297–7316. [Google Scholar] [CrossRef]
- Do, J.; Woo, J. From Gut to Brain: Alteration in Inflammation Markers in the Brain of Dextran Sodium Sulfate-induced Colitis Model Mice. Clin. Psychopharmacol. Neurosci. 2018, 16, 422. [Google Scholar] [CrossRef]
- Grathwohl, S.; Quansah, E.; Maroof, N.; Steiner, J.A.; Spycher, L.; Benmansour, F.; Duran-Pacheco, G.; Siebourg-Polster, J.; Oroszlan-Szovik, K.; Remy, H.; et al. Experimental colitis drives enteric alpha-synuclein accumulation and Parkinson-like brain pathology. bioRxiv 2018, 505164. [Google Scholar] [CrossRef]
- Sroor, H.M.; Hassan, A.M.; Zenz, G.; Valadez-Cosmes, P.; Farzi, A.; Holzer, P.; El-Sharif, A.; Gomaa, F.A.-Z.M.; Kargl, J.; Reichmann, F. Experimental colitis reduces microglial cell activation in the mouse brain without affecting microglial cell numbers. Sci. Rep. 2019, 9, 20217. [Google Scholar] [CrossRef]
- Gampierakis, I.-A.; Koutmani, Y.; Semitekolou, M.; Morianos, I.; Polissidis, A.; Katsouda, A.; Charalampopoulos, I.; Xanthou, G.; Gravanis, A.; Karalis, K.P. Hippocampal neural stem cells and microglia response to experimental inflammatory bowel disease (IBD). Mol. Psychiatry 2021, 26, 1248–1263. [Google Scholar] [CrossRef]
- He, X.F.; Li, L.L.; Xian, W.B.; Li, M.Y.; Zhang, L.Y.; Xu, J.H.; Pei, Z.; Zheng, H.Q.; Hu, X.Q. Chronic colitis exacerbates NLRP3-dependent neuroinflammation and cognitive impairment in middle-aged brain. J. Neuroinflamm. 2021, 18, 153. [Google Scholar] [CrossRef] [PubMed]
- Talley, S.; Valiauga, R.; Anderson, L.; Cannon, A.R.; Choudhry, M.A.; Campbell, E.M. DSS-induced inflammation in the colon drives a proinflammatory signature in the brain that is ameliorated by prophylactic treatment with the S100A9 inhibitor paquinimod. J. Neuroinflamm. 2021, 18, 263. [Google Scholar] [CrossRef] [PubMed]
- Gil-Martínez, A.-L.; Estrada, C.; Cuenca, L.; Cano, J.-A.; Valiente, M.; Martínez-Cáceres, C.-M.; Fernández-Villalba, E.; Herrero, M.-T. Local Gastrointestinal Injury Exacerbates Inflammation and Dopaminergic Cell Death in Parkinsonian Mice. Neurotox. Res. 2019, 35, 918–930. [Google Scholar] [CrossRef]
- Houser, M.C.; Caudle, W.M.; Chang, J.; Kannarkat, G.T.; Yang, Y.; Kelly, S.D.; Oliver, D.; Joers, V.; Shannon, K.M.; Keshavarzian, A.; et al. Experimental colitis promotes sustained, sex-dependent, T-cell-associated neuroinflammation and parkinsonian neuropathology. Acta Neuropathol. Commun. 2021, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The firmicutes/bacteroidetes ratio: A relevant marker of gut dysbiosis in obese patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Stojanov, S.; Berlec, A.; Štrukelj, B. The influence of probiotics on the firmicutes/bacteroidetes ratio in the treatment of obesity and inflammatory bowel disease. Microorganisms 2020, 8, 1715. [Google Scholar] [CrossRef]
- Yu, L.C.-H. Microbiota dysbiosis and barrier dysfunction in inflammatory bowel disease and colorectal cancers: Exploring a common ground hypothesis. J. Biomed. Sci. 2018, 25, 79. [Google Scholar] [CrossRef]
- Pawłowska, B.; Sobieszczańska, B.M. Intestinal epithelial barrier: The target for pathogenic Escherichia coli. Adv. Clin. Exp. Med. Off. Organ Wroc. Med. Univ. 2017, 26, 1437–1445. [Google Scholar] [CrossRef]
- Iyer, N.; Corr, S.C. Gut Microbial Metabolite-Mediated Regulation of the Intestinal Barrier in the Pathogenesis of Inflammatory Bowel Disease. Nutrients 2021, 13, 4259. [Google Scholar] [CrossRef] [PubMed]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.; Faber, K.N.; Hermoso, M.A. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.-N.; Wang, M.; Guo, J.; Wang, J.-P. Role of intestinal microbiota and metabolites in inflammatory bowel disease. Chin. Med. J. 2019, 132, 1610–1614. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef]
- Johansson, M.E.; Gustafsson, J.K.; Holmén-Larsson, J.; Jabbar, K.S.; Xia, L.; Xu, H.; Ghishan, F.K.; Carvalho, F.A.; Gewirtz, A.T.; Sjövall, H.; et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2014, 63, 281–291. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Pallone, F.; Monteleone, G. Mechanisms of tissue damage in inflammatory bowel disease. Curr. Opin. Gastroenterol. 2001, 17, 307–312. [Google Scholar] [CrossRef]
- O’Sullivan, S.; Gilmer, J.F.; Medina, C. Matrix Metalloproteinases in Inflammatory Bowel Disease: An Update. Mediat. Inflamm. 2015, 2015, 964131. [Google Scholar] [CrossRef]
- Barichello, T.; Generoso, J.S.; Collodel, A.; Petronilho, F.; Dal-Pizzol, F. The blood-brain barrier dysfunction in sepsis. Tissue Barriers 2021, 9, 1840912. [Google Scholar] [CrossRef]
- Van Dyken, P.; Lacoste, B. Impact of Metabolic Syndrome on Neuroinflammation and the Blood-Brain Barrier. Front. Neurosci. 2018, 12, 930. [Google Scholar] [CrossRef]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Varatharaj, A.; Liljeroth, M.; Cramer, S.; Stuart, C.; Zotova, E.; Darekar, A.; Larsson, H.; Galea, I. Systemic inflammation and blood–brain barrier abnormality in relapsing–remitting multiple sclerosis. Lancet 2017, 389, S96. [Google Scholar] [CrossRef]
- Edelblum, K.L.; Turner, J.R. The tight junction in inflammatory disease: Communication breakdown. Curr. Opin. Pharmacol. 2009, 9, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Bergemalm, D.; Andersson, E.; Hultdin, J.; Eriksson, C.; Rush, S.T.; Kalla, R.; Adams, A.T.; Keita, Å.V.; D’Amato, M.; Gomollon, F. Systemic inflammation in preclinical ulcerative colitis. Gastroenterology 2021, 161, 1526–1539.e1529. [Google Scholar] [CrossRef]
- Rojo, Ó.P.; Román, A.L.S.; Arbizu, E.A.; de la Hera Martínez, A.; Sevillano, E.R.; Martínez, A.A. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 269–277. [Google Scholar] [CrossRef]
- Reichmann, F.; Hassan, A.M.; Farzi, A.; Jain, P.; Schuligoi, R.; Holzer, P. Dextran sulfate sodium-induced colitis alters stress-associated behaviour and neuropeptide gene expression in the amygdala-hippocampus network of mice. Sci. Rep. 2015, 5, 9970. [Google Scholar] [CrossRef]
- Kiely, C.J.; Pavli, P.; O’Brien, C.L. The microbiome of translocated bacterial populations in patients with and without inflammatory bowel disease. Intern. Med. J. 2018, 48, 1346–1354. [Google Scholar] [CrossRef]
- Gutierrez, A.; Holler, E.; Zapater, P.; Sempere, L.; Jover, R.; Perez-Mateo, M.; Schoelmerich, J.; Such, J.; Wiest, R.; Frances, R. Antimicrobial peptide response to blood translocation of bacterial DNA in Crohn’s disease is affected by NOD2/CARD15 genotype. Inflamm. Bowel Dis. 2011, 17, 1641–1650. [Google Scholar] [CrossRef]
- Linares, R.; Francés, R.; Gutiérrez, A.; Juanola, O. Bacterial Translocation as Inflammatory Driver in Crohn’s Disease. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Kumar, M.; Leon Coria, A.; Cornick, S.; Petri, B.; Mayengbam, S.; Jijon, H.B.; Moreau, F.; Shearer, J.; Chadee, K. Increased intestinal permeability exacerbates sepsis through reduced hepatic SCD-1 activity and dysregulated iron recycling. Nat. Commun. 2020, 11, 483. [Google Scholar] [CrossRef]
- Rahman, M.T.; Ghosh, C.; Hossain, M.; Linfield, D.; Rezaee, F.; Janigro, D.; Marchi, N.; van Boxel-Dezaire, A.H.H. IFN-γ, IL-17A, or zonulin rapidly increase the permeability of the blood-brain and small intestinal epithelial barriers: Relevance for neuro-inflammatory diseases. Biochem. Biophys. Res. Commun. 2018, 507, 274–279. [Google Scholar] [CrossRef]
- Wood Heickman, L.K.; DeBoer, M.D.; Fasano, A. Zonulin as a potential putative biomarker of risk for shared type 1 diabetes and celiac disease autoimmunity. Diabetes/Metab. Res. Rev. 2020, 36, e3309. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A. Intestinal permeability and its regulation by zonulin: Diagnostic and therapeutic implications. Clin. Gastroenterol. Hepatol. 2012, 10, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Matejuk, A.; Ransohoff, R.M. Crosstalk Between Astrocytes and Microglia: An Overview. Front. Immunol. 2020, 11, 1416. [Google Scholar] [CrossRef]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell. Neurosci. 2021, 15, 344. [Google Scholar] [CrossRef]
- Bonaz, B.; Sinniger, V.; Pellissier, S. Vagus nerve stimulation: A new promising therapeutic tool in inflammatory bowel disease. J. Intern. Med. 2017, 282, 46–63. [Google Scholar] [CrossRef]
- Ito, G.; Ariga, H.; Nakagawa, Y.; Iwatsubo, T. Roles of distinct cysteine residues in S-nitrosylation and dimerization of DJ-1. Biochem. Biophys. Res. Commun. 2006, 339, 667–672. [Google Scholar] [CrossRef]
- Kiss, R.; Zhu, M.; Jójárt, B.; Czajlik, A.; Solti, K.; Fórizs, B.; Nagy, É.; Zsila, F.; Beke-Somfai, T.; Tóth, G. Structural features of human DJ-1 in distinct Cys106 oxidative states and their relevance to its loss of function in disease. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2017, 1861, 2619–2629. [Google Scholar] [CrossRef]
- Wilson, M.A. The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxid. Redox Signal. 2011, 15, 111–122. [Google Scholar] [CrossRef]
- Takahashi-Niki, K.; Inafune, A.; Michitani, N.; Hatakeyama, Y.; Suzuki, K.; Sasaki, M.; Kitamura, Y.; Niki, T.; Iguchi-Ariga, S.M.M.; Ariga, H. DJ-1-dependent protective activity of DJ-1-binding compound no. 23 against neuronal cell death in MPTP-treated mouse model of Parkinson’s disease. J. Pharmacol. Sci. 2015, 127, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, J.; Wang, J.; Yang, B.; He, Q.; Weng, Q. Role of DJ-1 in Immune and Inflammatory Diseases. Front. Immunol. 2020, 11, 994. [Google Scholar] [CrossRef] [PubMed]
- Canet-Avilés, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar] [CrossRef] [PubMed]
- Nagakubo, D.; Taira, T.; Kitaura, H.; Ikeda, M.; Tamai, K.; Iguchi-Ariga, S.M.M.; Ariga, H. DJ-1, a Novel Oncogene Which Transforms Mouse NIH3T3 Cells in Cooperation withras. Biochem. Biophys. Res. Commun. 1997, 231, 509–513. [Google Scholar] [CrossRef]
- Maita, C.; Tsuji, S.; Yabe, I.; Hamada, S.; Ogata, A.; Maita, H.; Iguchi-Ariga, S.M.M.; Sasaki, H.; Ariga, H. Secretion of DJ-1 into the serum of patients with Parkinson’s disease. Neurosci. Lett. 2008, 431, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Jin, W. Novel Insights into PARK7 (DJ-1), a Potential Anti-Cancer Therapeutic Target, and Implications for Cancer Progression. J. Clin. Med. 2020, 9, 1256. [Google Scholar] [CrossRef] [PubMed]
- He, X.Y.; Liu, B.Y.; Yao, W.Y.; Zhao, X.J.; Zheng, Z.; Li, J.F.; Yu, B.Q.; Yuan, Y.Z. Serum DJ-1 as a diagnostic marker and prognostic factor for pancreatic cancer. J. Dig. Dis. 2011, 12, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, H.; Zhang, L.; Liu, X.; Zhang, C.; Wang, Y.; He, Q.; Zhang, Y.; Li, Y.; Chen, Q.; et al. DJ-1 promotes colorectal cancer progression through activating PLAGL2/Wnt/BMP4 axis. Cell Death Dis. 2018, 9, 865. [Google Scholar] [CrossRef]
- Antipova, D.; Bandopadhyay, R. Expression of DJ-1 in Neurodegenerative Disorders. In DJ-1/PARK7 Protein; Ariga, H., Iguchi-Ariga, S., Eds.; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Vasseur, S.; Afzal, S.; Tomasini, R.; Guillaumond, F.; Tardivel-Lacombe, J.; Mak, T.W.; Iovanna, J.L. Consequences of DJ-1 upregulation following p53 loss and cell transformation. Oncogene 2012, 31, 664–670. [Google Scholar] [CrossRef]
- Qin, L.-X.; Tan, J.-Q.; Zhang, H.-N.; Rizwana, K.; Lu, J.-H.; Tang, J.-G.; Jiang, B.; Shen, X.-M.; Guo, J.-F.; Tang, B.-S.; et al. BAG5 Interacts with DJ-1 and Inhibits the Neuroprotective Effects of DJ-1 to Combat Mitochondrial Oxidative Damage. Oxid. Med. Cell. Longev. 2017, 2017, 5094934. [Google Scholar] [CrossRef]
- Choi, D.-H.; Hwang, O.; Lee, K.-H.; Lee, J.; Beal, M.F.; Kim, Y.-S. DJ-1 cleavage by matrix metalloproteinase 3 mediates oxidative stress-induced dopaminergic cell death. Antioxid. Redox Signal. 2011, 14, 2137–2150. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.L.; Wang, H.B.; Yong, J.K.; Zhong, J.; Li, Q.H. MiR-128-3p overexpression sensitizes hepatocellular carcinoma cells to sorafenib induced apoptosis through regulating DJ-1. Eur. Rev. Med. Pharm. Sci. 2018, 22, 6667–6677. [Google Scholar]
- Du, S.L.; Xu, L.Y.; Gao, P.; Liu, Q.S.; Lu, F.F.; Mo, Z.H.; Fan, Z.Z.; Cheng, X.L.; Dong, Z.H. MiR-203 regulates DJ-1 expression and affects proliferation, apoptosis and DDP resistance of pancreatic cancer cells. Eur. Rev. Med. Pharm. Sci. 2019, 23, 8833–8840. [Google Scholar]
- Xiong, R.; Wang, Z.; Zhao, Z.; Li, H.; Chen, W.; Zhang, B.; Wang, L.; Wu, L.; Li, W.; Ding, J.; et al. MicroRNA-494 reduces DJ-1 expression and exacerbates neurodegeneration. Neurobiol. Aging 2014, 35, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Cholez, E.; Debuysscher, V.; Bourgeais, J.; Boudot, C.; Leprince, J.; Tron, F.; Brassart, B.; Regnier, A.; Bissac, E.; Pecnard, E.; et al. Evidence for a protective role of the STAT5 transcription factor against oxidative stress in human leukemic pre-B cells. Leukemia 2012, 26, 2390–2397. [Google Scholar] [CrossRef]
- Hwang, J.; Pallas, D.C. STRIPAK complexes: Structure, biological function, and involvement in human diseases. Int. J. Biochem. Cell Biol. 2014, 47, 118–148. [Google Scholar] [CrossRef]
- Tanti, G.K.; Goswami, S.K. SG2NA recruits DJ-1 and Akt into the mitochondria and membrane to protect cells from oxidative damage. Free Radic. Biol. Med. 2014, 75, 1–13. [Google Scholar] [CrossRef]
- Tanti, G.K.; Pandey, S.; Goswami, S.K. SG2NA enhances cancer cell survival by stabilizing DJ-1 and thus activating Akt. Biochem. Biophys. Res. Commun. 2015, 463, 524–531. [Google Scholar] [CrossRef]
- DJ-1/PARK7 Protein: Parkinson’s Disease, Cancer and Oxidative Stress-Induced Diseases; Iguchi-Ariga, H.A.M.M., Ed.; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Kahle, P.J.; Waak, J.; Gasser, T. DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free Radic. Biol. Med. 2009, 47, 1354–1361. [Google Scholar] [CrossRef]
- Andres-Mateos, E.; Perier, C.; Zhang, L.; Blanchard-Fillion, B.; Greco, T.M.; Thomas, B.; Ko, H.S.; Sasaki, M.; Ischiropoulos, H.; Przedborski, S.; et al. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc. Natl. Acad. Sci. USA 2007, 104, 14807–14812. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Ishimori, C.; Takahashi-Niki, K.; Taira, T.; Kim, Y.-C.; Maita, H.; Maita, C.; Ariga, H.; Iguchi-Ariga, S.M.M. DJ-1 binds to mitochondrial complex I and maintains its activity. Biochem. Biophys. Res. Commun. 2009, 390, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P.Y. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef]
- Im, J.-Y.; Lee, K.-W.; Woo, J.-M.; Junn, E.; Mouradian, M.M. DJ-1 induces thioredoxin 1 expression through the Nrf2 pathway. Hum. Mol. Genet. 2012, 21, 3013–3024. [Google Scholar] [CrossRef] [PubMed]
- Haybrard, J.; Simon, N.; Danel, C.; Pinçon, C.; Barthélémy, C.; Tessier, F.J.; Décaudin, B.; Boulanger, E.; Odou, P. Factors Generating Glucose Degradation Products In Sterile Glucose Solutions For Infusion: Statistical Relevance Determination of Their Impacts. Sci. Rep. 2017, 7, 11932. [Google Scholar] [CrossRef] [PubMed]
- Byun, K.; Yoo, Y.; Son, M.; Lee, J.; Jeong, G.-B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol. Ther. 2017, 177, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Bekker, P.; Tsimikas, S. Advanced Glycation End Products and Diabetic Cardiovascular Disease. Cardiol. Rev. 2012, 20, 177–183. [Google Scholar] [CrossRef]
- He, Y.; Zhou, C.; Huang, M.; Tang, C.; Liu, X.; Yue, Y.; Diao, Q.; Zheng, Z.; Liu, D. Glyoxalase system: A systematic review of its biological activity, related-diseases, screening methods and small molecule regulators. Biomed. Pharmacother. 2020, 131, 110663. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, D.; Chen, L.; Li, J.; Yuan, G.; Yang, G.; Zhang, H.; Guo, X.; Zhang, J. Glycine increases glyoxalase-1 function by promoting nuclear factor erythroid 2-related factor 2 translocation into the nucleus of kidney cells of streptozotocin-induced diabetic rats. J. Diabetes Investig. 2019, 10, 1189–1198. [Google Scholar] [CrossRef]
- Zhou, W.; Freed, C.R. DJ-1 Up-regulates Glutathione Synthesis during Oxidative Stress and Inhibits A53T α-Synuclein Toxicity*. J. Biol. Chem. 2005, 280, 43150–43158. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Song, J.; Kwon, K.; Jang, S.; Kim, C.; Baek, K.; Kim, J.; Park, C. Human DJ-1 and its homologs are novel glyoxalases. Hum. Mol. Genet. 2012, 21, 3215–3225. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Rao, S.P.; Kalivendi, S.V. The deglycase activity of DJ-1 mitigates α-synuclein glycation and aggregation in dopaminergic cells: Role of oxidative stress mediated downregulation of DJ-1 in Parkinson’s disease. Free Radic. Biol. Med. 2019, 135, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Kimura, M.; Queliconi, B.B.; Kojima, W.; Mishima, M.; Takagi, K.; Koyano, F.; Yamano, K.; Mizushima, T.; Ito, Y.; et al. Parkinson’s disease-related DJ-1 functions in thiol quality control against aldehyde attack in vitro. Sci. Rep. 2017, 7, 12816. [Google Scholar] [CrossRef] [PubMed]
- Mencke, P.; Boussaad, I.; Romano, C.D.; Kitami, T.; Linster, C.L.; Krüger, R. The Role of DJ-1 in Cellular Metabolism and Pathophysiological Implications for Parkinson’s Disease. Cells 2021, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.E.; Mouradian, M.M. Cytoprotective mechanisms of DJ-1 against oxidative stress through modulating ERK1/2 and ASK1 signal transduction. Redox Biol. 2018, 14, 211–217. [Google Scholar] [CrossRef]
- Zhang, F.; Peng, W.; Zhang, J.; Wang, L.; Dong, W.; Zheng, Y.; Wang, Z.; Xie, Z.; Wang, T.; Wang, C.; et al. PARK7 enhances antioxidative-stress processes of BMSCs via the ERK1/2 pathway. J. Cell. Biochem. 2021, 122, 222–234. [Google Scholar] [CrossRef]
- Shendelman, S.; Jonason, A.; Martinat, C.; Leete, T.; Abeliovich, A. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2004, 2, e362. [Google Scholar] [CrossRef]
- Zhou, W.; Zhu, M.; Wilson, M.A.; Petsko, G.A.; Fink, A.L. The Oxidation State of DJ-1 Regulates its Chaperone Activity toward α-Synuclein. J. Mol. Biol. 2006, 356, 1036–1048. [Google Scholar] [CrossRef]
- Kim, J.-H.; Choi, D.-J.; Jeong, H.-K.; Kim, J.; Kim, D.W.; Choi, S.Y.; Park, S.-M.; Suh, Y.H.; Jou, I.; Joe, E.-H. DJ-1 facilitates the interaction between STAT1 and its phosphatase, SHP-1, in brain microglia and astrocytes: A novel anti-inflammatory function of DJ-1. Neurobiol. Dis. 2013, 60, 1–10. [Google Scholar] [CrossRef]
- Peng, L.; Zhou, Y.; Jiang, N.; Wang, T.; Zhu, J.; Chen, Y.; Li, L.; Zhang, J.; Yu, S.; Zhao, Y. DJ-1 exerts anti-inflammatory effects and regulates NLRX1-TRAF6 via SHP-1 in stroke. J. Neuroinflamm. 2020, 17, 81. [Google Scholar] [CrossRef]
- Chen, L.; Luo, M.; Sun, X.; Qin, J.; Yu, C.; Wen, Y.; Zhang, Q.; Gu, J.; Xia, Q.; Kong, X. DJ-1 deficiency attenuates expansion of liver progenitor cells through modulating the inflammatory and fibrogenic niches. Cell Death Dis. 2016, 7, e2257. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, J.-M.; Jang, H.-J.; Choi, S.Y.; Park, S.-A.; Kim, I.S.; Yang, Y.R.; Lee, Y.H.; Ryu, S.H.; Suh, P.-G. DJ-1 contributes to adipogenesis and obesity-induced inflammation. Sci. Rep. 2014, 4, 4805. [Google Scholar] [CrossRef] [PubMed]
- Inden, M.; Kitamura, Y.; Takahashi, K.; Takata, K.; Ito, N.; Niwa, R.; Funayama, R.; Nishimura, K.; Taniguchi, T.; Honda, T.; et al. Protection Against Dopaminergic Neurodegeneration in Parkinson’s Disease–Model Animals by a Modulator of the Oxidized Form of DJ-1, a Wild-type of Familial Parkinson’s Disease–Linked PARK7. J. Pharmacol. Sci. 2011, 117, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Sajjad, M.U.; Green, E.W.; Miller-Fleming, L.; Hands, S.; Herrera, F.; Campesan, S.; Khoshnan, A.; Outeiro, T.F.; Giorgini, F.; Wyttenbach, A. DJ-1 modulates aggregation and pathogenesis in models of Huntington’s disease. Hum. Mol. Genet. 2014, 23, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Lang, A.; van de Warrenburg, B.P.; Sue, C.M.; Tabrizi, S.J.; Bertram, L.; Mercimek-Mahmutoglu, S.; Ebrahimi-Fakhari, D.; Warner, T.T.; Durr, A.; et al. Nomenclature of genetic movement disorders: Recommendations of the international Parkinson and movement disorder society task force. Mov. Disord. 2016, 31, 436–457. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Fu, Y.; Halliday, G.M.; Sue, C.M. PARK Genes Link Mitochondrial Dysfunction and Alpha-Synuclein Pathology in Sporadic Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 1755. [Google Scholar] [CrossRef]
- Ariga, H.; Takahashi-Niki, K.; Kato, I.; Maita, H.; Niki, T.; Iguchi-Ariga, S.M.M. Neuroprotective Function of DJ-1 in Parkinson’s Disease. Oxid. Med. Cell. Longev. 2013, 2013, 683920. [Google Scholar] [CrossRef]
- Martinat, C.; Shendelman, S.; Jonason, A.; Leete, T.; Beal, M.F.; Yang, L.; Floss, T.; Abeliovich, A. Sensitivity to Oxidative Stress in DJ-1-Deficient Dopamine Neurons: An ES- Derived Cell Model of Primary Parkinsonism. PLoS Biol. 2004, 2, e327. [Google Scholar] [CrossRef]
- Taira, T.; Saito, Y.; Niki, T.; Iguchi-Ariga, S.M.M.; Takahashi, K.; Ariga, H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004, 5, 213–218. [Google Scholar] [CrossRef]
- Yanagisawa, D.; Kitamura, Y.; Inden, M.; Takata, K.; Taniguchi, T.; Morikawa, S.; Morita, M.; Inubushi, T.; Tooyama, I.; Taira, T.; et al. DJ-1 Protects against Neurodegeneration Caused by Focal Cerebral Ischemia and Reperfusion in Rats. J. Cereb. Blood Flow Metab. 2007, 28, 563–578. [Google Scholar] [CrossRef]
- Sun, S.-Y.; An, C.-N.; Pu, X.-P. DJ-1 protein protects dopaminergic neurons against 6-OHDA/MG-132-induced neurotoxicity in rats. Brain Res. Bull. 2012, 88, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, S.; Yanagida, T.; Nunome, K.; Ishikawa, S.; Inden, M.; Kitamura, Y.; Nakagawa, S.; Taira, T.; Hirota, K.; Niwa, M.; et al. DJ-1-binding compounds prevent oxidative stress-induced cell death and movement defect in Parkinson’s disease model rats. J. Neurochem. 2008, 105, 2418–2434. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, T.; Kitamura, Y.; Yamane, K.; Takahashi, K.; Takata, K.; Yanagisawa, D.; Yasui, H.; Taniguchi, T.; Taira, T.; Honda, T.; et al. Protection Against Oxidative Stress-Induced Neurodegeneration by a Modulator for DJ-1, the Wild-Type of Familial Parkinson’s Disease-Linked PARK7. J. Pharmacol. Sci. 2009, 109, 463–468. [Google Scholar] [CrossRef]
- Angeloni, C.; Zambonin, L.; Hrelia, S. Role of Methylglyoxal in Alzheimer’s Disease. BioMed Res. Int. 2014, 2014, 238485. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Nicholson, L.F.B. Advanced glycation end products induce in vitro cross-linking of α-synuclein and accelerate the process of intracellular inclusion body formation. J. Neurosci. Res. 2008, 86, 2071–2082. [Google Scholar] [CrossRef]
- Cheng, L.; Zhang, W. DJ-1 affects oxidative stress and pyroptosis in hippocampal neurons of Alzheimer’s disease mouse model by regulating the Nrf2 pathway. Exp. Med. 2021, 21, 557. [Google Scholar] [CrossRef]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef]
- Finkbeiner, S. Huntington’s Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a007476. [Google Scholar] [CrossRef]
- Aleyasin, H.; Rousseaux, M.W.; Phillips, M.; Kim, R.H.; Bland, R.J.; Callaghan, S.; Slack, R.S.; During, M.J.; Mak, T.W.; Park, D.S. The Parkinson’s disease gene DJ-1 is also a key regulator of stroke-induced damage. Proc. Natl. Acad. Sci. USA 2007, 104, 18748–18753. [Google Scholar] [CrossRef]
- Molcho, L.; Ben-Zur, T.; Barhum, Y.; Offen, D. DJ-1 based peptide, ND-13, promote functional recovery in mouse model of focal ischemic injury. PLoS ONE 2018, 13, e0192954. [Google Scholar] [CrossRef]
- Yamane, K.; Kitamura, Y.; Yanagida, T.; Takata, K.; Yanagisawa, D.; Taniguchi, T.; Taira, T.; Ariga, H. Oxidative Neurodegeneration Is Prevented by UCP0045037, an Allosteric Modulator for the Reduced Form of DJ-1, a Wild-Type of Familial Parkinson’s Disease-Linked PARK7. Int. J. Mol. Sci. 2009, 10, 4789–4804. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, D.; Chander, V.; Bandyopadhyay, A. PARIS-DJ-1 Interaction Regulates Mitochondrial Functions in Cardiomyocytes, Which Is Critically Important in Cardiac Hypertrophy. Mol. Cell. Biol. 2020, 41, e00106-20. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Nicholson, C.K.; Polavarapu, R.; Pantner, Y.; Husain, A.; Naqvi, N.; Chin, L.S.; Li, L.; Calvert, J.W. Role of DJ-1 in Modulating Glycative Stress in Heart Failure. J. Am. Heart Assoc. 2020, 9, e014691. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, H.; Du, A.; Li, Y. DJ-1 alleviates anoxia and hypoglycemia injury in cardiac microvascular via AKT and GSH. Mol. Cell. Probes 2020, 53, 101600. [Google Scholar] [CrossRef]
- Amatullah, H.; Maron-Gutierrez, T.; Shan, Y.; Gupta, S.; Tsoporis, J.N.; Varkouhi, A.K.; Teixeira Monteiro, A.P.; He, X.; Yin, J.; Marshall, J.C.; et al. Protective function of DJ-1/PARK7 in lipopolysaccharide and ventilator-induced acute lung injury. Redox Biol. 2021, 38, 101796. [Google Scholar] [CrossRef]
- Liu, X.-W.; Ma, T.; Cai, Q.; Wang, L.; Song, H.-W.; Liu, Z. Elevation of Serum PARK7 and IL-8 Levels Is Associated With Acute Lung Injury in Patients With Severe Sepsis/Septic Shock. J. Intensive Care Med. 2017, 34, 662–668. [Google Scholar] [CrossRef]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef]
- Cheng, Y.-T.; Ho, C.-Y.; Jhang, J.-J.; Lu, C.-C.; Yen, G.-C. DJ-1 plays an important role in caffeic acid-mediated protection of the gastrointestinal mucosa against ketoprofen-induced oxidative damage. J. Nutr. Biochem. 2014, 25, 1045–1057. [Google Scholar] [CrossRef]
- Di Narzo, A.F.; Brodmerkel, C.; Telesco, S.E.; Argmann, C.; Peters, L.A.; Li, K.; Kidd, B.; Dudley, J.; Cho, J.; Schadt, E.E.; et al. High-Throughput Identification of the Plasma Proteomic Signature of Inflammatory Bowel Disease. J. Crohns Colitis 2019, 13, 462–471. [Google Scholar] [CrossRef]
- Dubois, P.C.A.; Trynka, G.; Franke, L.; Hunt, K.A.; Romanos, J.; Curtotti, A.; Zhernakova, A.; Heap, G.A.R.; Adány, R.; Aromaa, A.; et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010, 42, 295–302. [Google Scholar] [CrossRef]
- Shi, S.Y.; Lu, S.-Y.; Sivasubramaniyam, T.; Revelo, X.S.; Cai, E.P.; Luk, C.T.; Schroer, S.A.; Patel, P.; Kim, R.H.; Bombardier, E.; et al. DJ-1 links muscle ROS production with metabolic reprogramming and systemic energy homeostasis in mice. Nat. Commun. 2015, 6, 7415. [Google Scholar] [CrossRef] [PubMed]
- Singh, Y.; Chen, H.; Zhou, Y.; Föller, M.; Mak, T.W.; Salker, M.S.; Lang, F. Differential effect of DJ-1/PARK7 on development of natural and induced regulatory T cells. Sci. Rep. 2015, 5, 17723. [Google Scholar] [CrossRef]
- Yang, J.; Kim, M.J.; Yoon, W.; Kim, E.Y.; Kim, H.; Lee, Y.; Min, B.; Kang, K.S.; Son, J.H.; Park, H.T.; et al. Isocitrate protects DJ-1 null dopaminergic cells from oxidative stress through NADP+-dependent isocitrate dehydrogenase (IDH). PLoS Genet. 2017, 13, e1006975. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharm. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Raninga, P.V.; Di Trapani, G.; Tonissen, K.F. The Multifaceted Roles of DJ-1 as an Antioxidant. In DJ-1/PARK7 Protein; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Moschen, A.R.; Gerner, R.R.; Wang, J.; Klepsch, V.; Adolph, T.E.; Reider, S.J.; Hackl, H.; Pfister, A.; Schilling, J.; Moser, P.L. Lipocalin 2 protects from inflammation and tumorigenesis associated with gut microbiota alterations. Cell Host Microbe 2016, 19, 455–469. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal Model of IBD | Effect on the CNS | Refs. |
|---|---|---|
| TNBS-induced colitis in rabbit | Increased blood-brain barrier permeability | Hathaway et al. [32] |
| TNBS-induced colitis in rat | Elevated blood-brain barrier permeability and reduced endothelial barrier antigen expression | Natah et al. [33] |
| Increased interleukin IL-6 expression in the hypothalamus and cerebral cortex | Wang et al. [34] | |
| DSS-induced colitis in mouse | Elevated TNF-α, IL-1ß, and IL-6 expression in the substantia nigra | Villarán et al. [35] |
| Increased TNF-α and IL-6 expression in the cortex and decreased TJ protein occludin and claudin-5 in the brain | Han et al. [36] | |
| Increased nigral level of IL-1ß and dopaminergic neuron death | Garrido Gil et al. [37] | |
| Increased COX-2 expression in the hippocampus and hypothalamus | Do et al. [38] | |
| α-syn aggregation in the midbrain | Grathwohl et al. [39] | |
| Microglial polarization into M1 and M2 phenotype in the medial prefrontal cortex | Sroor et al. [40] | |
| Increased IL-1ß, IL-6, TNF-α and IL-10 expression in the hippocampus | Gampierakis et al. [41] | |
| NLRP3 activation, amyloid plaque accumulation, and apoptosis in hippocampus, Cortex | He et al. [42] | |
| Elevated IL-1ß and TNF-α expression in the brain | Talley et al. [43] | |
| Increased microglia and astrocyte activation and loss of dopaminergic neurons in the substantia nigra pars compacta after PD inducing MPTP treatment | Gil-Martínez et al. [44] | |
| Increased neurotoxic effect of MPTP treatment | Houser et al. [45] |
| Molecule | Effect on PARK7/DJ-1 | Tissue or Cell Type | Refs. |
|---|---|---|---|
| Negative regulators of PARK7/DJ1 | |||
| H2O2 | Overoxidation | Human brain | [80,81] |
| p53 | Reduced expression | mouse embryonic fibroblasts | [91] |
| BAG5 | Decreased stability | HEK293 human embryonic kidney | [92] |
| MMP-3 | Proteomic fragmentation | CATH.a mouse neuronal | [93] |
| LPS | Reduced expression | HT-29 human colonic adenocarcinoma | [11] |
| TNF-α | Reduced expression | HT-29 human colonic adenocarcinoma | [11] |
| TGF-β | Reduced expression | HT-29 human colonic adenocarcinoma | [11] |
| miR-128-3p | Reduced expression | Human hepatocellular carcinoma | [94] |
| miR-494 | Reduced expression | 3T3-L1 mouse adipocytes and Neuro-2a neuroblastoma | [96] |
| miR-203 | Reduced expression | SW1990/DDP human pancreatic cancer cells | [95] |
| Positive regulators of PARK7/DJ1 | |||
| STAT5A | Increased expression | human leukemic pre-B | [97] |
| SG2NA | Protection from degradation | Neuro2a neuroblastoma | [99,100] |
| IL-17 | Increased expression | HT-29 human colonic adenocarcinoma | [11] |
| H2O2 | Increased expression | HT-29 human colonic adenocarcinoma | [11] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pap, D.; Veres-Székely, A.; Szebeni, B.; Vannay, Á. PARK7/DJ-1 as a Therapeutic Target in Gut-Brain Axis Diseases. Int. J. Mol. Sci. 2022, 23, 6626. https://doi.org/10.3390/ijms23126626
Pap D, Veres-Székely A, Szebeni B, Vannay Á. PARK7/DJ-1 as a Therapeutic Target in Gut-Brain Axis Diseases. International Journal of Molecular Sciences. 2022; 23(12):6626. https://doi.org/10.3390/ijms23126626
Chicago/Turabian StylePap, Domonkos, Apor Veres-Székely, Beáta Szebeni, and Ádám Vannay. 2022. "PARK7/DJ-1 as a Therapeutic Target in Gut-Brain Axis Diseases" International Journal of Molecular Sciences 23, no. 12: 6626. https://doi.org/10.3390/ijms23126626
APA StylePap, D., Veres-Székely, A., Szebeni, B., & Vannay, Á. (2022). PARK7/DJ-1 as a Therapeutic Target in Gut-Brain Axis Diseases. International Journal of Molecular Sciences, 23(12), 6626. https://doi.org/10.3390/ijms23126626